organic papers
Acta Cryst.(2006). E62, o1139–o1140 doi:10.1107/S1600536806003734 Myintet al. C
10H16O2
o1139
Acta Crystallographica Section EStructure Reports Online
ISSN 1600-5368
Adamantane-1,2-diol
Mo Aung Myint, Jason R. Price* and Eng Wui Tan
Department of Chemistry, University of Otago, PO Box 56, Dunedin, New Zealand
Correspondence e-mail: [email protected]
Key indicators
Single-crystal X-ray study T= 83 K
Mean(C–C) = 0.002 A˚ Rfactor = 0.061 wRfactor = 0.176
Data-to-parameter ratio = 26.1
For details of how these key indicators were automatically derived from the article, see http://journals.iucr.org/e.
Received 25 January 2006 Accepted 31 January 2006
#2006 International Union of Crystallography
All rights reserved
In the crystal structure of the title compound, C10H16O2, the
hydroxyl groups are involved in both intra- and intermolecular hydrogen bonding. Molecules are arranged in discrete layers
propagated by a network of O—H O hydrogen-bonding
interactions. The asymmetric unit comprises one chiral molecule but the presence of a crystallographic centre of inversion leads to racemic crystals.
Comment
Being a rigid cage-like molecule with two substituents which are capable of hydrogen bonding, adamantane-1,2-diol, (I), promises to be an interesting candidate as a template for molecular imprinting. There is a chiral centre at position C-2; however, the title compound crystallizes as the racemate.
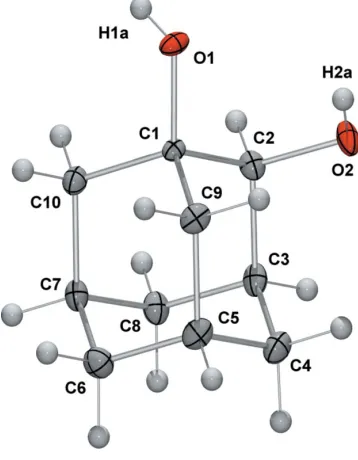
The molecular structure of (I) is illustrated in Fig. 1. In the
[image:1.610.300.361.358.428.2] [image:1.610.240.419.487.717.2]molecule, there is an intramolecular hydrogen bond
Figure 1
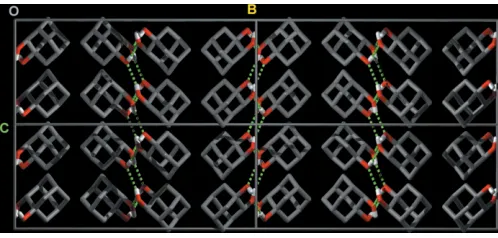
[O2 O1 = 2.8688 (15) A˚ ]. In the crystal structure (Fig. 2), molecules are arranged in discrete two-dimensional layers that lie parallel to the crystallographic ac plane. Intermolecular hydrogen-bonding interactions (Table 1) form a two-dimen-sional scaffold supporting bilayers of molecules of (I).
Experimental
Adamantane-1,2-diol was synthesized via a four-step synthesis according to the methods of McKerveyet al.(1971) and Janjatovicet al.(1980). After repeated recrystallization from methanol at room temperature, colourless block-shaped crystals of (I) were obtained.
Crystal data
C10H16O2
Mr= 168.23
Orthorhombic,Pccn a= 9.6159 (3) A˚
b= 20.6781 (7) A˚
c= 8.3921 (2) A˚
V= 1668.67 (9) A˚3
Z= 8
Dx= 1.339 Mg m
3
MoKradiation Cell parameters from 2252
reflections
= 3.4–32.6 = 0.09 mm1
T= 83 (2) K Block, colourless 0.360.240.10 mm
Data collection
Bruker Kappa-APEX-II area-detector diffractometer
’and!scans
Absorption correction: multi-scan (SADABS; Bruker, 2004)
Tmin= 0.684,Tmax= 1.000
41081 measured reflections
2845 independent reflections 2252 reflections withI> 2(I)
Rint= 0.059
max= 32.7
h=13!13
k=30!31
l=11!12
Refinement
Refinement onF2
R[F2> 2(F2)] = 0.061
wR(F2) = 0.176
S= 1.03 2845 reflections 109 parameters
H-atom parameters constrained
w= 1/[2(F
o2) + (0.0791P)2
+ 1.9937P]
whereP= (Fo2+ 2Fc2)/3
(/)max= 0.001
max= 0.84 e A˚
3
min=0.37 e A˚
3
Table 1
Hydrogen-bond geometry (A˚ ,).
D—H A D—H H A D A D—H A
O2—H2A O1 0.84 2.45 2.8688 (15) 112 O1—H1A O2i 0.84 2.02 2.8625 (15) 176 O2—H2A O1ii
0.84 2.10 2.8633 (14) 152
Symmetry codes: (i)x1
2;yþ1;zþ 1
2; (ii)x;yþ1;z.
H atoms were included in idealized positions and refined using a riding model, with tertiary and secondary C—H bond lengths fixed at 0.99 and 1.00 A˚ , respectively, and the O—H bonds fixed at 0.84 A˚.
Uiso(H) values were fixed at 1.2Ueqof the parent C and O atoms.
Data collection:APEXII(Bruker 2004); cell refinement:APEXII
andSAINT(Bruker 2004); data reduction:SAINT; program(s) used to solve structure:SHELXS97(Sheldrick, 1997); program(s) used to refine structure:SHELXL97(Sheldrick, 1997); molecular graphics:
ORTEP-3(Farrugia, 1997) and POV-RAY (Persistence of Vision, 2004); software used to prepare material for publication: enCIFer
(Allenet al., 2004).
The authors thank Fonterra Co-operative Group and the New Zealand Foundation for Research Science and Tech-nology for their support. Assistance from Professor Sally A. Brooker and Professor Jim Simpson, University of Otago, is also appreciated.
References
Allen, F. H., Johnson, O., Shields, G. P., Smith, B. R. & Towler, M. (2004).J. Appl. Cryst.37, 335–338.
Bruker (2004). APEXII (Version 1.017), SAINT (Version 7.12A) and
SADABS(Version 2004/1). Bruker AXS Inc., Madison, Wisconsin, USA. Farrugia, L. J. (1997).J. Appl. Cryst.30, 565.
Janjatovic, J. & Majerski, Z. (1980).J. Org. Chem.45, 4892–4898.
McKervey, M. A., Cuddy, B. D. & Grant, D. (1971).J. Chem. Soc. C,19, 3173– 3179.
Persistence of Vision (2004).POV-RAY. Version 3.6. Persistence of Vision Pty. Ltd, Williamstown, Victoria, Australia.
[image:2.610.315.564.70.189.2] [image:2.610.314.565.250.404.2]Sheldrick, G. M. (1997).SHELXL97. University of Go¨ttingen, Germany.
Figure 2
Crystal packing diagram of (I), viewed along theaaxis. Key: C grey, H white and O red. Hydrogen bonds are shown as green dashed lines (H atoms not involved in hydrogen bonding have been omitted for clarity). The molecules are arranged in layers parallel to theacplane.
Figure 3
supporting information
sup-1 Acta Cryst. (2006). E62, o1139–o1140
supporting information
Acta Cryst. (2006). E62, o1139–o1140 [https://doi.org/10.1107/S1600536806003734]
Adamantane-1,2-diol
Mo Aung Myint, Jason R. Price and Eng Wui Tan
adamantane-1,2-diol
Crystal data
C10H16O2 Mr = 168.23
Orthorhombic, Pccn
Hall symbol: -P 2ab 2ac
a = 9.6159 (3) Å
b = 20.6781 (7) Å
c = 8.3921 (2) Å
V = 1668.67 (9) Å3 Z = 8
F(000) = 736
Dx = 1.339 Mg m−3
Mo Kα radiation, λ = 0.71073 Å Cell parameters from 2252 reflections
θ = 3.4–32.6°
µ = 0.09 mm−1 T = 83 K Block, colourless 0.36 × 0.24 × 0.10 mm
Data collection
Bruker Kappa-APEX-II area-detector diffractometer
Radiation source: fine-focus sealed tube Graphite monochromator
φ and ω scans
Absorption correction: multi-scan (SADABS; Bruker, 2004)
Tmin = 0.684, Tmax = 1.000
41081 measured reflections 2845 independent reflections 2252 reflections with I > 2σ(I)
Rint = 0.059
θmax = 32.7°, θmin = 2.0° h = −13→13
k = −30→31
l = −11→12
Refinement
Refinement on F2
Least-squares matrix: full
R[F2 > 2σ(F2)] = 0.061 wR(F2) = 0.176 S = 1.03 2845 reflections 109 parameters 0 restraints
Primary atom site location: structure-invariant direct methods
Secondary atom site location: difference Fourier map
Hydrogen site location: inferred from neighbouring sites
H-atom parameters constrained
w = 1/[σ2(F
o2) + (0.0791P)2 + 1.9937P]
where P = (Fo2 + 2Fc2)/3
(Δ/σ)max = 0.001
Δρmax = 0.84 e Å−3
Δρmin = −0.37 e Å−3
Special details
Refinement. Refinement of F2 against ALL reflections. The weighted R-factor wR and goodness of fit S are based on F2,
conventional R-factors R are based on F, with F set to zero for negative F2. The threshold expression of F2 > σ(F2) is used
only for calculating R-factors(gt) etc. and is not relevant to the choice of reflections for refinement. R-factors based on F2
are statistically about twice as large as those based on F, and R- factors based on ALL data will be even larger.
Fractional atomic coordinates and isotropic or equivalent isotropic displacement parameters (Å2)
x y z Uiso*/Ueq
O1 −0.13529 (11) 0.53630 (5) 0.09461 (12) 0.0133 (2)
H1A −0.2032 0.5204 0.1438 0.020*
O2 0.13649 (12) 0.51341 (5) 0.22384 (13) 0.0182 (3)
H2A 0.1086 0.5052 0.1312 0.027*
C1 −0.06880 (14) 0.58392 (6) 0.19276 (15) 0.0095 (2)
C2 0.03234 (15) 0.55025 (7) 0.30695 (16) 0.0134 (3)
H2B −0.0214 0.5206 0.3782 0.016*
C3 0.10813 (15) 0.60078 (7) 0.41011 (16) 0.0146 (3)
H3A 0.1749 0.5786 0.4833 0.017*
C4 0.18742 (16) 0.64722 (8) 0.30032 (18) 0.0173 (3)
H4A 0.2563 0.6227 0.2367 0.021*
H4B 0.2382 0.6795 0.3653 0.021*
C5 0.08549 (16) 0.68195 (7) 0.18849 (17) 0.0163 (3)
H5A 0.1376 0.7126 0.1184 0.020*
C6 −0.02213 (17) 0.71906 (7) 0.28638 (18) 0.0179 (3)
H6A −0.0885 0.7409 0.2140 0.021*
H6B 0.0250 0.7526 0.3509 0.021*
C7 −0.10082 (15) 0.67278 (7) 0.39624 (17) 0.0144 (3)
H7A −0.1707 0.6974 0.4604 0.017*
C8 0.00006 (16) 0.63765 (7) 0.50756 (17) 0.0155 (3)
H8A 0.0472 0.6694 0.5773 0.019*
H8B −0.0520 0.6071 0.5760 0.019*
C9 0.01004 (15) 0.63076 (7) 0.08472 (16) 0.0148 (3)
H9A −0.0558 0.6524 0.0114 0.018*
H9B 0.0788 0.6067 0.0199 0.018*
C10 −0.17528 (15) 0.62165 (7) 0.29281 (17) 0.0140 (3)
H10A −0.2271 0.5914 0.3622 0.017*
H10B −0.2428 0.6432 0.2214 0.017*
Atomic displacement parameters (Å2)
U11 U22 U33 U12 U13 U23
supporting information
sup-3 Acta Cryst. (2006). E62, o1139–o1140
C7 0.0134 (6) 0.0161 (6) 0.0136 (6) 0.0034 (5) 0.0010 (5) −0.0005 (5) C8 0.0161 (6) 0.0187 (6) 0.0117 (6) 0.0029 (5) 0.0009 (5) −0.0006 (5) C9 0.0152 (6) 0.0186 (6) 0.0104 (6) −0.0024 (5) 0.0000 (5) 0.0020 (5) C10 0.0115 (6) 0.0172 (6) 0.0133 (6) 0.0004 (5) 0.0003 (5) −0.0016 (5)
Geometric parameters (Å, º)
O1—C1 1.4343 (15) C5—C6 1.528 (2)
O1—H1A 0.8400 C5—C9 1.551 (2)
O2—C2 1.4387 (17) C5—H5A 1.0000
O2—H2A 0.8400 C6—C7 1.529 (2)
C1—C9 1.5279 (18) C6—H6A 0.9900
C1—C2 1.5327 (19) C6—H6B 0.9900
C1—C10 1.5369 (19) C7—C8 1.530 (2)
C2—C3 1.540 (2) C7—C10 1.544 (2)
C2—H2B 1.0000 C7—H7A 1.0000
C3—C8 1.526 (2) C8—H8A 0.9900
C3—C4 1.534 (2) C8—H8B 0.9900
C3—H3A 1.0000 C9—H9A 0.9900
C4—C5 1.535 (2) C9—H9B 0.9900
C4—H4A 0.9900 C10—H10A 0.9900
C4—H4B 0.9900 C10—H10B 0.9900
C1—O1—H1A 109.5 C9—C5—H5A 109.7
C2—O2—H2A 109.5 C5—C6—C7 110.18 (12)
O1—C1—C9 108.40 (10) C5—C6—H6A 109.6
O1—C1—C2 109.27 (10) C7—C6—H6A 109.6
C9—C1—C2 110.13 (11) C5—C6—H6B 109.6
O1—C1—C10 111.43 (11) C7—C6—H6B 109.6
C9—C1—C10 109.45 (11) H6A—C6—H6B 108.1
C2—C1—C10 108.17 (10) C6—C7—C8 110.58 (12)
O2—C2—C1 112.28 (10) C6—C7—C10 108.60 (12)
O2—C2—C3 107.60 (11) C8—C7—C10 108.19 (11)
C1—C2—C3 110.09 (11) C6—C7—H7A 109.8
O2—C2—H2B 108.9 C8—C7—H7A 109.8
C1—C2—H2B 108.9 C10—C7—H7A 109.8
C3—C2—H2B 108.9 C3—C8—C7 109.98 (11)
C8—C3—C4 110.34 (12) C3—C8—H8A 109.7
C8—C3—C2 108.53 (12) C7—C8—H8A 109.7
C4—C3—C2 108.80 (11) C3—C8—H8B 109.7
C8—C3—H3A 109.7 C7—C8—H8B 109.7
C4—C3—H3A 109.7 H8A—C8—H8B 108.2
C2—C3—H3A 109.7 C1—C9—C5 109.36 (11)
C3—C4—C5 110.04 (12) C1—C9—H9A 109.8
C3—C4—H4A 109.7 C5—C9—H9A 109.8
C5—C4—H4A 109.7 C1—C9—H9B 109.8
C3—C4—H4B 109.7 C5—C9—H9B 109.8
H4A—C4—H4B 108.2 C1—C10—C7 110.23 (11)
C6—C5—C4 109.80 (12) C1—C10—H10A 109.6
C6—C5—C9 109.14 (12) C7—C10—H10A 109.6
C4—C5—C9 108.82 (12) C1—C10—H10B 109.6
C6—C5—H5A 109.7 C7—C10—H10B 109.6
C4—C5—H5A 109.7 H10A—C10—H10B 108.1
O1—C1—C2—O2 58.47 (14) C5—C6—C7—C8 58.37 (15)
C9—C1—C2—O2 −60.49 (14) C5—C6—C7—C10 −60.20 (15)
C10—C1—C2—O2 179.94 (11) C4—C3—C8—C7 57.96 (15)
O1—C1—C2—C3 178.32 (11) C2—C3—C8—C7 −61.17 (15)
C9—C1—C2—C3 59.36 (14) C6—C7—C8—C3 −57.91 (15)
C10—C1—C2—C3 −60.21 (14) C10—C7—C8—C3 60.90 (15)
O2—C2—C3—C8 −176.45 (11) O1—C1—C9—C5 −178.82 (11)
C1—C2—C3—C8 60.90 (14) C2—C1—C9—C5 −59.32 (14)
O2—C2—C3—C4 63.46 (14) C10—C1—C9—C5 59.47 (14)
C1—C2—C3—C4 −59.19 (14) C6—C5—C9—C1 −60.02 (14)
C8—C3—C4—C5 −58.53 (15) C4—C5—C9—C1 59.79 (15)
C2—C3—C4—C5 60.44 (15) O1—C1—C10—C7 −179.64 (10)
C3—C4—C5—C6 58.54 (16) C9—C1—C10—C7 −59.76 (14)
C3—C4—C5—C9 −60.85 (15) C2—C1—C10—C7 60.24 (14)
C4—C5—C6—C7 −58.44 (16) C6—C7—C10—C1 59.48 (14)
C9—C5—C6—C7 60.76 (15) C8—C7—C10—C1 −60.59 (14)
Hydrogen-bond geometry (Å, º)
D—H···A D—H H···A D···A D—H···A
O2—H2A···O1 0.84 2.45 2.8688 (15) 112
O1—H1A···O2i 0.84 2.02 2.8625 (15) 176
O2—H2A···O1ii 0.84 2.10 2.8633 (14) 152