F O R U M
Open Access
Non-model model organisms
James J. Russell
1, Julie A. Theriot
2*, Pranidhi Sood
3, Wallace F. Marshall
3*, Laura F. Landweber
4, Lillian Fritz-Laylin
5,
Jessica K. Polka
6, Snezhana Oliferenko
7,8, Therese Gerbich
9, Amy Gladfelter
9, James Umen
10, Magdalena Bezanilla
5,
Madeline A. Lancaster
11, Shuonan He
12, Matthew C. Gibson
12,13, Bob Goldstein
14, Elly M. Tanaka
15,
Chi-Kuo Hu
16and Anne Brunet
16,17Abstract
Model organisms are widely used in research as accessible and convenient systems to study a particular area or question in biology. Traditionally only a handful of organisms have been widely studied, but modern research tools are enabling researchers to extend the set of model organisms to include less-studied and more unusual systems. This Forum highlights a range of 'non-model model organisms' as emerging systems for tackling questions across the whole spectrum of biology (and beyond), the opportunities and challenges, and the outlook for the future.
Introduction—model organisms for understanding biology
Wallace F. Marshall
The transition in biology from description to mechanis-tic understanding during the 20th century was due in large part to a conscious decision to employ model or-ganisms. The idea of a model organism is that if one wants to study a particular aspect of biology, it makes sense to employ a simple, tractable organism that facili-tates experimental work. Bacteriophage, bacteria, corn, and yeast revealed most of what we know about basic molecular biology of the central dogma, while flies, worms, Arabidopsis, and mice played a similar role in the study of development. The choice of these systems was not arbitrary—they typically were chosen because they were smaller, simpler, and faster growing than more complex organisms such as humans or trees. The term “model organism” was used to indicate a simplified, tractable system that could be used to study a larger
* Correspondence:[email protected];[email protected]
2
Departments of Biochemistry and of Microbiology & Immunology, Howard Hughes Medical Institute Stanford University, Stanford, CA 94305, USA
3
Department of Biochemistry & Biophysics, University of California San Francisco, 600 16th St, San Francisco, CA 94158, USA
Full list of author information is available at the end of the article
theme of biology, and indicated not so much a feature of the system itself, as an attitude on the part of the re-searcher. The“phage group”was not primarily interested in how bacteriophage worked as an end in itself, but ra-ther as a means to a larger end of understanding gene regulation. Bacteriophage were simply a convenient model for studying the bigger question. An experiment could be done hundreds or thousands of times more quickly and cheaply using bacteriophage than human cells, so it is hardly surprising that research in simpler systems rapidly outpaced work in humans. Likewise, flies have been studied for a century not so much because so many people find flies themselves interesting, but be-cause flies made genetic analysis of development easy and fast. In some cases the simplest systems are so sim-ple that they lack key features of interest—for example, bacteria and bacteriophage do not employ the full range of regulatory mechanisms that eukaryotes do—requiring the use of more complex model systems such as yeast for the study of chromatin, meiosis, and other eukaryotic-specific parts of the central dogma. The term model or-ganism was used to describe these systems and conveyed the meaning of“an organism that is inherently convenient to study a particular area of biology”.
Because these model organisms were so convenient, and made progress so rapid, researchers flocked to use them. This led to the development of tools and re-sources specifically for these organisms. Rere-sources in-clude infrastructure, such as databases and strain collections, as well as molecular toolkits and extensive collections of techniques and methods, accumulated over the years by legions of researchers. The develop-ment of these resources happened for model organisms because so many people were working on them, and because they were already so convenient. Why spend time developing methods for a less convenient system? As a result, model organisms began to outpace other systems not only in terms of their inherent convenience, but also in terms of the availability of infrastructure to
study them. This difference was highlighted by the early genome projects, which for obvious reasons focused on model organisms. Once the yeast, Drosophila and Cae-norhabditis elegans genomes were available, it made even less sense to work on anything else. The gap in methodology and resources between the select few model organisms and everything else led to a gradual linguistic shift in how the term “model organism” was understood, so that now many people, when they say model organism, use it not in its original sense, but in-stead in the sense of“an organism for which a wealth of tools and resources exist”.
But it was always appreciated that the major model or-ganisms, while convenient for studying many aspects of biology, weren’t necessarily the best systems for all pos-sible questions. None of the standard models were that good at regenerating, for example, and the extremely sparse coverage of biodiversity represented by standard models meant that evolutionary questions had to be handled very carefully. Model organisms were known for many of these hard-to-reach areas of biology, but they were only model organisms in the original sense (convenient for the study of a biological process) but not in the newer sense (possessing infrastructure and resources). Fortunately, the continual decrease in cost of genomic sequencing has now made it feasible to determine a genome sequence for these classic but under-supported models. Even if, as is often the case, established genome centers refuse to take on a new or-ganism, citing lack of a large community of researchers, it is now possible for individual labs to assemble their own sequences. Once a genome sequence is in hand, many methods, such as RNA sequencing, can be imme-diately applied, and other methods such as CRISPR come into range for development. As a result, there has been an explosion of interest in extending the set of model organisms to include both classic systems long known to be excellent models for particular areas of biology, as well as completely novel systems that have never been explored experimentally but which pose fas-cinating challenges for mechanistic understanding. We will refer to organisms that are models in the original sense, but not yet in the newer sense, as “non-model model organisms”(NMMO).
The present Forum describes the opportunities cre-ated by several such non-model model organisms, as well as the challenges faced in developing methods and resources to study them. The use of genomic informa-tion is a common thread, as is the emphasis on Biology writ large. The organisms discussed here were picked up because of their inherent advantages for studying key biological questions, including pattern formation (diatoms, Stentor), branching morphogenesis ( Physco-mitrella, Ashbya), regeneration (Stentor, axolotl), and
aging (killifish). The diversity of life addressable using NMMO provides new opportunities for studying the evolution of multicellular life (Volvox), body plans (Nematostella, tardigrades, cerebral organoids), and cell biological processes (Oxytricha, Naegleria, Physcomi-trella, fission yeasts). Other questions now being asked using NMMO are more on the sci-fi end of the spectrum, including suspended animation (tardigrades, killfish), phase transitions (Ashbya), and nanobiotech-nology (R bodies, diatoms). All of these examples have one thing in common—exploiting the unique biological features of a special organism to address questions of general importance. These organisms aren’t being stud-ied because they are weird, or because of a fondness for biodiversity, but because they make it easier to ask central questions about biology that have remained un-answered to this day.
Diatoms are ready for their close-up James J. Russell and Julie A. Theriot
Diatoms are unicellular eukaryotes abundant in aquatic environments. Their photosynthesis represents a sig-nificant fraction of global primary productivity and oceanic carbon sequestration [1]. Among cell biologists, however, diatoms are best known for their extraordin-ary and beautifully nano-patterned cell walls, made of silicon dioxide—that is, glass [2] (Fig. 1a). Many of us first encountered diatoms in the form of isolated glass cell walls, known as frustules, mounted on slides used as a measure of resolving power, for dark field alignment of microscopes, or in scanning electron micrographs invoking alien-like architecture (Fig. 1b–d). Their exquis-ite complexity is reminiscent of high-magnification images of snowflakes; however, the diatom frustules are created by genetically encoded developmental programs, and as such are highly reproducible and characteristic for many of the 10,000 to 100,000 estimated species [3]. The variety found even in a single environmental sample can be suffi-cient to inspire endlessly fascinating but very tiny art [4]. How do these cells design and build their glass houses?
ways that enhance their material properties and de-termine their characteristic larger-scale architectures [6, 7]. For diatoms, the fundamental building block of the glass frustule is a near-spherical silicon dioxide nodule about 40 nm in diameter [8]. These precipitated nodules can be formed from soluble silicic acid by sev-eral characterized diatom proteins, notably the silaffins [9]. However, the mechanisms by which the diatom cells assemble these simple structural precursors into highly regular nanoscale and microscale patterns in the valve of the frustule are largely unknown. While
subcel-lular microtubule and actin distributions show
intriguing correlations with some frustule features [10] and pharmacological disruption of microtubules can lead to defects in pattern determination [11], there is essentially no molecular information available about the mechanisms of pattern formation.
Why do we know so little about the cell biological mechanisms of these lovely organisms? One major bar-rier has been the lack of useful classic genetics in any diatom species. All characterized diatoms grow vegeta-tively with diploid genomes, making random mutagen-esis strategies difficult, and while individuals of many species have been observed to undergo a sexual cycle in
nature [2], they have proved to be shy about reliably mating in the test tube. Despite the lack of classic gen-etics, several recent advances have made possible the examination of cell biological questions in diatoms using reverse genetic and post-genomic approaches. The first complete genome sequences for two widely cultivated diatoms, Thalassiosira pseudonana and
Phaeodactylum tricornutum, were released in 2004 and 2008, respectively [12, 13], and several additional se-quenced diatom genomes have been annotated and made publicly available, belonging primarily to species difficult to culture [14, 15]. Although diatoms are phylogenetically distant from the opisthokonts, including fungi and metazoans, where we have developed our most complete understanding of the mechanisms of regulation of eukaryotic gene expression, nevertheless the structure of diatom genomes appears to be sufficiently similar to our familiar model species that it has been possible to generate robustly annotated genomes, with support from diatom EST libraries and RNA-sequencing data.
Critically, several model diatom species have been shown to be genetically transformable by either electro-poration [16] or bacterial conjugation [17], and capable of expressing tagged transgenic protein constructs, including GFP fused to integral components of the glass frustule [18]. In addition, CRISPR-Cas9 genome editing has re-cently become feasible in model diatoms [19, 20], and sev-eral diatom viruses have been sequenced [21] which may provide useful resources for building tools as so many ani-mal viruses have done before them. Owing to the relative ease of adapting these modern genetic tools to diatoms, several labs are engaging additional tools with promising results, including proximity proteomics, live cell micros-copy, and super-resolution fluorescence microscopy [22].
What is next for the study of pattern formation in dia-toms? Unfortunately, the two model diatoms whose ge-nomes have been robustly annotated are not among the more charismatic of this clade. They are both small and structurally simple; indeed Phaeodactylum tricornutum
is poorly silicified and does not produce clear nano-patterns, and the tiny valve ofThalassiosira pseudonana
displays only a rudimentary silica pore structure. A few more annotated genomes for a few elaborately struc-tured but still rapidly growing laboratory strains would be particularly useful; one enticing candidate is Cylin-drotheca fusiformis, a large (~50μm length), motile dia-tom with gracious long arms and a dramatic helical twist along its valvar (cell division) axis (Fig. 1e).C. fusiformis
is amenable to electroporation-mediated genetic trans-formation (unpublished data). In addition to the intrinsic value of diatoms as a case for studying pattern formation and biomineralization, diatoms have also attracted atten-tion as sources for large-scale biomolecule producatten-tion (including lipids for fuel or nutrition) [23], and further
development of molecular methods for diatoms could enable genetic optimization for this purpose. Diatoms are easy to grow and wondrous to observe, and now is an ideal time to apply modern approaches to reexamine the ancient mystery of how diatoms achieve their nano-scale elegance.
Stentor coeruleusas a model for single-cell regeneration
Pranidhi Sood and Wallace F. Marshall
Individual cells can exhibit a great deal of cellular com-plexity in the organization of subcellular features and organelles. These subcellular patterns must be estab-lished and maintained to ensure a cell functions proper-ly—for example, the apico-basal polarity of epithelial cells is required for them to correctly organize in sheets [24]. Cells are not small and amorphous, therefore, but can display complex and invariable internal organization. In fact, some even rival the size and complexity of multi-cellular embryos. How is morphological complexity cre-ated and regulcre-ated within a single membrane bound sac of cytoplasm?
Understanding how analogously complex structures arise in multicellular organisms formed the basis of the field of developmental biology. To study these problems before the availability of genetic tools, early researchers took advantage of systems that could regenerate, for example, the planarian flatworm. Similarly, studying the regeneration of cells can provide a window into the origins of cell geometry by decoupling assembly of structures from the normal growth processes in the cell. Historically, the analysis of regeneration in cells was in large part car-ried out using the giant ciliateStentor coeruleus(Stentor) as an experimental system (Fig. 2). Stentor is a freshwater pond organism, notable for its bright blue coloration and the fact that a single cell can grow to be well over a milli-meter in length. Each cell has an invariable, complex anat-omy with an oral apparatus at one end, a holdfast at the other, and longitudinal stripes of blue pigment, separated by rows of cilia subtended by microtubule bundles, run-ning down the length of the cell.
the fact that its prominent organelles provide a clearly visible, built-in coordinate system. For example, a cell’s entire surface is covered with visible features including long, oriented blue pigment stripes. These provide a frame of reference to determine if a cell has been cor-rectly re-formed or if different parts of the cell are in the correct relative positions. Without these naturally occurring fiducial markers, it would be far more diffi-cult to assess the progress of regeneration. It is known that the nucleus [27, 28] and transcription [29–32] are re-quired for most regeneration processes in Stentor. How-ever, there have been comparatively few molecular studies of regeneration in Stentor, owing in part to the challenges of growing large quantities. Stentor divides with a doub-ling time of several days, and it can take a long time to grow biochemically useful quantities.
Modern genomic technologies remove the need for growing huge numbers of cells and these tools can poten-tially shed light on the molecular mechanisms of regeneration. The key pre-requisite is to have the genome sequence. This was a major challenge in developing Sten-tor as a model system, because genome centers and se-quencing programs proved unwilling to sequence an organism that didn’t already have a large community of researchers studying it. In the end, we took a DIY ap-proach, sequencing and assembling the genome in our own lab in collaboration with the DeRisi lab at UCSF, and then enlisting a team of experts to analyze the resulting
genome. Through teamwork, we were recently able to publish the first Stentor genome [33].
With the Stentor genome in hand, we can begin to de-cipher the molecular networks behind cellular level regen-eration, using techniques such as RNA-seq. We know that transcription is a key requirement for regeneration from foundational biochemical studies [29–32, 34], though spe-cific transcripts driving regeneration were not identified. Also, there is evidence that transcripts synthesized during regeneration can become physically associated with newly formed organelles [32, 35], suggesting that RNA localization might play a role in patterning the cell as it does in the Drosophila melanogaster embryo. In the lab, we have recently developed an RNAi methodology for Stentor [36] that will allow us to functionally test the role of any genes that appear to be specifically induced during regeneration.
We expect that our molecular studies of regeneration and re-patterning in Stentor will reveal fundamental principles of how cells generate and regulate morph-ology, a general phenomenon relevant to the survival of all living systems. Cancer cells, for example, are marked by their loss of subcellular organization and recent studies have linked pathways that regulate polarity to those that suppress tumors [37]. How an individual cell establishes and maintains its subcellular organization is therefore a vital area of study in the initiation of tumori-genesis. Additionally, these studies could inform future
a
b
technology development ranging from novel regenera-tive therapies that reactivate pathways in damaged cells to the creation of self-healing cellular robots.
Life with 16,000 chromosomes:Oxytrichaas a model system to study genome biology, epigenetic inheritance, and somatic differentiation
Laura F. Landweber
The unicellular eukaryote Oxytricha with its extreme genomic architecture, provides a model system for many studies, including chromosome biology, post-zygotic de-velopment, epigenetics, and genome rearrangement.
Oxytricha is a ciliated protist, and like other ciliate gen-era, including Stentor (see the preceding section from Sood and Marshall in this Forum) and the classic
models Tetrahymena and Paramecium, Oxytricha
shares the feature of nuclear dimorphism—the coexist-ence of two types of nuclei in one cell. The archival mi-cronucleus is mostly transcriptionally silent but houses the complete diploid germline genome, which is large at over 500 Mb on at least 75 chromosomes [38], and it produces haploid micronuclei for cell mating. The sec-ond type of nucleus, the somatic macronucleus, is a highly differentiated organelle devoted to gene expression. It develops from a copy of the new zygotic micronucleus after mating. Hence, nuclear differentiation in Oxytricha
offers a microcosm of animal development in a unicellular model, as though development progresses to a sophisti-cated two-cell stage, with differentiated germline and soma, but without cell division. This results in a single protist cell with multiple nuclei. Additionally, some ciliate cells contain more than one nucleus of either type.
The process of forming a new macronucleus involves massive DNA elimination, rearrangement, and amplifica-tion [39]. Remarkably, approximately one-fifth of all Oxy-trichagene loci are scrambled in the germline [38]. These loci require a combination of translocation and/or inver-sion of DNA segments, in addition to DNA deletion, to assemble the expressed macronuclear versions (Fig. 3d). The combination of removal of nearly all intergenic DNA and loss of all satellites and transposons results in a som-atic genome comprising over 16,000 tiny chromosomes that average approximately 3 kb, as well as a much smaller genome size (approximately 50 Mb) [40]. Macronuclear chromosomes lack centromeres but are capped with their own telomeres and telomere binding proteins, and thus classically Oxytricha was one of the first model systems for studies of telomeres and their associated proteins [41, 42]. Amplification of these “nanochromosomes” to an average copy number of ~1900 in the macronucleus [39] creates a DNA-rich and physically enormous (10–30 mi-cron) macronucleus [43] (Fig. 3c).
A phenomenal genome editor, Oxytricha reorganizes its zygotic genome by stitching together over 200,000
germline DNA segments, requiring a nearly equal number of programmed DNA breakage and joining events [38]. These are accompanied by RNA-guided DNA repair [44]. Noncoding RNA molecules are at the heart of orchestrating all these complex events, with long, noncoding, maternal RNA transcripts of the pre-vious generation’s MAC genome supplying templates for chromosome rearrangements [44, 45] and small RNAs marking the specific germline regions to be retained in the new somatic genome [46]. Therefore,
Oxytricha provides a paragon for studies of DNA and chromosome dynamics, noncoding RNA-chromosome in-teractions, DNA breakage, recombination, and repair, and transposon participation [47]. The much reduced size of
Oxytricha’s somatic nanochromosomes also makes them a unique platform for basic studies of chromatin biology (Behet al., unpublished data) as well as gene regulation, genome annotation, and gene discovery [48].
The cytoplasmic mixing that occurs during mating (Fig. 3d), coupled to the fact that the cytoplasm and cell surface material of exconjugant cells explicitly derive from the parental cells, make ciliates excellent model organisms to study epigenetic inheritance (reviewed in [49]). RNA molecules are among the contents that can be directly passed on from parent to exconjugant daughter cell, and RNA-mediated transgenerational inheritance has been demonstrated via injection of foreign long or small RNAs that reprogram genome rearrangement pathways [44, 46]. These approaches for RNA-guided gene editing, facilitated by the natural machinery in the cell, also provide tools for creating somatic gene knockouts or fusion genes [50]. For example, the programmed retention of short genomic re-gions that interrupt reading frames [46] can introduce premature stop codons and lead to the construction of la-boratory strains (that can be stored as frozen cysts) with an inability to express a gene that is normally found in the macronucleus. Additional tool development is underway and still more is needed, for example, to permit parallel screens, but Oxytricha is emerging as a powerful and unique model system to probe features of complex eukaryotic cells and chromatin within the confines of a single cell.
Naegleria gruberi: one cell with two extreme forms of motility
Lillian Fritz-Laylin
noninfectiousNaegleria gruberi(Naegleria) takes on two extremely different forms during its lifecycle: an amoeba that crawls using actin, and a flagellate that swims with two flagella (Fig. 4). The rapid differentiation between these forms makes Naegleria a prime model for under-standing both types of cell motility [52, 53].
Naegleria amoebae crawl and divide without any ob-served cytoplasmic microtubules [53, 54]. Not only does Naegleria undergo closed mitosis (the mitotic spindle is always contained within the nuclear envelope), but the barrel-shaped mitotic spindle lacks centrioles [54] and is thought to be built from divergent tubulin expressed prior to mitosis and degraded after the completion of cytokinesis [55]. Therefore, and remarkably, all cellular functions except mitosis are likely achieved without microtubules—including crawling at speeds topping 120 microns per minute [56]. This makes Naegleria the fastest crawling cell that I am aware of, at least twice as fast as fish keratocytes, Dictyostelium discoideum, and human neutrophils [57–59]. Chemical inhibitor data from other organisms suggest that rapid actin-based cell migration may not require microtubules [60, 61], and Naegleria provides biologically relevant corroboration of this hypothesis. Furthermore, there is a growing appreci-ation that there are multiple modes of cell migrappreci-ation,
each driven by distinct molecular mechanisms [62–64]. Unpublished data clearly indicate that Naegleria, like D. discoideumand some human cells [65, 66], migrates both by using actin-filled pseudopods (a mode we call alpha-motility) and by blebbing (a delamination of the plasma membrane from the underlying actin cortex) (Fig. 4).
The differentiation of Naegleria from crawling amoebae to swimming flagellates involves assembling an entire microtubule cytoskeleton de novo, including two basal bodies (centrioles), the two flagella (cilia) that they pattern, and an entire cortical microtubule array [53, 67–71], as well as coordinating this new cytoskeleton with the pre-existing actin cytoskeleton. The process of differentiation includes transcribing and translating all of the micro-tubule cytoskeletal components—including tubulin—yet takes only 60–90 minutes [53, 67, 69, 72–74].
Differentiation is easily synchronized, with >90% of cells assembling basal bodies de novo within a 5–10-minute window [54, 67, 71]. (In contrast, mammalian cells take on the order of 24 hours to assemble centrioles de novo, typically after large experimental perturbations [75–77].) Recent evidence indicates that only one basal body is formed de novo, with a second in quick succession by mentored (previously “templated”) assembly [78]. The speed and synchrony of Naegleria differentiation makes it
a useful model for studying centriole assembly, and in fact it was the organism in which de novo centriole assembly was first widely accepted [54]. Naegleria is also well suited for understanding how cells build motile flagella, and tran-scriptional synchrony of differentiation has been used to identify novel centriole and flagellar components widely conserved among eukaryotes, including humans [74].
Clearly, Naegleria is an organism uniquely positioned to reveal new insights into both crawling and swimming mo-tility. A completely sequenced genome and publicly avail-able transcriptional profiling of differentiation provide first steps toward harnessing this potential [51, 74]. The greatest roadblock remains the lack of usable molecular transformation and gene manipulation techniques, a hur-dle we and others are actively attempting to overcome.
R bodies: simple, dynamic protein lattices Jessica K. Polka
Long-range biological motion is typically the product of nucleotide-dependent motors. For example, actomyosin contraction, the bacterial flagellum, and intracellular transport along microtubules all rely on nucleotide-dependent processes carried out by complex assemblies of proteins that can be difficult to reconstitute and engineer. Therefore, if we wish to control biological motion for bio-technological applications (for example, in delivering therapeutic cargoes across membrane barriers), we should instead look for simpler systems.
Some force-generating biological machines are com-posed of dynamic lattices of proteins that amplify, through polymerization, the nanoscale conformational changes of their protomers to create large scale motion. Unrelated to one another in sequence or structure, these lattices are present in multiple domains of life. They include forisomes (biological “stop-cocks” that can expand to occlude fluid flow in the sieve tubes of plants [79, 80]), spasmonemes (the motile elements in the stalk ofVorticellathat rapidly contract to withdraw the ciliate to its attachment site [81]), and R bodies (coiled structures formed in the cytoplasm of bacteria that extend, when triggered with low pH, to break membranes). Each of these structures undergoes large scale motion without relying on nucleotide hydrolysis. Because R bodies are large, genetically simple, and chemically ro-bust, they constitute a model system to study the mecha-nisms of controlled self-assembly and conformational rearrangements that drive functionally related protein ma-chines. Furthermore, they have the potential to act as a powerful chassis for engineering actuators for a variety of biotechnology applications.
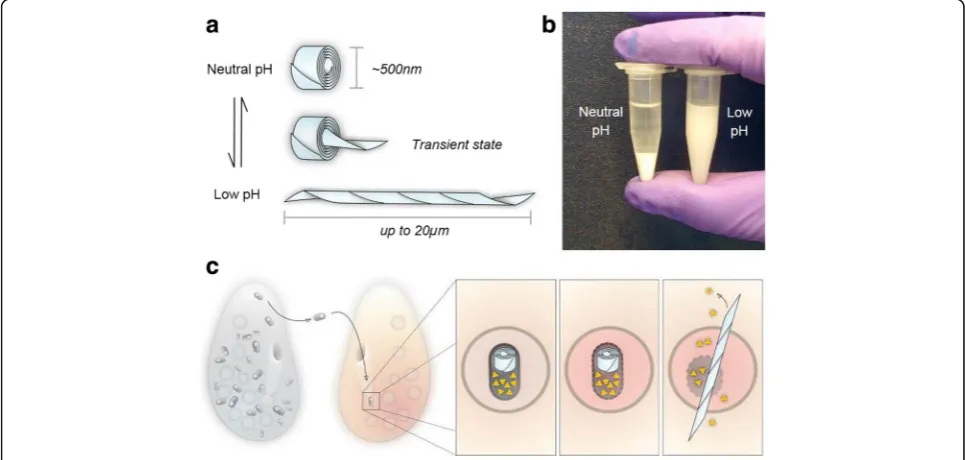
In nature, R bodies are coiled ribbons of protein approximately 500 nm in diameter that bacteria use to de-liver toxins to eukaryotes. Upon exposure to a trigger (such as low pH) they rapidly extend in a telescopic fashion to form lancets tens of microns long. This violent extension is driven by protonation, does not require nucleotide fuel sources, and is fully reversible (Fig. 5). Sequences encoding
R bodies are widespread [82], but they were first discovered inCaedibacter taeniospiralis, an obligate bacterial endosym-biont of Paramecium tetraurelia [83, 84]. Strains of the paramecium that carryC. taeniospiralisare capable of kill-ing paramecium strains that do not carry the endosymbiont. Bacteria containing the R bodies are shed into the environ-ment, where they are consumed by sensitive strains of para-mecium. In the acidic environment of the paramecium phagosome, the R bodies extend to puncture the membrane and mix its contents with the cytoplasm (Fig. 5c) [85]. Feed-ing a diet of purifiedC. taeniospiralisis lethal to these sensi-tive strains, but importantly, feeding purified R bodies is not [86]. This suggests that the R body itself is not toxic; rather, it acts as a delivery system for a co-expressed toxin.
The mechanisms of R body assembly and actuation in response to low pH are largely unknown. Fortunately, R bodies are extremely simple and eminently tractable: one operon encoding four ORFS (each <120 amino acids) is sufficient for their formation inEscherichia coli[87, 88]. By an unknown mechanism, two of these small, nanometer-scale polypeptides can self-assemble into an organized structure many orders of magnitude larger than their indi-vidual size [87]. This process is representative of a broad biological challenge facing all cells: producing long-range order from components that interact at short length scales. By understanding the assembly processes, we may enable the production of actuators of specified mechanical proper-ties and materials with tunable geometries.
These assembled R bodies are resistant to sonication, detergents, and diverse buffer conditions, making them stable and robust force-generating machines that could perform work in a variety of micron-scale devices. At the same time, their behavior can be probed with a simple vis-ual assay: under appropriate buffer conditions, contracted R bodies sediment, while extended R bodies remain in so-lution [89]. This change can be easily seen with the naked eye as well as measured quantitatively by absorbance in a plate reader (Fig. 5b). This assay enables R bodies to be studied in a high-throughput fashion and enabled the identification of mutant R bodies that can transition at lower or higher pH than wild type [89].
R bodies’amenability to engineering suggests that they could be used to deliver biologically active payloads across biological barriers. For example, R bodies could be conjugated to cargo such as DNA, siRNA, labeled proteins, and other chemicals. This strategy could also be used to transform recalcitrant cell types or to deliver high-value cargoes with improved efficiency.
The awesome power of comparative fission yeast genetics
Snezhana Oliferenko
Working on a “non-model” organism can be exception-ally rewarding because of the promise of new biology,
new insights into old problems, and a whole set of new questions to solve. It might be especially tempting to venture to understudied branches of the evolutionary tree to capture the widest possible range of biological di-versity. Yet, based on our experience studying mitotic division in two fission yeasts, Schizosaccharomyces pombe and Schizosaccharomyces japonicus, I want to make a case for exploring the cell biology of closely re-lated species. Such a comparative approach is comple-mentary to the development of new “stand-alone” systems discussed elsewhere in this Forum and I would like to argue that it can be particularly powerful if one of the two species is an established model organism.
Eukaryotes have evolved a staggering variety of mitotic mechanisms. Different species and even different cell types within the same organism may take various routes to mitotic spindle assembly [90], nuclear envelope (NE) remodeling [91], and cytokinesis [92]. For example, all dividing eukaryotic cells must remodel the nuclear enve-lope (NE) to allow chromosome segregation and forma-tion of the daughter nuclei. This invariably involves major rearrangements of the NE–endoplasmic reticulum system coordinated with mitotic spindle dynamics. How-ever, the strategies used to achieve the net result vary from fission of a seemingly intact mother NE into two daughters (“closed” mitosis) to a virtual loss of NE iden-tity in prophase followed by its reassembly around the segregated genomes (“open” mitosis) and several strat-egies in between [91]. Although work by many groups provided detailed insights into the mechanisms under-lying NE remodeling in a number of organisms [93, 94], we understand very little about how these circuitries evolve. Investigating this process in closely related ex-perimentally tractable systems may explain how vari-ation arises in evolution, probe how mitotic nuclear dynamics intersects with the rest of cellular physiology, and inform our understanding of basic NE biology and nuclear origins.
acid phosphatase Lipin supports the differences in mi-totic NE surface area control [98]. These observations may be a starting point in linking the underlying meta-bolic properties of the cell to the emergence of a par-ticular mode of mitosis.
Another important point of divergence between the two sister species relates to differences in regulating chromatin–NE interactions during mitosis, with unex-pected links to nucleolar dynamics. We have shown that although chromosomes must detach from the NE for the duration of mitosis in organisms with closed nuclear division [99, 100],S. japonicushas evolved an anaphase-specific mechanism supporting association between the nuclear pore complexes (NPCs) and chromatin [101]. These interactions executed by the inner nuclear mem-brane protein of the LEM domain family Man1 ensure equal partitioning of the nuclear membrane and effi-cient inheritance of the NPCs by the daughter nuclei, which essentially co-partition with segregating chromo-somes (Fig. 6). It remains to be seen if variations on this mechanism function in other cell types with rela-tively early NE reassembly, for example, during karyo-mere formation in embryonic divisions in some animals [102, 103]. Yet another LEM domain protein, Lem2, functions in supporting timely NE breakage and refor-mation inS. japonicus [96]. Thus, this organism can be used as a simple model to elucidate the poorly un-derstood molecular mechanisms responsible for func-tions of the evolutionarily conserved LEM proteins in maintaining nuclear structure and integrity across eukaryotes [104–106]. Perhaps more surprisingly, in S. japonicus chromatin–NE interactions appear to pro-mote disassembly of the nucleolus that takes place in cells where NE integrity is lost during mitosis but not in organisms with closed nuclear division [101].
The examples above illustrate how comparing related organisms may illuminate evolutionary innovations
required for attaining specific functions or identify con-served elements obscured by grossly different molecular toolkits of distant species. Knowing one of the model systems well—in our case,S. pombe—allows for an easier transition to a related organism, in terms of both recog-nizing interesting phenotypes and adapting existing technical tools. Another important advantage of work-ing in closely related systems is the relative ease of pin-pointing the divergent nodes in otherwise conserved networks supporting cell biological processes, and ret-roengineering the processes with novel properties in the sister species. We have been using the latter ap-proach in our studies of mitotic NE dynamics but also to investigate division plane positioning in the two yeasts. Cells of both species divide in the middle but our studies suggest that S. pombe, a popular model for cytokinesis research, has evolved an unusual medial division ring assembly mechanism based on neofunctio-nalization of one of the recently duplicated anillin para-logs [107]. Importantly, unlikeS. pombethat assembles the actomyosin ring in metaphase and requires a mech-anism preventing its precocious constriction, S. japoni-cusinitiates ring assembly only at mitotic exit, similarly to animal cells [107, 108] (Fig. 6). In general, the metazoan-like properties of S. japonicus division ring assembly combined with mitotic NE breakdown make it an attractive new model for studying regulation and mechanisms of cytokinesis [109].
genetic tools [110, 111] and Nick Rhind who spear-headed the fission yeast clade genomes project. Beyond its utility studying mitotic NE dynamics and other as-pects of mitotic division, S. japonicus could become a great system for investigating the cell biology of hyphal transition [112, 113], energy metabolism [114], and centromere biology [95, 115]. Yet, it is capitalizing on the“experiment of Nature”and using the two sister spe-cies alongside each other that offers conceptually new possibilities in cell biology by expanding itsevolutionary
dimension.
Ashbya gossypiias a model for cytoplasm organization
Therese Gerbich and Amy Gladfelter
All cells face challenges in spatial organization of their contents. One solution used by eukaryotic cells is to create individual membrane-bound compartments for specialized cellular functions. But cells also need to be able to organize all the cytosolic spaces between these compartments so that biochemistry, signaling, and pro-tein production can be tightly regulated. Gradients are one example of organization that is widely observed from micron-sized bacteria to developing insect em-bryos [116, 117]. How cytosolic patterns are established and maintained in spite of the dissipative power of dif-fusion is an area of active investigation in a variety of systems. However, the problem is especially striking in syncytial cells where many nuclei are enclosed in a large, single cytoplasm. Syncytia are found in diverse contexts, including human muscle and placental cells, many fungi, developing insects, and plant tissues. These special cell types face even greater challenges in organ-izing their cytosolic contents, making them a powerful place to study fundamental principles of cytoplasmic organization.
A non-traditional syncytial model system that has been enormously useful for uncovering principles of cytosolic
organization is the filamentous fungus Ashbya gossypii
(Ashbya)(Fig. 7, left). Ashbya is an ascomycete that is closely related to Saccharomyces cerevisiae, but with a rather different lifestyle [118–120]. It has a small gen-ome with ~4000 genes, tolerates replicating plasmids, is readily transformed, and is amenable to molecular genet-ics via gene targeting [121–123]. It has been a valuable system for understanding highly polarized growth and nuclear movement, and is even used as an industrial producer of riboflavin. What makes the system notable for cytoplasmic organization studies is that the many nu-clei in the continuous cytoplasm go through the nuclear division cycle asynchronously [124]. This is remarkable because one would expect that all nuclei would go through the cell cycle together, as global levels of each cyclin protein rise and fall in the common cytoplasm. In studying this paradoxical cell cycle, new modes of cyto-solic organization have been revealed.
Ashbya nuclei create local zones within the cytoplasm to insulate neighboring nuclei from one another so that their division cycles don’t entrain. One way these territories of cytosol form is through an RNA-binding protein that self-associates and positions mRNA tran-scripts of a G1 cyclin near nuclei [125] (Fig. 7, middle and left). The protein contains a large polyQ tract that enables it to form phase-transitioned assemblies that then trap cyclin transcripts in the vicinity of nuclei. If this protein can no longer undergo a phase transition and position cyclin transcripts, nuclei in the shared cytoplasm divide more synchronously [125, 126]. While the ability of proteins to undergo liquid phase transi-tions in vivo and in vitro had been observed previously, studies in Ashbya are one of the best connections of a biological function (positioning of cyclin transcripts to establish and maintain nuclear asynchrony) to regulated protein phase transition.
Cytosolic compartments are not just important for nu-clear cycling in Ashbya but also in cell polarity. Ashbya
grows exclusively in a polarized manner at cell tips such that many polarity axes coexist in a shared cytoplasm and new growth sites have to be established throughout the cell. The same RNA-binding protein that acts near nuclei forms physically distinct liquid compartments at incipient and established growth sites. These liquid droplets are important for positioning RNAs involved in polarized growth and potentially locally regulating their translation [127]. Future work in this organism will be important for understanding how different liquid com-partments form, coexist, and function within a shared cytoplasm. By taking advantage of the special features that the biology of these cells offer, study of Ashbya can identify mechanisms of cytoplasmic organization rele-vant to all eukaryotes. A key lesson from this and all un-conventional systems is that it is important to embrace biological paradoxes and try to figure them out. We have only just begun to tap into understanding the diverse ways cells and tissues solve the problems of staying alive.
Volvox: revealing the origins of multicellularity and germ–soma division of labor
James Umen
Multicellularity evolving from unicellular ancestors is considered one of the major evolutionary transitions [128], with at least two dozen independent occurrences among five major eukaryotic super-clades [129–131]. Approaches aimed at understanding the origins of multi-cellularity, particularly for plants (embryophytes) and an-imals (metazoans), are challenged by the difficulties associated with reconstructing ancient events based on deeply divergent extant multicellular and unicellular lineages. Volvox and its close relatives (the volvocine green algae) are an alternative model for investigating multicellularity, including the early origins of traits such as cell adhesion and intercellular connections, cell-type differentiation with dedicated germ cells and terminally differentiated somatic cells, asymmetric cell divisions, morphogenetic patterning, and sexual dimorphism—all of which are found in more complex multicellular taxa. What differentiates volvocine algae from other taxa and makes them a unique model is their simplicity and their relatively recent transition to multicellularity, with sev-eral well-characterized genera that capture successive in-creases in morphological complexity [132, 133] (Fig. 8a). Conveniently, a close relative of all multicellular volvo-cine algae is the well-studied unicellular model organism
Chlamydomonas reinhardtii (Chlamydomonas) [134, 135] that serves as an outgroup and a proxy for the an-cestral state of the lineage.
Volvox carteri (Volvox) has been used as a “non-mainstream” model for development for several de-cades [136, 137], and belongs to a genus with a distin-guished history that dates back to some of the earliest
recorded light microscopic observations that were made by van Leeuwenhoek [138]. Vegetatively propa-gated Volvox individuals have a spheroidal shape and exhibit a streamlined body plan composed of just two cell types: ~2000 small terminally differentiated somatic cells arranged on the spheroid exterior with flagella ori-ented outward to provide motility, and ~16 large repro-ductive cells called gonidia located on the interior (Fig. 8b–e). All cells are embedded within an extensive clear extracellular matrix that occupies ~99% of the spheroid volume. Volvox somatic cells are similar in size and overall structure to Chlamydomonas cells (Fig. 8f ), though they possess several unique derived features that distinguish them from Chlamydomonas cells in form and function [139] (Fig. 8a).
The appeal of Volvox as a model for investigating the evolutionary and mechanistic bases of multicellularity derives not just from the potential to build on several decades of detailed developmental and genetic studies but also from increasing information on related genera whose genome sequences are enabling the history of developmental innovations and their genetic origins to be reconstructed [140–142]. Intriguingly, all of the developmental regulators identified so far in Volvox (as yet only a handful) have either Chlamydomonas ortho-logs or are members of protein families whose origins can be traced to related families in Chlamydomonas [132]. In some cases orthologs are interchangeable between the two species, raising unanticipated ques-tions about ancestral gene function when the trait gov-erned by the Volvox gene has no obvious parallel in Chlamydomonas (for example, tissue morphogenesis or asymmetric cell division [143]).
As with any experimental system, the questions one can ask are dictated by available tools and resources, some of which have been reviewed recently [135]. The focus here is on Volvox, but it should be understood that Chlamydomonas has available an even more exten-sive molecular genetic toolkit, making it an ideal partner species for integrated and comparative cellular, develop-mental, and evolutionary studies. Three volvocine ge-nomes are now published and publicly accessible—those of Volvox carteri, Chlamydomonas reinhardtii, and
which can be tricky [146–148], but there are newer iso-lates of Volvox that perform well in crosses [149], and in my opinion classic genetic approaches such as UV or chemical mutagenesis followed by screening, outcross-ing, and whole genome re-sequencing will be the pre-ferred way to characterize and identify mutants going forward. Volvox is transformable with exogenous DNA that integrates randomly into its haploid gen-ome, and a variety of transgenes have been expressed including fluorescently tagged proteins, antibiotic re-sistance markers, and endogenous genes [150–154].
Hairpin and antisense-based gene expression knock-downs can also be done, making reverse genetics feasible [149, 155, 156]. While CRISPR-Cas9 editing has not been reported yet for Volvox, it has been successful in Chlamydomonas and could be developed for other volvocine species [157, 158].
As a developmental system Volvox has some ap-pealing features, including organismal size and clarity that make it well suited to live-cell 3D imaging methods, including selective plane illumination mi-croscopy (SPIM) [159]. The chlorophyll and other
(b)
(c)
g
a
Gonium pectorale
Pandorina morum
Eudorina elegans
Pleodorina starrii
Volvox carteri • Genetic modulation of
cell number
• Incomplete cytokinesis • Organismal polarity and
basal body rotation
• Partial inversion
• Cell-cell attachments
• Full inversion • ECM expansion • Partial germ-soma division of labor • Anisogamy
• Oogamy
• Complete germ-soma division of labor • Asymmetric cell
division • Bifurcated
embryonic cell cycle Chlamydomonas
reinhardtii
100 µM 100 µM
b
c
d
e
f
25 µM 5 µM 5 µM
pigments that are in Volvox cells can interfere with live-cell fluorescence detection methods just as in plants, but more discriminating confocal microscopy technology and sensitive detection systems have helped to mitigate this issue [160]. Vegetative Volvox is easy to mass culture and will synchronize under a two-day diurnal cycle (Fig. 8g). In addition, the individual spheroids are large enough that rapid visual screens for developmental mutants can be performed using only a dissecting microscope and micropipette to pick out candi-date mutants. Embryonic cleavage follows a stereotyped pattern, and the lineage relationships between cells during normal development are known; but interestingly, cell-size is the ultimate determinant of germ–soma differenti-ation for post-embryonic cells [161]. Many fascinating and potentially valuable developmental mutants of Volvox that affect specific multicellular and developmental traits have been described [137], and some causative genes have been identified [132], but most mutants are no longer in culture (as yet there is not a routine way to freeze Volvox cultures, though there has been some success reported [162]).
The pieces are in place to implement a promising ap-proach for investigating multicellular innovations and their origins in Volvox using a combination of forward and reverse genetics, and making use of Chlamydomo-nas and other volvocine species to interrogate ancestral gene functions and origins. Descriptions of previously isolated mutants, including several that alter germ and somatic cell fates, provide an indication of the untapped riches of Volvox development [132], and with a se-quenced genome and relatively inexpensive sequencing technology it is now possible to go from mutant pheno-type to causative mutation in a matter of weeks. Once a mutant is identified and verified, its function can be studied not only in Volvox, but also in Chlamydomonas and other volvocine species where the causative gene is likely to have a 1:1 ortholog (or at least a homolog). In some cases Volvox and Chlamydomonas orthologs will be interchangeable in function, and in other cases not; but either result can be informative for understanding the relationships between ancestral and derived traits in the volvocine lineage. A similar combined comparative genomics and experimental approach for investigating evolutionary divergence of mitotic mechanisms in fission yeasts is described by Snezhana Oliferenko elsewhere in this Forum.
The approach outlined above is only one of several productive ways in which Volvox can be used to ask about the origins of multicellular trait innovations, and is meant to stimulate thinking about the new possibil-ities that genomics and other recent technologies add to this model system. Most importantly, the opportun-ities for exciting discoveries far outnumber the re-searchers who are currently using Volvox and its
relatives. A great way to learn more about Volvox and volvocine algae and to tap into this research commu-nity is to attend a meeting [163, 164], or to visit a la-boratory that uses these intriguing microcosms of multicellularity and experience first-hand their beauty and the scientific wonder they inspire.
Physcomitrella patens: harnessing anatomical simplicity to investigate the cellular basis of tissue morphology
Magdalena Bezanilla
Living organisms use their genome as a blue print to build intricately complex and beautiful structures. Within an organism, where every cell has the same blue print, simply controlling how the blueprint is read leads to the formation of different body parts. However, even single cells establish and maintain unique shapes, evi-denced by the vast morphological diversity amongst unicellular organisms. In many organisms, cell shape stems from restrictions imposed by the extracellular en-vironment. Eukaryotes control this by building a wide variety of extracellular structures. For example, animals build bones and shells, plants build polysaccharide walls, and diatoms construct silica-based frustules as described in this Forum by Russell and Theriot. Extra-cellular structures, which often are patterned over macroscopic scales, impose constraints on both cellular and tissue morphology. Yet, individual cells are respon-sible for depositing extracellular matrix. Thus, how or-ganisms control this large-scale patterning of their extracellular matrices is an open question.
To gain insight into this question, it would be ideal to work on an organism whose body plan enables im-aging of individual cells within tissues, and that builds a complex extracellular matrix in the context of a variety of tissues throughout development. Although land plants, with their polysaccharide walls and their indeterminate growth, certainly satisfy the latter cri-terion, access to individual cells within all tissues is challenging in the vast majority of vascular plants. In contrast, the moss Physcomitrella patens (Physcomi-trella) satisfies both criteria. The Physcomitrella body plan is simple, with most tissues only a single cell layer thick, thereby providing an excellent system with which to dissect intracellular control in pattern-ing of the extracellular matrix.
events, whereby a subapical cell undergoes an asym-metric division generating a new apical stem cell that gives rise to a daughter filament. As the plant matures, the apical cells differentiate into a second cell type known as caulonemal cells (Fig. 9), which are character-ized by faster growth and obliquely positioned cell plates. Caulonemal cells are developmentally distinct as they can grow in the absence of light whereas chlorone-mal cells cannot [165].
The filamentous network is the juvenile state of the plant and establishes a radial symmetry (Fig. 9). This network begins to mature into the adult plant by under-going an additional developmental transition character-ized by the emergence of buds (Fig. 9) from subapical cells. Buds represent a switch from two- to three-dimensional growth. The bud initially resembles a new branch but the apex of the cell is more rounded and the first division is oblique (Fig. 9), establishing the apical basal axis. Both daughter cells divide perpendicularly to
the first oblique division [166]. This division generates the apical stem cell in the bud. The bud eventually de-velops into gametophores that have leaf-like structures known as phyllids (Fig. 9). Phyllids emerge regularly off the gametophore and are only a single cell layer thick.
Within the vegetative state of Physcomitrella, both ju-venile and adult tissues are a single cell layer thick and thus readily accessible to microscopic observation. Fur-thermore, these tissues grow via distinct mechanisms. The juvenile state grows two-dimensionally by polarized secretion of extensible cell wall material to the tip of the apical stem cell, enabling turgor-driven cell expansion only at the cell apex. In contrast, the adult state switches to three-dimensional growth characterized by diffuse cell expansion. Strikingly, the gametophore is generated from a single apical stem cell [166], a dramatic simplifi-cation in comparison to seed plants, which have an ap-ical domain known as the meristem comprised of several layers of undifferentiated stem cells.
Branch
Bud Bud Bud
Phyllid Caulonemal Cell
e r o h p o t e m a G a
t a m e n o t o r P
Chloronemal cell
50 µm
20 µm
20 µm
20 µm 20 µm 50 µm
Even with the relatively simple anatomy of Physcomi-trella, continuous imaging over developmental time of events that occur in the denser regions of the filament-ous network, such as caulonemal cell maturation and bud formation, has been challenging. Phyllid expansion that occurs in the air and in three dimensions was also not accessible to high-resolution imaging. Recently these limitations have been largely overcome by the ability to grow Physcomitrella in custom-made microfluidic im-aging chambers for weeks [167], providing the unique opportunity to observe protonemal tissue differentiation, bud formation, and phyllid expansion at cellular and subcellular resolutions.
Another feature that makes Physcomitrella a particu-larly useful model system is the ability to propagate the plant vegetatively. Upon mechanical disruption, cells in the damaged tissue de-differentiate into chloronemal cells, re-establishing a new plant. This effectively enables indefinite propagation of any Physcomitrella line, which is especially useful for mutant strains with developmen-tal defects. As an extreme example of vegetative propa-gation, it is possible to remove the cell wall from Physcomitrella tissue enzymatically, generating a suspen-sion of protoplasts, which given appropriate osmotic conditions then rebuild their walls and generate a new plant resembling one germinated from spores.
Protoplasts are also easily transformable with DNA [168] opening the door to genetic manipulations. Among these is the ability to use homologous recom-bination for gene targeting [169–171], a feature unique to mosses amongst land plants, which has made it possible to generate lines that express native proteins fused to reporter genes [172] or fluorescent proteins expressed from their own genomic context [173]. Most recently CRISPR-Cas9-mediated gene targeting has also been shown to generate genomic lesions effectively [174, 175]. Because juvenile and adult tissues are hap-loid, a genomic lesion immediately results in a mutant. Additionally, RNA interference (RNAi), which can tar-get multiple genes simultaneously, can be performed transiently [176, 177] or inducibly [178], enabling loss-of-function studies of whole gene families. Finally, since whole-genome sequencing has become routine, it is also possible to identify genomic lesions introduced by random mutagenesis [179].
The extensive genetic tool box coupled with facile imaging of single cells within the context of whole tissues uniquely positions Physcomitrella among land plants as an excellent model organism. In addition to interrogating how molecules within individual cells pattern extracellular matrix over macroscopic length scales, Physcomitrella provides the opportunity to an-swer key questions in plant cell and developmental biology.
Cerebral organoids model human brain development and disease
Madeline A. Lancaster
For centuries, the human brain has been one of the most difficult organs to study. The brain is what makes us unique, both as individuals and as a species. But for this very reason, its particular features are impossible to study in other organisms, and ethical and methodo-logical limitations prevent us from directly studying it mechanistically. So while animal models have provided insight into what it is to be a vertebrate, a mammal, or even a primate, there still remain many questions sur-rounding what it is to be human. For example, while neural stem cells behave in much the same fashion in all vertebrates, their neurogenic potential is greatly in-creased in humans, giving rise to over a thousand times the number of neurons in a mouse brain [180], and a brain that is over three times larger than our closest rel-atives, chimpanzees and bonobos [181]. Furthermore, there are important differences in cytoarchitectonic organization, such as the presence of grey matter mini-columns [181] and numerous unique interneuron popu-lations in the cortex [182, 183], and overall denser, more complex dendritic trees and spines [184].
Because of these unique features, it has proven diffi-cult to recapitulate many human neurological disorders accurately in mouse models. For example, primary microcephaly (small head size) in humans is caused by homozygous null mutations in any of a number of cen-trosomal or DNA repair genes, yet when these muta-tions have been introduced in mice, the effect on brain size is minimal [185]. Likewise, mouse models of human mutations seen in neurodegenerative conditions fail to display the full range of defects, such as both plaques and tangles seen in brains of patients with Alz-heimer’s disease [186]. These are just a couple of the numerous failures to model human neurological con-ditions in traditional animal models, which unfortu-nately has led to a drying up of the drug pipeline in this area, and a lack of further interest on the part of the pharmaceutical industry [187].
for generation of larger, more complex rosettes as a result of culturing in 3D before plating tissues in 2D. Building upon these studies, in 2013, we developed a completely 3D model system of human brain deve-lopment: so-called cerebral organoids [192]. Because of their reliance on endogenous signals, cerebral organoids are capable of remarkable self-organization resulting in complex tissues containing a variety of interconnected brain regions. That same year, Kadoshima et al. estab-lished a 3D method for generation of forebrain tissues [193], and in 2015, Paşca et al. developed a method for generating spheroids containing cortical rosettes [194].
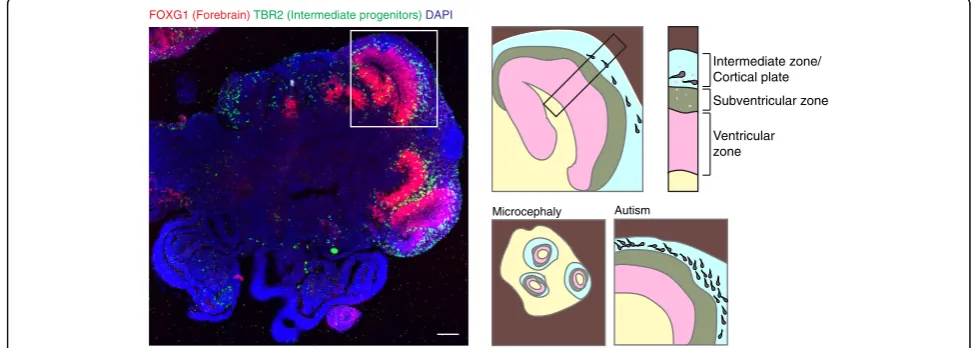
Overall, the methods that have arisen in the past 5 years have revealed the remarkable ability of stem cells to self-organize and form tissues reminiscent of the early developing brain. While cerebral organoids contain a variety of brain regions with remarkable complexity, spheroids generated with exogenous patterning factors and small molecules more reproducibly generate fore-brain and cortical rosettes [195]. But one thing all the methods have in common is the ability to accurately model the behavior of neural stem cells and their organization into discrete progenitor zones highly rem-iniscent of the tissue architecture in vivo. Because of their organization, species-specific differences in tissue architecture and stem cell behavior can be detected in neural organoids (Fig. 10). For example, human cerebral organoids display large numbers of outer radial glia [192, 193], an extra population of neural stem cells that is highly abundant in the developing primate brain, but limited in mice. Furthermore, differences in both neural
stem cell division dynamics and fate have recently been described between human and non-human primate organoids [196, 197].
The fact that brain organoids display human-specific features holds great promise for their use in modeling neurological disorders. Indeed, despite their very recent development, neural organoids have already been dem-onstrated to model features of microcephaly [192], aut-ism [198], lissencephaly [199], and even Zika virus infection [200, 201]. A further testament to their utility is the increasing adoption of these methods in numerous independent laboratories. As with many novel technolo-gies, widespread adoption takes time and so from the cerebral organoid paper in 2013 through 2015 only four publications made use of 3D neural organoids. But last year alone this number jumped to 19 and there is no sign of slowing in the immediate future. While it is still early days, the hope is that the application of brain orga-noid methodologies to the study of an increasing num-ber of neurological syndromes will provide a treasure trove of new insight into disorders of this previously en-igmatic organ.
Nematostella vectensis: born to be a starlet Shuonan He and Matthew C. Gibson
Cnidarians have long attracted attention from biologists and it is easy to see why. From Abraham Trembley’s classic illustrations of regenerating hydra to Ernst Haeckel’s vivid depiction of discomedusae and sea anem-ones inArt Forms in Nature, these delicate creatures ex-hibit an exotic beauty [202, 203]. For contemporary
Ventricular zone
Microcephaly Autism
Subventricular zone Intermediate zone/ Cortical plate
FOXG1 (Forebrain) TBR2 (Intermediate progenitors) DAPI
studies of evolutionary cell and developmental biology, cnidarians have begun to offer much more than simple visual appeal. Widely accepted as the sister group to bilaterian animals, cnidarians possess apparent radial symmetry, lack definitive mesoderm, and have only a single opening that functions as both mouth and anus [204, 205] (Fig. 11). Beyond aesthetic intrigue, these morphological distinctions indicate key evolutionary transitions in the bilaterian lineage after the split of both phyla from their common ancestor, making cnidarian biology central to our understanding of animal evolu-tion. Nevertheless, more than 250 years after Trembley’s pioneering work, we still know surprisingly little about the molecular mechanisms that dictate the distinguish-ing morphological features of cnidarians. One major obstacle has been the absence of a singular cnidarian species that is equally tractable for developmental, cellu-lar, and genomic analysis.
Addressing this issue, the starlet sea anemone Nema-tostella venctensis (Nematostella) has emerged at the forefront of cnidarian model systems with the potential to serve broad research interests.
Nematostella is an estuarine, burrowing sea anemone, first described and named by Thomas Stephenson in 1935 [206]. In the wild, they can be found in brackish ponds or marshes along the coast with recorded salin-ities ranging from 8.96 to 51.54% and water tempera-tures from−1 to 28 °C [207, 208]. Adaptation to such an
ever-changing habitat might explain why Nematostella is exceptionally easy to culture in the laboratory compared with most other cnidarian species. Nematostella belongs to the class Anthozoa, which consists of corals, sea anemones, and sea pens. Phylogenic analysis based on morphology, rRNA, 18S rDNA, and mtDNA data placed Anthozoa at a more basal position within the cnidarian phylum [204, 209–212]. Indeed, it has been proposed that the Anthozoan life history (with only a sessile polypoid adult form) represents the ancestral state of cnidarians [213–215]. If the polyp-first hypothesis is cor-rect, comparative studies using Nematostella are ideal for reconstructing morphological traits of the putative bilateria–cnidaria common ancestor.
Nematostella is a dioecious species. Although sexual plasticity has been reported in other Anthozoans, this phenomenon has not been observed in Nematostella [216, 217]. In the lab, spawning can be induced easily by subjecting sexually mature animals to a combination of light and heat shock [208, 218, 219]. During spawning, females produce gelatinous egg masses, each containing hundreds of eggs, while males release sperm directly into the water. This highly controllable spawning process en-ables access to large quantities of synchronized develop-ing embryos that are amenable for further experimental manipulations. Nematostella has a simple Anthozoan life history with no medusa stage. The fertilized egg under-goes a series of“chaotic”cleavages, and quickly forms a
? ?
? ?
Deuterostomia
Protostomia
Hexacorollia
Octocorollia
Staurozoa
Cubozoa
Scyphozoa
Hydrozoa
Porifera
Ctenophora (Nematostella)
Anthozoa
Medusazoa
Bilateria
Cnidaria
Mesenteries or Septum
b
a
Physa
Mouth
0.5mm Pharynx
Tentacles
well-organized single epithelial layer. Epithelial polarity is established in early cleavage stages, providing a perfect system to study epithelial formation, growth, and mor-phogenesis during early embryogenesis. Embryos gastru-late around 20 hours post fertilization, developing only two germ layers, the ectoderm and the endoderm (also referred to as the entoderm or endomesoderm). Within 48 hours post-fertilization, the embryo develops into a fully ciliated, free-swimming planula larva and starts to escape from its surrounding gelatin matrix. By day seven, elongated planulae settle down and metamorph-ose into polyps bearing four tentacles [220, 221]. Under optimal conditions, it takes 2 to 3 months for a juvenile polyp to reach sexual maturity. Once sexually mature, spawning can be induced every 2 to 3 weeks year round without damaging the animals. Nematostella can also undergo asexual reproduction, which usually occurs via transverse fission through the body column, and can be triggered by extensive feeding, even prior to sexual mat-uration [222]. Interestingly, the life span of Nematostella remains undetermined as it apparently exceeds the “life span”of a PhD student or postdoc.
Nematostella was the first cnidarian to have its whole genome sequenced. The high-quality genome sequence revealed the presence of the majority of the gene re-pertoire for bilateria development and biochemical processes in the eumetazoan ancestor [223]. More strikingly, at the genomic level, vertebrates share more similarities with Nematostella than with ecdysozoans (including, for example, fruit flies and nematodes) [223–226]. Despite the notable conservation of intra-genic sequence and gene structures, the conservation of function as well as regulation of these genes remain poorly explored. Fortunately, a rapidly expanding Nematostella toolkit will fulfill this purpose. Morpho-lino and mRNA delivery via microinjection has proven to be a powerful approach to manipulate gene expres-sion level [227]. Meganuclease-mediated transgenesis is also well established, which helped the generation of several tissue- and cell lineage-specific reporter lines [228–230]. Genomic approaches such as ChIP-seq enable the identification of potential enhancer/promoter regions for certain genes and allow a careful dissection of the gene regulatory network [231]. Most importantly, to our know-ledge, Nematostella is the only cnidarian system where TALEN- and CRISPR/Cas9-mediated genome editing has been reported [232, 233]. The ability to generate knock-out as well as knock-in mutants opens up new possibilities and finally permits sophisticated genetic analysis of gene functions in a cnidarian species.
Over the past decade, studies on Nematostella have shed light on a few fundamental innovations of bila-terian evolution, including the determination of body axis [233–236], the origin of mesoderm [237, 238],
and the emergence of a centralized nervous system [229, 230, 239, 240]. Through these studies, a surpris-ing new picture is emergsurpris-ing of a morphologically and genomically complex eumetaozoan ancestor. Paralleled by progress in other cnidarian model systems [241–244], future research using Nematostella will provide new in-sights into common molecular mechanisms behind the di-versity of life and promises to reshape our understanding of animal evolution.
Water bears: evolution of body forms and survival of extremes
Bob Goldstein
In May 1997, a new molecular phylogeny of the animals revealed that C. elegans and Drosophila were much more closely related than had been thought [245]. Pre-vious phylogenies had placed the nematodes (which include C. elegans) and arthropods (which include
Drosophila) so distantly from each other that arthro-pods were thought to be even more closely related to
us than they were to nematodes. But the new work revealed that these two groups were united along with a handful of hard-to-pronounce animal phyla: ony-chophorans, kinorhynchs, priapulids, nematomorphs, tardigrades, and later the loriciferans as well. I thought that this branch of the tree of life would be a terrific place to look for new models for comparative biology that could take advantage of the two strong model sys-tems nearby. I was especially interested in studying how developmental mechanisms evolved in ways that produced diverse animal forms, and I figured that C. elegans and Drosophila could be rich and ongoing sources of information for comparative studies.
Tardigrades, better known as“water bears”, are eight-legged microscopic animals (Fig. 12). These animals live just about everywhere, and remarkably, they survive desiccation, so they can be found readily by placing clean biological substrates such as mosses or lichens in spring water. We were fortunate that amateur scientist Bob McNuff had been growing water bear cultures con-tinuously in his home for two decades [246], apparently overcoming historical difficulties with keeping cultures long-term [247], and he generously shared his culture methods. And another lab had begun to collect se-quence data [248]. The species we had chosen, which Roberto Bertolani kindly identified for us as Hypsibius dujardini[246], has a short, two-week generation time, and is easy to keep as living cultures in the lab or as frozen stocks [246].


![Fig. 3. Oxytrichaversion; segment 2 is inverted; telomeres are shown ascell degrades, leaving telomere-to-telomere RNA transcripts behind to guide rearrangement [44, 45]](https://thumb-us.123doks.com/thumbv2/123dok_us/8907788.1835042/7.595.59.538.84.558/oxytrichaversion-inverted-telomeres-degrades-telomere-telomere-transcripts-rearrangement.webp)