Ch a p e r o n i n 60: Mo l e c u l a r, Ce l l u l a r a n d St r u c t u r a l St u d ie s
TO De h n e it s Ro l e a s a n In f o r m a t i o n Mo l e c u l e
T h e s is s u b m itte d b y
Sa h a r Kh a n
fo r th e d e g re e o f
Do c t o r o f Ph il o s o p h y
in th e
F acu lty o f M e d ic in e
U n iv e rs ity o f L o n d o n
D e p a rtm e n t o f O ra l a n d M ax illo fa cia l S u rg e ry
E a stm a n D e n ta l In s titu te fo r O ra l H e a lth C are S cien ces
U n iv e rs ity C o lleg e L o n d o n
256 G ra y 's I n n R o a d
L o n d o n , W C1X-8LD
-
2000
All rights reserved
INFORMATION TO ALL USERS
The quality of this reproduction is dependent upon the quality of the copy submitted.
In the unlikely event that the author did not send a complete manuscript and there are missing pages, these will be noted. Also, if material had to be removed,
a note will indicate the deletion.
uest.
ProQuest U156832
Published by ProQuest LLC(2016). Copyright of the Dissertation is held by the Author.
All rights reserved.
This work is protected against unauthorized copying under Title 17, United States Code. Microform Edition © ProQuest LLC.
ProQuest LLC
789 East Eisenhower Parkway P.O. Box 1346
DECLARATION
I hereby certify that the work embodied in this thesis is the result of
This thesis owes its existence to a dedicated group of scientists, namely, Prof. Brian
H enderson, Dr Sean Nair, Dr Peter Tabona, Dr M onika Preuss and Dr Anita Skinner.
Their continuous support, eissistance and inspiration played a pivotal role in bringing
this project to fruition. I feel extremely fortunate to have been given the opportunity to
be tau g h t by, and to w ork w ith such a talented group of individuals.
Thank you very much.
I w ould also like to take the opportunity to thank the collaborators of this project w ho
diligently w orked along side me, especially Prof. Tony Coates of St. George's H ospital
Medical School, London, Dr Mike Keller of Imperial College London and Ms Angela
Paul of the Institute of Cancer Research, C hester Beatty Laboratories, London.
The last three years also enabled m e to make some w onderful friends. In addition to
the nam es listed above, I w ould like to express m y gratitude to Lindsay Richez, Mike
Stevens, D r Julia Ranford, Julia Banks, Samantha Packer and Dr M anipreet Bhatti.
Of course, 250 pages of text and diagrams could not be w ritten w ithout technical
failures and software inadequacies. Fortunately, Steven H artland and Eric Fosdike
w ere constantly at hand to tackle and solve these m inor disasters. I am indebted to
both of you.
Finally, I w ould like to thank m y m entor and m y best friend Nicholas Shepperd for his
This w ork is dedicated to m y parents, Salim & Raana and to m y sisters,
Experimental evidence obtained in this thesis establishes th at the purified,
recombinant, E. coli chaperonin 60 protein, GroEL, is a potent stim ulator of cytokine
production by hum an peripheral blood m ononuclear cells (PBMCs) and th a t this
cytokine inducing activity is not due to LPS, or to the proteins th a t contam inate
'purified' GroEL. Suprisingly, trypsinization of GroEL failed to inhibit the cytokine-
inducing activity of this protein, and by use of reverse phase HPLC, MALDI-MS and
N-term inal sequencing, the bioactive GroEL tryptic peptides w ere identified. The
question as to w hether physical interaction was required betw een GroEL and PBMCs
in order to activate the cells was answ ered by flow cytometry, w hich dem onstrated
th at leukocytes responding to GroEL, bind the protein w hereas cells unresponsive to
GroEL do not. Single- and double-labelling experim ents revealed th at only a
proportion of CD-14 positive monocytes in PBMCs bind GroEL. GroEL w as also show n
to stim ulate m acrophage cells and cell lines, w hich suggests th at the proportion of
monocytes observed to bind GroEL m ay have a m ore macrophage-like m orphology.
Affinity chrom atography of surface biotinylated PBMCs on GroEL affinity m atrices
indicates a 100-150kDa and a 35-40kDa band as possible receptors for GroEL, or as p art
of the GroEL receptor complex. It has also been dem onstrated that GroEL does not
stim ulate cells via CD14 (a know n LPS receptor) as this protein rem ains active in the
presence of anti-CD14 antibodies and is unable to stim ulate the CD14 positive cell line
M ono Mac-6.
These studies support the hypothesis th at chaperonin 60 is a bacterial virulence factor
T
a b l e
o f
C
o n t e n t s
D e c l a r a t i o n ... 2
A c k n o w l e d g e m e n t s ... 3
A b s t r a c t ...5
L i s t o f F i g u r e s ... 12
L i s t o f T a b l e s ... 15
A b b r e v i a t i o n s ... 16
C h a p t e r 1 ...17
I n t r o d u c t i o n...17
1.1 History of the heat shock response... 17
1.2 The heat shock or stress resp o n se...18
1.3 The molecular chaperones...19
1.3.1 Definition of molecular chaperones...19
1.3.2 Classification of the molecular chaperones...20
1.4 The ch ap eronins... 22
1.4.1 Introduction and classification...22
1.4.2 Structure of GroE chaperonins...23
1.5 The Escherichia coli chaperonin 60, GroEL...25
1.5.1 Structure of the GroEL oligom er...28
1.5.2 GroEL m ediated protein folding...29
1.6 Chaperonins in disease... 32
1.6.1 Diseases caused by chaperonin m ediated protein fo ld in g... 32
1.6.2 Diseases caused by non-folding biological actions of chaperonins... 32
1.6.2.1 Cellular location of chaperonins... 33
1.6.2.2 Chaperonins induce cytokine production and upregulate adhesion molecules 35 1.6.2.3 Cell m ediated im m unity to chaperonins... 37
1.6.2.4 Role of chaperonins in the pathogenesis of arterial diseases (atherosclerosis).. 38
1.6.2.5 Chaperonins and autoim m une diseases...40
1.6.2.6 Bone resorption by chaperonins... 43
1.7 S u m m ary... 44
Ch a p t e r 2 ... 46
Ma t e r ia l s a n d Me t h o d s...46
2.1 M aterials... 46
2.1.1 Chemical reagents and k its ... 46
2.1.2 E quipm ent... 48
2.1.3 C onsum ables... 49
2.1.4 Commercial antibodies... 49
2.2 Isolation and purification of GroEL in Escherichia coli...49
2.2.1 Cloning and expression of GroEL in E. coli... 49
2.2.2 Growth of E. coli recom binant strain TG2 / p A M l...50
2.2.3 Cell lysis by sonication... 50
2.2.4 Removal of nucleic acids and protein precipitation...51
2.2.5 Gel filtration chrom atography... 51
2.2.6 Anion exchange chrom atography... 52
2.2.7 Concentration and dialysis of purified GroEL...52
2.2.8 Storage of G roEL... 52
2.3 Protein assays... 53
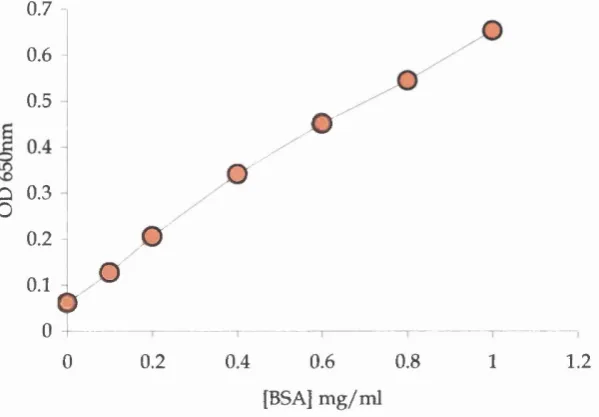
2.3.1 Bio-Rad detergent compatible (DC) protein a s sa y ...53
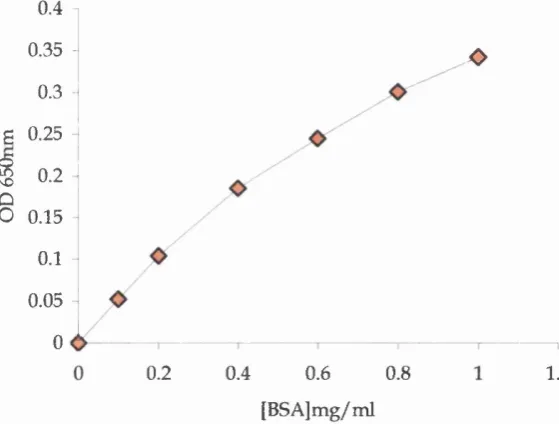
2.3.2 The Bradford assay... 55
2.3.3 Estim ating GroEL concentration using its extinction coefficient... 56
2.3.4 S u m m ary ... 56
2.4 Protein detection... 57
2.4.1 SDS polyacrylam ide gel electrophoresis (SDS-PAGE)...57
2.4.2 Gel sta in in g ... 59
2.4.2.1 Coomassie b lu e ... 59
2.4.2.2 Colloidal Coomassie Blue... 60
2.4.2.3 Silver sta in ... 60
2.5 W estern im m unoblotting... 60
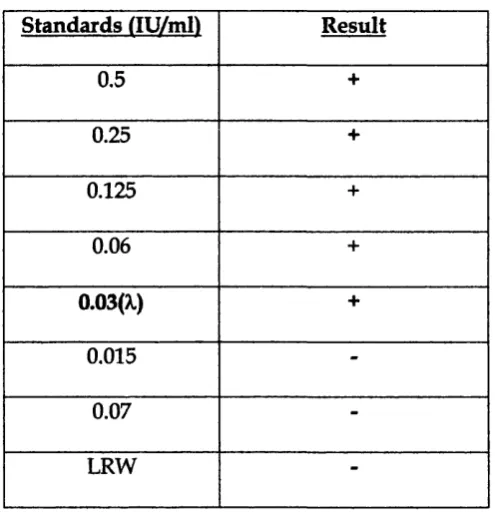
2.6 The Limulus amoebocyte lysate (LAL) assay ...64
2.6.1 Preparing standards and samples for the Limulus a ssa y ...64
2.6.2 Setting up the LAL assay...64
2.6.3 Results and interpretation... 65
2.7 Cell c u ltu re ... 67
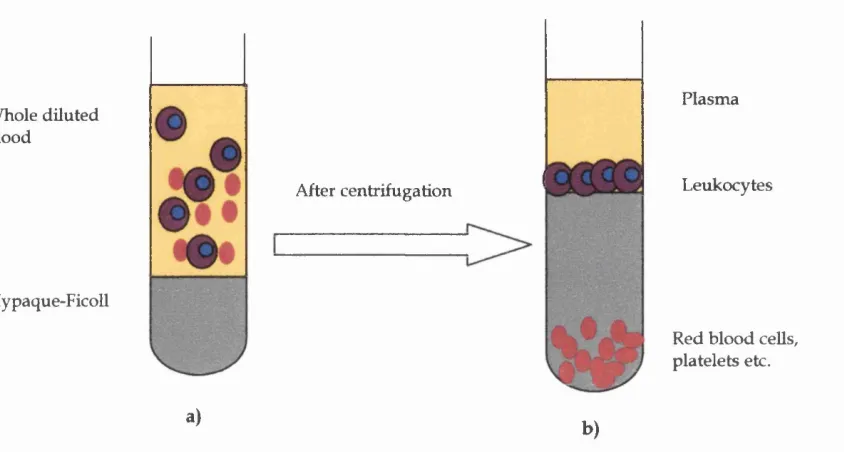
2.7.1 Preparation of peripheral blood m ononuclear cells from buffy coat b lo o d ... 67
2.7.2 Culture of anti-CD14 producing m ouse hybridom as 26ic and 60bca... 70
2. 8 Im m unological assay of cytokines...71
2.8.1 Enzyme Linked im m unosorbent assay (ELISA)...71
2.8.2 ELISA to detect levels of IL-6... 73
2.8.3 ELISA to detect levels of IL-1(3...75
2.8.4 ELISA to detect levels of T N F -a...75
2.8.5 ELISA to detect levels of IL-8...76
2.8.6 ELISA to detect levels of IFN-y... 77
2.8.7 ELISA to detect levels of lL-10...78
2.8.8 ELISA to detect levels of GM-CSF...78
2.8.9 ELISA to detect levels of IL-12...79
2.8.10 M urine TNF- a ELISA...79
Table of contents
Ch a p t e r s... 8 i Pr e p a r a t i o n o f Ho m o g e n e o u s Re c o m b i n a n t Gr o e la n d As s e s s m e n t o fit s
Bio l o g ic a l Ac t i v i t y... 8i
3.1 In tro d u ctio n ... 81
3.2 A im ... 82
3.3 Materials and m eth o d s... 82
3.3.1 Purification of GroEL to hom ogeneity on reactive red affinity m atrices...82
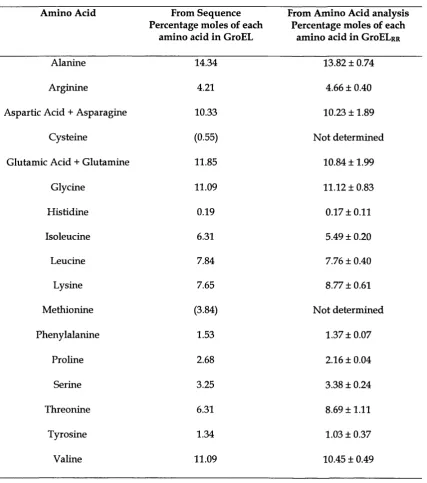
3.3.2 Characterization of the GroELpR... 83
3.3.3 Assay of the endotoxin content of GroELRR... 84
3.3.4 Assay of cytokine stim ulatory activity of GroELRR... 85
3.3.5 Assay of cytokine stim ulatory activity of GroELRR in the presence of the LPS inhibitor polymyxin B or the anti-CD14 antibody MY4... 85
3.3.6 Cytokine a ssa y s...85
3.4 Results... 86
3.4.1 Characterization of GroELRR... 86
3.4.2 Endotoxin content of GroELRR... 87
3.4.3 Cytokine stim ulatory activity of GroELRR... 88
3.4.4 Cytokine stim ulatory activity of GroELRR in the presence of the anti-CD14 antibody MY4... 90
3.4.5 Cytokine stim ulatory activity of GroEL contam inants eluted from the reactive red affinity colum n... 91
3.5 Discussion... 93
3.6 Conclusion... 96
C h a p t e r 4 ...97
G r o e l S t i m u l a t i o n o f M y e l o i d C e l l C y t o k i n e S y n t h e s i s...97
4.1 In tro d u ctio n ... 97
4.2 A im ... 98
4.3 Materials and m eth o d s... 98
4.3.1 GroEL purification from £. coli and assay of its endotoxin co n te n t...98
4.3.2 GroEL stim ulation of peripheral blood m ononuclear cells... 98
4.3.3 GroEL stim ulation of PBMC in the presence of anti-CD14 antibodies...99
4.3.4 GroEL stim ulation of hum an and m ouse m onocyte/ m acrophage cell lines 100 4.3.4.1 H um an m onocyte/m acrophage cell lines THP-1, HL-60 and U-937...100
4.3.4.2 H um an m onocyte/ m acrophage cell line Mono-Mac 6 (MM6)...101
4.3.4.3 Mouse m acrophage ceU line J774... 102
4.3.5 GroEL stim ulation of m ouse bone m arrow derived m acrophages...103
4.3.6 Cytokine A ssays...104
4.4 Results... 104
4.4.1 Endotoxin content of purified GroEL...104
4.4.2 GroEL stim ulation of PBM Cs... 104
4.4.2.1 Pro-inflam matory cytokines IL-ip, lL-6 and lL-8 produced by peripheral blood cells in response to GroEL...105
4.4.2.2 GroEL stim ulation of PBMCs to produce TNF-a and lL-10... 108
4.4.2.3 GroEL stim ulation of PBMCs to produce interferon - gam m a (IFN-y)... 112
4.4.2 5 GroEL stimulation of PBMCs to produce GM-CSF...115
4.4.3 GroEL stimulation of PBMCs in the presence of anti-CD14 antibodies...116
4.4.3.1 Anti-CD14 antibody MY4...116
4.4.3.2 Anti-CD14 antibody 60bca ...117
4.4.4 GroEL stimulation of human monocyte/macrophage cell lines...118
4.4.5 GroEL stimulation of mouse macrophage cells and cell lines... 119
4.4.5.1 TNF-a response of J774 cells stimulated with GroEL... 119
4.4.5.2 IL-6 response of mouse bone marrow derived macrophages stimulated with GroEL in the presence of polymyxin B... 120
4.5 Discussion... 121
4.6 Conclusion... 128
C h a p t e r s ...129
SXRUCTURE-AcnVITY STUDIES OF GROEL... 129
5.1 Introduction...129
5.2 A im ...130
5.3 M aterials and m ethods...130
5.3.1 Trypsin digestion of GroEL...130
5.3.2 Separation of GroEL tryptic fragm ents by gel filtration chrom atography...131
5.3.3 Isolation of GroEL tryptic peptides by reverse phase chrom atography...133
5.3.4 Stim ulation of PBMCs w ith digested GroEL and GroEL tryptic fractions separated by various chrom atographic m eth o d s...134
5.3.5 Cytokine assays...135
5.3.6 LAL assay of active GroEL fractions...135
5.3.7 Mass spectrom etry and N-terminal sequencing of active GroEL peptides...136
5.3.7.1 Mass spectrom etry...136
5.3.7.2 Edm an sequencing of GroEL derived peptides...137
5.3.8 Chemical synthesis of GroEL derived p e p tid e s...138
5.4 Results... 140
5.4.1 Cytokine stim ulatory activity of trypsin digested GroEL...140
5.4.2 Cytokine stim ulatory activity of GroEL tryptic fragm ents separated by gel filtration chrom atography...142
5.4.3 Cytokine stim ulatory activity of GroEL tryptic fragm ents isolated by reverse phase chrom atography... 146
5.4.3.1 Reverse phase chrom atography of GroEL tryptic fragm ents...146
5.4.3.2 D eterm ination of bioactivity of fractions obtained from reverse p h a se ... chrom atography of digested GroEL...147
5.4.3.3 Identification of active GroEL tryptic p e p tid e s...148
5.4.3.4 M odelling the identified active GroEL peptides to the crystal structure of GroEL ...152
5.4.3.5 Cytokine stim ulatory activity of the synthetic GroEL tryptic peptides...155
5.4.4 Cytokine stim ulatory activity of Mycobacterial cpn 60 derived p e p tid e s ...155
5.4.4.1 C pn 60.1 peptide 195-219... 156
5.5 D iscussion...158
Table of contents
Chapter 6 ... 167
IDENTIFICATION OF LEUKOCYTE SURPOPULATIONS BINDING TO GROEL... 167
6.1 Introduction... 167
6.2 Aim...168
6.3 Materials and methods...169
6.3.1 Flow cytometry...169
6.3.2 Surface labelling of PBMCs... 170
6.3.3 Flow cytometry of PBMCs... 174
6.3.4 Double labelling of PBMCs... 174
6.4 Results... 175
6.4.1 GroEL binding to PBMCs... 175
6.4.2 Flow cytometry of PBMCs isolated from an individual unresponsive to GroEL 180 6.4.3 Double labelling of monocytes... 181
6.5 Discussion...184
6.5 Conclusion...187
CHAPTER?... 188
Physicochem ical Analysisofthe Putative Groel Receptor... 188
7.1 Introduction...188
7.2 Aim...189
7.3 Materials and methods...189
7.3.1 Biotinylation of GroEL...189
7.3.2 Biotinylation of surface proteins of PBMCs...191
7.3.3 CeU lysis...192
7.3.4 Immunoprécipitation of PBMCs surface proteins binding to GroEL...193
7.3.5 Affinity chromatography...
1%
7.3.5.1 GroEL affinity column...196
7.3.5.2 Affinity chromatography of surface biotinylated proteins... 198
7.4 Results... 201
7.4.1 Biotinylation of GroEL...201
7.4.2 Immunoprécipitation of the GroEL receptor on the surface of PBMCs...202
7.4.3 Identification of biotinylated surface proteins of PBMCs binding to a GroEL affinity column...204
7.5 Discussion...208
Ch a p t e r s... 2i5
D i s c u s s io n a n d F u tu r e W o r k ... 215
8.1 Cytokine-inducing activity of contaminants found associated with GroEL...217
8.2 Cytokine-inducing activity of GroEL... 218
8.3 Flow cytometry to establish GroEL binding to PBMCs...220
8.4 Isolation and identification of the putative GroEL receptor... 221
8.5 Isolation and identification of active GroEL-derived peptides...223
8.6 Is GroEL a bacterial virulence factor?... 225
8.7 Questions for the future... 226
Biblio g raph y... 227
L
i s t
o f
F
i g u r e s
Ch a pte r 1
Fig 1.1 The double ring (GroE) chaperonin 60 ohgom er 24
Fig 1.2 Aerial view of the seven subunit ring of GroEL 26
Fig 1.3 EM image of the GroEL oligomer w ith a single subunit expanded 27
Fig 1.4 GroEL interaction w ith ATP and GroES 30
Ch a p t e r 2
Fig 2.1 A typical standard curve for the Bio-Rad DC assay 53
Fig 2.2 A typical standard curve for the Bradford assay 55
Fig 2.3 Electrophoretic transfer of proteins 63
Fig 2.4 Setting u p the LAL assay 65
Fig 2.5 Reading the LAL assay 65
Fig 2.6 Density gradient separation of peripheral blood m ononuclear cells 68
Fig 2.7 A rrangem ent of antibodies for ELISA 72
Ch a pte r 3
Fig 3.1 SDS-PAGE of standard GroEL and GroELRR 86
Fig 3.2 PBMCs stim ulated w ith GroEL and GroELRR to produce lL-6 88
Fig 3.3 Dose response of LPS-induced IL-6 production from PBMCs 89
Fig 3.4 GroELRR stim ulation of PBMCs in the presence of MY4 and polym yxin B 91 Fig 3.5 Cytokine stim ulatory activity of GroEL, GroELRR & its contam inants 92
Ch a pte r 4
Fig 4.1 GroEL stim ulation of hum an PBMCs IL-1|3 synthesis 105
Fig 4.2 GroEL stim ulation of hum an PBMCs lL-6 synthesis 106
Fig 4.3 GroEL stim ulation of hum an PBMCs lL-8 synthesis 107
Fig 4.4 TNF-a and IL-10 production by GroEL stim ulated PMBCs of donors 1& 2 109 Fig 4.5 TNF-a and IL-10 production by GroEL stim ulated PMBCs of donors 3& 4 111
Fig 4.6 GroEL stim ulation of hum an PBMCs IFN-y synthesis 112
Fig 4.7 IFN-y production by PBMCs of five donors w hen stim ulated w ith GroEL 113
Fig 4.8 GroEL stim ulation of hum an PBMCs IL-12 synthesis 114
Fig 4.9 GroEL stim ulation of hum an PBMCs GM-CSF synthesis 115
Fig 4.10 GroEL stim ulation of PBMCs in the presence of MY4 116
Fig 4.11 GroEL stim ulation of PBMCs in the presence of 60bca 117
Fig 4.12 GroEL stim ulation of Mono-Mac cells 118
Fig 4.13 GroEL stim ulation of J774 cells 119
Fig 4.14 GroEL stim ulation of m ouse bone m arrow -derived macrophages 120
Ch a pt e r 5
Fig 5.1 Diagrammatic representation of MALDI-MS 137
Fig 5.2 SDS-PAGE of trypsinized GroEL 140
Fig 5.3 Cytokine stim ulatory activity of digested GroEL 141
Fig 5.4 Size exclusion chrom atography of digested GroEL (cut off 20kDa) 142 Fig 5.5 IL-6 stim ulation by GroEL fractions separated by size-exclusion HPLC (20kDa) 143 Fig 5.6 Size exclusion chrom atography of digested GroEL (cut off 200kDa) 144 Fig 5.7 IL-6 stim ulation by GroEL fractions separated by size exclusion HPLC (200kDa) 145
Fig 5.8 Reverse phase chrom atography of trypsinized GroEL 147
Fig 5.9 IL-6 stim ulation by trypsinized GroEL fractions separated by RP-HPLC 148
Fig 5.10 MALDI-MS of fraction 17 149
Fig 5.11 MALDI-MS of fraction 24 150
Fig 5.12 MALDI-MS of fraction 28 151
Fig 5.13 MALDI-MS of fraction 30 151
Fig 5.14(a) M apping the GroEL tryptic peptides to the GroEL m onom er 153
Fig 5.14(b) A rrangem ent of the GroEL monomers in the oligomeric structure 153 Fig 5.15(a) M odelling the peptide 404-420 on the top ring of the GroEL oligomer 154 Fig 5.15(b) Side view of the lower GroEL ring w ith peptide 404-420 highlighted 154 Fig 5.16 Cytokine stim ulatory activity of the M. tuberculosis cpn 60.1 peptide 195-219 157
Ch a p t e r 6
Fig 6.1 Schematic representation of a flow cytometer 170
Fig 6.2 Incubation of PBMCs w ith GroEL, 4-3F and FITC conjugated IgG 171
Fig 6.3 Dot plot analysis of PBMCs 176
Fig 6.4 H istogram show ing staining of hum an PBMCs w ith an anti-CD14 antibody 176
Fig 6.5 H istogram show ing GroEL binding to PBMCs 177
Fig 6.6 Identifying T cells by staining PBMCs with m arkers specific for T cells 179
Fig 6.7 Histogram show ing GroEL binding to T cells 180
Fig 6.8 GroEL binding to monocytes isolated from unresponsive PBMCs 181
Fig 6.9 Dot plot show ing double labelling of PBMCs stained w ith isotype controls 182
List of Figures
Ch a pte r 7
Fig 7.1 Im m unoprécipitation of m em brane proteins binding to GroEL 194
Fig 7.2 Cytokine stim ulatory activity of biotinylated GroEL 202
Fig 7.3 Im m unoprécipitation of ceU surface proteins binding to GroEL 203
Table 1.1 Classification of the chaperonin family 23
Table 2.1 Confirming the vahdity of the LAL assay 66
Table 3.1 Analysis of amino acid composition of GroELpR 87
Table 5.1 Chaperonin 60 derived peptide synthesized by different groups 139
Table 5.2 Identification of bioactive tryptic peptides of GroEL 149
A
b b r e v i a t i o n s
Ax Absorbance at w avelength X
p-ME Beta-mercaptoethanol
APS A m m onium persulphate
ATP A denosine triphosphate
BSA Bovine serum albumin
cD N A Complementary de-oxyribonucleic acid
Cpn Chaperonin
DC D etergent compatible
DMEM D ulbecco's m odified essential m edium
DMSO D im ethyl sulphoxide
D N ase De-oxyribunuclease
DTT Dithiothreitol
ECL Enhanced chem ilum inescence
EDTA Ethylenediam ine tetraacetic acid
ELISA Enzym e linked im m unosorbent assay
EM Electron m icroscopy
FITC Fluorescein isothiocyanate
FPLC Fast protein liquid chromatography
g Gravitational force
GM-CSF Granulocyte m acrophage-colony stimulating
factor
HESS Hanks buffered salt solution
HIFCS H eat inactivated foetal calf serum
HPE H igh performance ELISA
HPLC H igh performance liquid chromatography
HRP H orseradish peroxidase
H sp H eat shock protein
IFN Interferon
Ig Im m unoglobulin
IL Interleukin
kDa Kilodalton
LAL L im u lu s am oebocyte lysate
LPS Lipopolysaccharide
LRW LAL reagent water
MALDI-MS Matrix assisted laser desorption ionization-m
spectrometry
M-CSE M acrophage-colony stim ulating factor
MEM M inim um essential m edium
MOPS M orpholinepropanesulfonic acid
NGS Norm al goat serum
ODx Optical density at w avelength X
OPD Orthophenylene diamine
PAGE Polyacrylam ide gel electrophoresis
PB Polym yxin B
PBMC Peripheral blood m ononuclear cell
PBS Phosphate buffered saline
PE Phycoerythrin
PMSF Phenyhnethylsulphonyl fluoride
PVDF Polyvinylidene fluoride
RP-HPLC Reverse phase- high performance liquid
chrom atography
RPMI Rockwell Park Memorial Institute
SDS Sodium d odecyl sulphate
TEMED Tetram ethylethylene diam ine
TEA Tri-fluoroacetic acid
Tris 2-amino-2 hydroxym ethyl propan-1,3-diol
UV Ultra violet
I
n t r o d u c t i o n
1.1 History of the heat shock response
The discovery of the heat shock response (now generically know n as the cell stress
response) w as a serendipitous event that occurred in the laboratory of F. Ritossa in
1962 [Ritossa, 1962]. The accidental, overnight incubation of Drosophila larvae at 30°C
rather than at the norm al physiological tem perature of 25°C, led to Ritossa noting that
the puffing patterns of the saHvary gland chromosomes of these larvae, differed from
the patterns of other larval cultures grow n at the norm al tem perature. W hen the larvae
w ere returned to grow th a t 25°C, the pattern of chromosomal puffs returned to normal.
In Ritossa's opinion [Ritossa, 1962], the accidental incubation of the larvae at
higher than norm al tem perature triggered the synthesis of a particular set of genes.
These genes, he predicted, were required by the larvae to w ithstand and survive
abnorm al tem peratures. In addition, this effect was reversible. These observations led
Ritossa to conclude th at adverse conditions, such as high tem perature, switched on in
built survival mechanisms in Drosophila, w hich protected the cells from further
Chapter 1 Introduction
damage. In light of these observations, Ritossa concluded his findings by introducing
the term, the heat shock response.
It is now know n that m ost organisms respond in a highly regulated m anner to
exposure to tem peratures greater than the norm al environm ental tem perature [Fayet et
al., 1989]. This response involves the upregulation of certain genes w ith the appearance
of their products in the cell [Ang et ah, 1991]. Genes activated in response to heat were
called heat shock genes and their encoded proteins were duly nam ed heat shock
proteins (hsp). As research progressed, it became apparent that the heat shock proteins
shared extensive sequence homology betw een species [Hemmingsen et ah, 1988;
McMullin and Hallberg, 1988; Picketts et ah, 1989; Jindal et ah, 1989; A ng et ah, 1991],
thus highlighting that there m ust be a common heat shock response mechanism.
Gradually, it w as noted that any form of stress was capable of initiating this response
and th at metabolic poisons, heavy metals, amino acid analogues, infection and m any
more stresses, upregulated the "heat shock" response [Fayet et ah, 1989; Ang et ah, 1991].
As a result, the heat shock proteins were renam ed stress proteins and their actions
defined as the stress response. Nowadays, both terms heat shock and stress (proteins,
response) are used interchangeably.
1.2 The heat shock or stress response
Protein aggregation appears to be the common initiator of the heat shock or stress
response. [Ananthan et ah, 1986] dem onstrate that adverse conditions such as high
tem perature lead to massive aggregation of newly-form ed and existing proteins and
that these denaturing cellular proteins initiate the stress response. The accumulation of
non-native protein switches on the stress response by activating the heat shock
HSF-1 is rapidly trim erized to its active form which in turn, binds to a consensus nucleotide
sequence, the heat shock element (HSE). This sequence is located in the promoter
region of the heat shock genes which are then rapidly transcribed. D uring the stress
response, stress proteins accumulate reaching high concentrations in the cell [Shinnick,
1991]. For example, the 60kDa hsp of Escherichia coli, GroEL, represents 1-2% of the
total protein content of a norm al cell [Hemmingsen et al., 1988]. U nder stress however,
the GroEL concentration has been found to increase five to ten fold [Hemmingsen et ah,
1988; Shinnick, 1991].
1.3 The m olecular chaperones
1.3.1 Definition of molecular chaperones
D uring the stress response, the major function of the heat shock (stress) proteins is to
prevent intracellular protein aggregation by reversing polypeptide unfolding and
encouraging norm al protein folding [Ziigel and Kaufmann, 1999]. Stress proteins
responsible for protein folding and assembly belong to the conserved family of protein
folding molecular chaperones. The term molecular chaperone was first used by Laskey
et al, (1978) to describe the properties of a protein called nucleoplasm in that was
essential for the correct assembly of DNA and histones during nucleosome formation
in Xenopus eggs [Laskey et ah, 1978]. This term was later used to describe a protein that
bound to the subunits of Rubisco (a carbon dioxide-fixing enzyme found in
chloroplasts of higher plants), to keep them soluble prior to their assembly [Musgrove
an d EUis, 1986; Ellis, 1996; Ellis, 1996]. Over the years, several classes of proteins were
identified in bacterial, fungal, plant and animal cells which m editated the assembly of
other proteins [EUis and Hemmingsen, 1989]. The concept of molecular chaperones
Chapter 1 Introduction
folding o f newly-formed proteins and the refolding of denatured proteins, without becoming
permanent components of these folding structures [Ellis, 1987; Ellis and Hemmingsen,
1989].
It is understandable that stressed cells need the assistance of molecular
chaperones to refold their dam aged and denatured proteins. However, it seems that
cells also require the assistance of these chaperones under norm al conditions as these
proteins are also foxmd in low concentrations in 'unstressed' cells [Fayet et al., 1989]. It
appears therefore, that the molecular chaperones play an im portant role in maintaining
the norm al intracellular environm ent and have in fact evolved to prevent intracellular
protein aggregation under all conditions [EUis, 1996].
It is im portant to note that during the stress response, not aU proteins produced
are molecular chaperones. In fact, only 5 of the 20 know n stress proteins of £. coli
function as molecular chaperones [Georgopoulos et al., 1990]. In a similar fashion, not
every molecular chaperone is a stress protein. The protein folding eukaryotic
chaperones TCP-1 and nucleoplasm in for example are not upregulated during the
stress response [ElUs, 1996].
1.3.2 C lassification of the m olecular chaperones
The molecular chaperones have been classified into several families [Ellis and van der
Vies, 1991]. Of these, the heat shock protein (hsp) 60, hsp70, and hsp90 famiUes are the
most studied and understood [Buchner, 1996; EUis and H artl, 1996]. The num ber
designated to each famUy refers to the average molecular m ass of its representative
chaperone in its monomeric state.
The folding of non-native proteins by m ost molecular chaperones is a complex
process th at involves recognition of hydrophobic residues [Fenton et al., 1994].
Generally, hydrophobic residues are buried in the interior of a fuUy folded protein. In
case of non-native proteins however, these hydrophobic sites are fully or partially
exposed [Zettlmeissl et al., 1979]. Protein folding by some molecular chaperones
involves a complex cycle of ATP binding and hydrolysis [Buchner, 1996; M artin and
H artl, 1997; Beissmger and Buchner, 1998].
Each molecular chaperone family has a characteristic w ay of recognizing
hydrophobic residues and non-native proteins. For example, members of the hsp70
family (DnaK in £. coli) recognize hydrophobic residues on extended polypeptide
chains and appear to stabilize polypeptide chains during their synthesis on ribosomes
[Buchner, 1996; Beissinger and Buchner, 1998]. U nder physiological conditions, the
hsp90 molecular chaperones do not possess refolding activity. Rather, they bind and
stabilize non-native proteins that have a high degree of secondary structure, and
prevent their prem ature aggregation, thus allowing them to be folded completely by
other chaperones [Buchner, 1996]. This protein appears to have a more supportive role,
yet it is vital for the viability of the cell [Nathan et al., 1997].
The hsp60 group of molecular chaperones is a family of sequence-related
oligomeric proteins w hich are globally referred to as the chaperonins [Hemmingsen et
al., 1988; Ellis and van der Vies, 1991]. Chaperonins have a distinctive double-donut
structure (Fig 1.1) w ithin which non-native proteins fold and refold [Hendrix, 1979].
In recent years it has emerged that the molecular chaperones have biological
actions in addition to those related to protein folding [Coates, 1996; Henderson et al.,
1996a; Coates and Henderson, 1998; Lewthwaite et ah, 1998; Coates et al., 1999]. The work
presented in this thesis focuses on such non-folding interactions. In particular,
attention will be given to the small family of molecular chaperones termed the
chaperonins (cpns). A brief discussion of the folding-actions of the chaperonins will be
followed by examples of diseases resulting from m isfolded proteins. The non-folding
interactions of the chaperonins and their role in disease will conclude the introduction.
Chapter 1 Introduction
1.4 The chaperonins
1.4.1 Introduction an d classification
The chaperonins (cpns) are a family of homologous proteins th at are found in all extant
species and are vital for the m aintenance of cell viability [Ellis and van der Vies, 1991].
The chaperonins w ere first identified as the major species of all induced proteins in
stressed cells [McMullin and Hallberg, 1988]. However, these proteins are also present
in cells un d er physiological conditions [Fayet et aL, 1989]. The cpn family is divided
into tw o subfamilies, the GroE subfamily and the TCP-1 subfamily (Table 1.1). The
sequence identity w ithin each subfamily is around 50%, b u t the identity betw een the
subfamilies is lower, at around 20% [EUis, 1996]. Cpns such as cpn60 (caUed GroEL in
E. coli) and cpnlO (GroES in E. coli) are found in bacteria and eukaryotic organeUes
such as m itochondria and chloroplasts [McMuUin and HaUberg, 1988; Jindal et aL, 1989;
Soltys and Gupta, 1997] while the eukaryotic cell cytosol contain cpns that belong to
TCP-1 (f-complex polypeptide 1) proteins [EUis and Hartl, 1996]. As the GroE
chaperonins are the focus of attention in this thesis, their structure and function wUl be
Table 1.1: Classification of the chaperonin family [Ellis, 1996]
Subclass I: GroE subclass Abbreviations Common name of chaperonin
Eubacterial chaperonin 60 Eu cpn60 GroEL (E. coli), hsp65 Eubacterial chaperonin 10 Eu cpnlO GroES (£. coli), co-chaperone
Mitochondrial chaperonin 60 Mt cpn60 Hsp60, m itonin
Mitochondrial chaperonin 10 Mt cpnlO HsplO, co-chaperone Chloroplast chaperonin 60 Ch cpn60 Rubisco subunit binding protein
Chloroplast chaperonin 10 Ch cpnlO Co-chaperone
Ch cpn21 Chaperonin21
Subclass II: TCP-lsubclass Abbreviations Common name of chaperonin
Cytosolic chaperonin 60 Cyt cpn60
TCP-1 f-complex polypeptide 1 CCT chaperonin-containing TCP-1
TRiC TCP-1 ring com plex
Archaebacterial chaperonin 60 Ar cpn60
TF55 Thermophilic factor 55
1.4.2 Structure of the GroE chaperonins
All cpns are single or double ringed structures. The double ringed chaperonins
comprise of tw o heptam eric rings, stacked back-to-back, form ing a large central cavity
w ithin which proteins fold and refold (Fig 1.1) [Hendrix, 1979; McMullin and Hallberg,
1988; Ellis and H artl, 1996]. D uring protein folding, the 60kDa GroE cpn requires the
assistance of a seven subunit dom e-shaped molecule called cpnlO, w hich forms a lid on
the top of one of the cpn60 rings [Ishii et ah, 1992]. No co-chaperones have yet been
identified in the TCP-1 subclass [Ranson et ah, 1998]. Both cpn60 and cpnlO in E. coli
are encoded by the same operon [Tilly et ah, 1981]. In Mycobacteria however, there is
more than one cpn60 gene, only one of which is found in the same operon as cpnlO
Chapter 1 Introduction
Studied in detail (for reviews see [Ranson et aL, 1998; Sigler et aL, 1998]). Binding and
hydrolysis of ATP by the cpn oligomer results in a conformational change in the
protein. This allows the binding of the co-chaperone GroES and results in the release
of the non-native proteins into the cavity for folding.
I
Cis ring.
60kDa subunit Tmns ring
Fig 1.1 The double ring (GroE) chaperonin 60 oligomer. The two heptameric rings (cis and trans) are arranged back-to-back. The binding of protein (shown in red) usually takes place in one ring at any one time which is capped with the lOkDa co-chaperone, allowing the protein to fold inside the cavity.
The number of subunits forming each ring varies w ithin the chaperonin family.
The GroE (bacterial cytosol cpn60 and mitochondrial cpn60) chaperonins have seven
identical 60kDa subunits in each ring (Fig 1.1), whereas the archaebacteria
(thermophilic factor-55) comprise of eight subunits in each ring and the TCP-1
(eukaryotic cytosol) chaperonins, are have nine different subunits in each ring
[Horwich and Willison, 1993]. GroEL is the subject for investigation in this thesis and
its structure and function will therefore be discussed in more detail in this
introduction.
1.5 The
Escherichia coli
chaperonin 60, GroELThe bacterial protein GroEL was originally identified in the early 1970s as a host
protein necessary for the replication of infecting bacteriophages X and T4
[Georgopoulos et al., 1972]. GroEL supervises the correct assembly of phage-encoded
protein subunits, which form an oligomer that joins the head to the tail of phage X.
Initially, GroEL w as studied exclusively as a phage-assembly protein and its role in the
functioning of the uninfected bacterium w as only revealed w hen significant sequence
identity was established betw een this oligomer and the chloroplast binding protein,
Rubisco [Hemmingsen et al., 1988]. A round the same time, a report w as published,
w hich revealed that GroEL cross-reacted w ith an antiserum th at was raised against the
58kDa heat-shock protein of Tetrahymena thermophila [McMuUin and Hallberg, 1988]. It
also shared structural and antigenic characteristics w ith 58-60kDa heat shock proteins
isolated from mitochondria of Saccharomyces cerevisiae, Zea mays and Homo sapiens
[McMuUin and Hallberg, 1988]. Extensive research of the protein folding capacity of
these m itochondrial proteins dom inated the late 80's and early 90's, and as a result the
concept of molecular chaperones was born, triggering a new w ave of research which
continues to this day. In addition, biological functions of cpns other than protein
folding w ere also revealed [Friedland et al, 1993; Cavanagh an d Morton, 1994; Kirby et
al., 1995; Wick et a l, 1995b; Tabona et al., 1998; C hen et a l, 1999]. It is these non-folding
Chapter 1 Introduction
7 ^
P
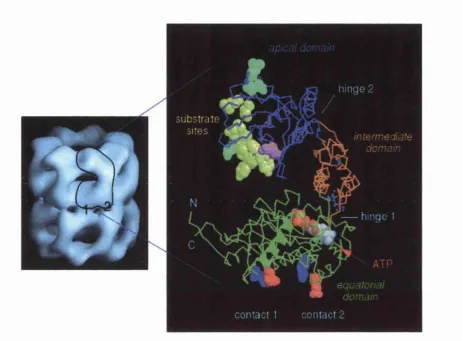
Fig 1.2 Aerial view of one seven subunit ring of GroEL. Orange residues extending into the central cavity of subunit number 2 represent the ATP binding site of the monomer. Red residues (alpha helices) shown in subunit 1 represent the section of the equatorial domain that makes contact with identical residues in the underlying ring. Blue residues on subunit 1 represent the apical domain that lines the opening of the central cavity. This figure was adapted from [Boisvert et al., 1996]. Reproduced with permission from the authors and Nature Structural Biology, copyright 1996 Macmillan Magazines Ltd.
J
Fig 1.3 Left: EM image of the GroEL oligomer. Right: A single subunit is expanded to show the domain organization of the GroEL monomer. The equatorial domain (green) contains the ATP binding site. Contacts 1 and 2 link the monomer to the subunit of the lower ring. The apical domain represented in blue contains the substrate binding sites (yellow) and the GroES binding site (cyan). The intermediate domain (orange) links the apical and the equatorial domains. This figure was adapted from [Roseman et ai, 1996]. Reproduced with permission from the authors
and Cell, copyright 1996 Cell Press.
Chapter 1 Introduction
1.5.1 Structure of th e GroEL oligom er
The structure of GroEL has been studied using cryo electron-microscopy [Chen et al.,
1994; Roseman et al., 1996] and X-ray crystallography [Braig et ah, 1994; Boisvert et ah,
1996]. The tw o-ringed 14-mer protein is slightly taller than it is wide, m easuring
approxim ately 146 Â in height and 137 Â in w idth, w ith a central cavity th at has been
reported to be 45Â in diam eter [Braig et ah, 1994]. The two heptam eric rings are
arranged back-to-back such that the external surfaces on both ends of the cylinder are
the same (Fig 1.3). The m ature GroEL subunit (monomer) is composed of 547 amino
acids [Hem mingsen et ah, 1988] and these are arranged in three clearly distinguishable
dom ains [Fenton et ah, 1994]. The equatorial domain is the largest of the three domains
and comprises both the N- and C- terminal sequences (residues 6-133 and 409-503) (Fig
1.3) [Braig et ah, 1994]. This region is rich in alpha-heHces and provides both the contact
sites betw een the rings and the side-to-side contacts betw een neighbouring subunits.
This dom ain also contributes m ost of the residues for the ATP binding site of GroEL
(Fig 1.2 & 1.3) [Roseman et ah, 1996].
The apical domain (residues 191-376) forms the opening at both ends of the
central cavity and also contains the putative GroES and polypeptide binding sites
[Fenton et ah, 1994; Buckle et ah, 1997]. This dom ain is said to be the m ost flexible and
disordered region of the protein (Fig 1.3) [Braig et ah, 1994]. The intermediate domain is
the sm allest of the three domains and links the equatorial dom ain to the apical dom ain
1.5.2 GroEL m ediated protein fold in g
GroEL-mediated protein folding is universally accepted to involve the cyclic binding
and release of substrate polypeptides. This ATP-dependent reaction has been critically
and comprehensively reviewed by Sigler et al (1998).
Phase I: Polypeptide Binding: Only one ring of the GroEL oligomer is involved
in polypeptide folding at any one time and there is no exchange of polypeptide from
one ring to the other [Rye et ah, 1997]. Crystal structure analysis estimates the volume
of each ring cavity to be around 85,000 [Xu et a l, 1997]. At this volume, the cavity
should be able to comfortably accommodate a native protein, of approximately 70kDa.
M utational studies have implicated that residues Y (tyrosine) 199, Y203, L (leucine) 234,
L237, L259, L263 and Y264 are im portant for polypeptide binding (Fenton et al, 1994).
Electron microscopy shows that these residues are p art of the apical dom ain and face
the central cavity, w here they form a ring of hydrophobic binding sites [Braig et al,
1994; Fenton et al., 1994]. Substrate protein, w ith its hydrophobic residues exposed
[Zettlmeissl et al., 1979], binds to these hydrophobic sites at the entrance of the central
cavity [Fenton et al., 1994].
In vitro binding assays have revealed that GroEL can interact w ith many
conformational arrangem ents of test peptides [Landry and Gierasch, 1991]. In E. coli,
GroEL appears to show a preference for a particular type of new ly translated protein
[Houry et al., 1999]. 300 of these recently identified proteins have been found to be
predom inantly (79%) smaller than 60kDa [Houry et al., 1999]. This observation
Chapter 1 Introduction
Phase II: GroES and Nucleotide Binding: As shown in Fig 1.3, the GroEL
oligomer is m ade up of two rings, and only one ring folds a polypeptide at any one
time [Rye et al., 1997]. For simplicity, polypeptide binding and folding w ithin one ring
only (cis ring) is explained here. Once bound to GroEL, the polypeptide must be
released into the cavity to enable its further refolding to its native state. ATP binding to
the cis ring results in a conformational change in the ring that enables GroES binding
[Xu et al, 1997]. GroES binding deprives the polypeptide of its binding sites on GroEL
thus pushing it into the cavity [Rye et al., 1997]. The GroEL and GroES rings are so
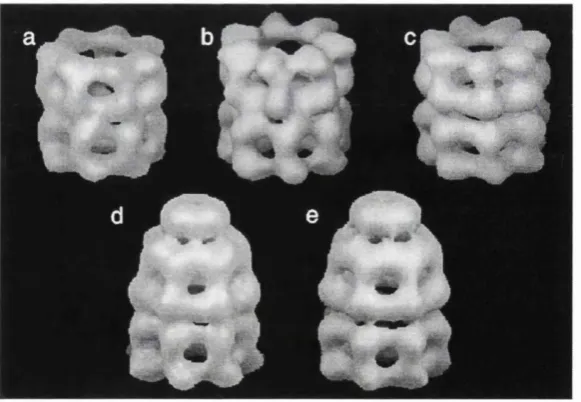
smoothly aligned that they nearly share an exact sevenfold rotational axis (Fig 1.4).
Their association appears as a dome shaped structure, the so-called Anfinsen's cage
[Roseman et al, 1996; Lorimer, 1997], where the apical domain forms the walls, and
GroES, the cap (Fig 1.4 (e)).
Fig 1.4 Conformational changes in GroEL on interaction with ATP and GroES. (a) GroEL, (b) GroEL-ADP, (c) GroEL-ATP, (d) GroEL-GroES-GroEL-ADP, (e) GroEL-GroES-ATP. GroES is the cap shaped structure closing the covering the cavity of the top ring (d, e). The binding of ATP (c) and GroES (e) results in the upivard movement of the apical domains of each monomer, which gives this complex a bidlet-shaped appearance (e). This figure is adapted from [Roseman et al., 1996], Reproduced with permission of the authors and Cell, copyright 1996 Cell Press.
Phase III: Protein Folding and Release of Ligands: Rye et al (1997) have show n that
alternate ATP binding to both cis and trans rings is essential for folding an d release of
the polypeptide from the GroEL-GroES complex. GroEL m utants have been used to
establish that 'cross talk' exists betw een the two rings at this stage [Rye et ah, 1997].
These have show n that ATP hydrolysis in the cis ring prim es the GroES-GroEL-
polypeptide complex for release, which is then disassem bled after ATP binds to the
trans ring.
GroEL-mediated protein folding is estim ated to take approxim ately 15 seconds.
The ATP on the cis ring undergoes hydrolysis 6-8 seconds after GroES binding. ATP
binding to the trans ring results in the release of the polypeptide and GroES from cis,
w ithin the next 6-7 seconds [Rye et al., 1997]. A 15 second cpn cycle m eans that the
substrate polypeptide is released from the complex regardless of w hether it is
completely folded. Only a fraction of proteins can reach their native state w ithin this
time period. Therefore, in order to avoid degradation by cell proteases, a num ber of
proteins have to undergo more than one cycle, either w ith GroEL, or w ith another
Chapter 1 Introduction
1 . 6 C h a p e r o n i n s i n d i s e a s e
1.6.1 D iseases caused by chaperonin m ediated protein fold in g
It is know n that a num ber of diseases arise from protein misfolding and aggregation
[Carrell and Lomas, 1997]. N eurodegenerative diseases such as Alzheimer's,
H untington's, and Parkinson's are all characterized by the presence of protein
aggregates in the brain [Carrell and Lomas, 1997; Coates et ah, 1999]. The involvement
of chaperones in the pathology of these diseases is currently unknow n.
However, a possible role of chaperones in m ediating the form ation of diseased
prion protein in transmissible spongiform encephalopathies (TSEs) has been suggested
[DebBurman et ah, 1997]. TSE is a fatal infectious disease of the m am m alian nervous
system which is caused by the conversion of a cellular protein, the prion protein, into
its diseased conformational state. In vitro experim ents have show n th at this conversion
of the norm al cellular prion protein is a tw o step process, w here the first step is
supervised by chaperones such as GroEL and hspl04 [DebBurman et ah, 1997]. At this
stage, the in vivo location of the prion protein conversion is unknow n. However, if
chaperones are involved in vivo, understanding their m ode of action may help in
combating this lethal disease.
1.6.2 D iseases caused by non-folding biological actions of chaperonins
The first evidence of a role of chaperonins in disease came from studies that revealed
that these proteins were potent im m unogens [Henderson et ah, 1996a; Coates, 1996;
Lewthwaite et ah, 1998; Coates et ah, 1999]. Bacterial cpns w hich share significant
sequence hom ology to m am m alian cpns [Picketts et al., 1989; Jindal et ah, 1989], have
been implicated in the pathogenesis of several diseases [Kirby et ah, 1995; Meghji et ah,
response, b u t w hat remains unknow n is w hether this induction is protective or
damaging. Cross-reactivity betw een bacterial and hum an cpns may lead to
autoim m unity [Lamb and Young, 1990]. Recent reports, how ever, suggest that
exposure to microbial hsp m ay be necessary to induce tolerance to self-hsp [van Eden,
2000], w hich themselves are also potent stim ulators of the im m une system [Chen et al.,
1999; K o le ta l, 2000].
1.6.2.1 C ellular location of chaperonins
C haperonins are generally located in the m itochondria and chloroplasts of eukaryotic
cells and the cytosol of bacteria [Gupta, 1996]. However, recent reports have revealed
that some cpns are secreted to the outer m em branes of some organisms during the
stress response [Ensgraber and Loos, 1992; Tang et al., 1997; Raulston et al., 1998; Belles
et ah, 1999]. This is a curious observation as cpns lack a leader sequence required for
secretion [Garduno et al., 1998].
Salmonella typhi dem onstrates an upregulation of the 58kDa (GroEL homologue)
and the 68kDa (DnaK homologue) proteins w hen heat shocked. U nder such conditions,
a high concentration of these cpns is found in the outer-m em brane fractions of the
disrupted cells [Tang et al., 1997]. Cpn60 expressed on the surface is thought to play the
role of a bacterial adhesin as suggested by Ensgraber et al., [1992], w ho show that the
cpn60 homologue from Salmonella typhimurium is responsible for binding of the
bacterium to the intestinal m ucus [Ensgraber and Loos, 1992]. Similarly Frisk et al
(1998) have reported that pre-incubation of Haemophilus ducreyi hsp60, w ith HELA
cells, inhibits the bacteria binding to these eukaryotic cells [Frisk et al., 1998]. The cpn60
of the oral opportunistic pathogen Actinobacillus actinomycetemcomitans has been
purified from a mixture of proteins found associated w ith the outer-surface of the
Chapter 1 Introduction
both intracellular and extracellular levels of cpn60 from A. actinomycetemcomitans,
increase substantially w hen the bacterium is heat shocked [Goulhen et al., 1998]. Cpn60
has also been locaHzed on the outer m em brane of Legionella pneumophila grow n under
norm al physiological conditions [Garduno et al., 1998]. Interestingly, w hen the cpn60
gene from L. pneumophila is cloned into E. coli, the recom binant protein is only found in
the cytoplasm of E. coli, suggesting that some bacteria (such as L. pneumophila) may
have mechanisms for transporting cpn60 to extracytoplasmic locations [Garduno et al.,
1998]. Intriguingly, if L. pneumophila cells are heat-shocked, or allow ed to infect HeLa
cells, the extracytoplasmic cpn60 levels are reduced. It is thought th at these stressful
conditions either provoke secretion of the cpn60 into the surrounding media, or
m ediate its association w ith the outer membrane, such th at the epitope recognized by
the anti-cpn60 antibody is blocked [Garduno et al., 1998].
A similar story is also em erging w ith eukaryotic cells. BALB/cByJ mice cells
infected w ith Listeria monocytogenes show increased levels of cpn60 on the plasma
m em branes of spleen and liver cells. This protein is no t detected on the surface of liver
and spleen cells taken from uninfected mice [Belles et al., 1999]. In addition, secretion of
chaperonins from eukaryotic cells has also been reported. Early pregnancy factor,
identified as a cpnlO homologue, is detected in the serum of pregnant w om en
[Cavanagh and Morton, 1994]. H um an cpn 60, on the other hand, has been localized in
the peritoneal fluids of w om en suffering from endometriosis [Kligman et ah, 1996].
W hat role do cpns play extracytoplasmically? It is unlikely th at they function as
protein folding molecules, as ATP (essential for protein folding) is absent in the
periplasm and outer-m em brane [Missiakas and Raina, 1997]. In some cases, especially
in microbes, cpn60 behave as adhesins, which makes them potentially im portant
virulence factors [Ensgraber and Loos, 1992]. O n the other hand, cell surface expression
suggested to be protective in nature as it is thought to signal danger to the immune
system [Matzinger, 1994]. H um an cpn60 is also thought to play a role in tum our
imm unogenecity as it is expressed on the surface of stim ulatory tum our cells, bu t is
absent from the surface of non-stim ulatory tum our cells [Wells and Malkovsky, 2000].
In addition, the protective T cell response to the hum an cpn60 expressing tum our cells
is blocked by antiserum raised against the hum an cpn60 (for review see [Wells and
M alkovsky, 2000]). Finally, cpn60 homologues m ay also play a role in stabilizing hpid
m em branes during heat shock, as they have been show n to interact and stabilize model
lipid m em branes via the C-terminus [Torok et al, 1997].
1.6.2.2 C haperonins induce cytokine production an d upregulate adhesion m olecules
The first clue that cpns m ay contribute to pathology came from the finding that these
proteins induce cytokine production [Friedland et al., 1993; Retzlaff et al, 1994;
M arcatih et al., 1997; Tabona et al., 1998; Yamaguchi et al., 1999; Chen et al., 1999].
Cytokines are proteinaceous effector molecules that bind to receptors on the surface of
cells and induce selective intracellular signalling events [Meager, 1998]. Produced by
both lym phoid and non-lym phoid cells, cytokines have a range of biological effects
such as [H enderson et al., 1996b]:
1. stim ulating cell proliferation, an essential process involved in tissue and organ
homeostasis, m orphogenesis and repair
2. control of physiological systems
3. defence of the organism by im m une mechanisms
Cytokines are generally inducible external stimuli. For example,
lipopolysaccharides (LPS) bind to their cell surface receptor complex (CD14 and Toll
like receptors), to trigger the synthesis of cytokines [Wright et al., 1990; Yang et al., 1998;
Chapter 1 Introduction
in response to infectious agents and cause inflam m ation which can result in host-
induced pathology [Henderson et ah, 1996b]. Inflamm atory events include increased
capillary permeability, heightened blood flow, and infiltration of im m une cells
(neutrophils, monocytes, T-cells etc.) to the site of infection [Male, 1993].
Bacterial cpns have been reported to stim ulate host cells to produce a range of
cytokines. GroEL and cpn60 homologues from other bacteria stimulate murine
m acrophages [Retzlaff et ah, 1994], kératinocytes [Marcatih et ah, 1997], peripheral
blood m ononuclear cells [Tabona et ah, 1998], and gastric epithelial ceUs [Yamaguchi et
ah, 1999] to secrete pro-inflam matory cytokines including IL-1(3, IL-6, IL-8 and TNF-a.
GroEL also upregulates the surface expression of the adhesion molecule, ICAM-1
(interceUular adhesion molecule-1) on kératinocytes [Marcatih et ah, 1997].
U pregulation of ICAM-1 is not blocked by antibodies to various pro-inflammatory
cytokines (also secreted in response to GroEL), im plicating that its increased surface
expression is a direct effect of GroEL [Marcatih et ah, 1997]. HUVECs (human umbilical
venous endothelial ceUs) express ICAM-1, E-selectin, and VCAM-1 (vascular ceUular
adhesion molecule-1) on their surface w hen stim ulated w ith GroEL [Galdiero et ah,
1997]. The mycobacterial cpn60.2 (hsp65) stim ulates monocytes, monocyte-derived
m acrophages and THP-1 cehs (human monocyte cell line) to synthesize pro-
inflam m atory cytokines [Friedland et ah, 1993; Peetermans et ah, 1994]. In addition,
hsp65 upregulates adhesion molecules on endothelial cehs w hich enables these cehs to
adhere to monocytes [Verdegaal et ah, 1996].
These findings suggest that cpns m ay play an im portant role in pathogenesis by
m ediating not only an inflammatory response b u t also by regulating the activation and
adhesion of monocytes and endothelial cehs. Recently, it has been show n that trypsin
digested GroEL retains its potential to induce inflam m ation [Tabona et ah, 1998].
therapeutic purposes (antagonizing the activity of cpn60 proteins) and is therefore one
of the m ain aims of this PhD thesis.
The precise m echanism by which cpns stim ulate cells rem ains yet unclear. The
m ost likely possibility is that they bind to a cell surface receptor. Retzlaff et al (1996)
suggest th at cpn 60 from Legionella pneumophila stimulates m ouse macrophages
probably by binding to a cell surface receptor [Retzlaff et al., 1996]. To determine if L.
pneumophila cpn60 was binding to the cells, latex beads coated w ith cpn60 were
incubated w ith macrophages in the presence of cytochalasins. Cytochalasins inhibit
microtubule form ation [Cooper, 1987] and block beads being phagocytosed, b u t allow
the particles to bind to cells. Control beads (latex beads and BSA coated beads) did not
stim ulate the ceUs. However, m acrophages incubated w ith cpn60 coated latex beads
show ed an increase in messenger RNA for IL-ip, thus im plying that cpn60 acted at the
cell surface and did not need to be internaUzed to stimulate m ouse peritoneal
m acrophages [Retzlaff et al., 1996]. Identification of the receptor (or receptors) for the
GroEL protein of E. coli, on the surface of peripheral blood m ononuclear cells, is one of
the aims of this PhD project.
Cpns are also involved in the upregulation of transcription factors which contribute
to the expression of adhesion proteins and cytokines. H um an and chlam ydial cpn 60
induce expression of NF-kB in endothelial cells in a m anner similar to LPS (Kol et al,
1999). C pns can, therefore, be viewed as intercellular proteins w ith protective functions
th at can also act as extracellular agonists w ith the capacity to stim ulate ceUs.
1.6.2.3 Cell m ed iated im m unity to chaperonins
T ceU and antibody responses to cpns, especially to cpn60 and cpnlO, are seen in a
range of infectious diseases [Coates, 1996; Ztigel an d Kaufmann, 1999]. Anti-hsp65
antibodies and T cells recognizing mycobacterial hsp65 (cpn60.2) have been repeatedly
Chapter 1 Introduction
reported in patients w ith tuberculosis and leprosy [Kaufmann, 1990; Shinnick, 1991]. yô
T cells recognizing the cpn60 of Listeria monocytogenes have been isolated from sites of
infection in mice and removal of these T cells by monoclonal antibodies enhanced
bacterial multiplication [Hiromatsu et al., 1992]. Interestingly, T cells reactive to hsp65
have also been isolated from healthy individuals [Kaufmann, 1990]. C ould it be that the
cpn60 reactive T cells recognize epitopes that are shared by cpns? Koga et al (1989)
have show n th at T cells raised against mycobacterial hsp65 recognize a cpn60
hom ologue expressed by stressed m urine macrophages. T cells that recognize shared
regions can obviously no t be used as an indicator of infection [Kaufmann, 1990].
However, hum an T cells which show specificity for mycobacterial hsp65 derived
peptides, and not autologous hsp65, have also been reported, w hich make these
peptides prim e targets for vaccine design and their presence a possible sign of infection
[Mustafa et al., 1996].
1.6.2.4 Role of chaperonins in th e pathogenesis of arterial diseases (atherosclerosis)
Endothelial cells line blood vessels and form a barrier betw een the flowing blood and
the underlying tissue [Schett et al., 1995]. These cells are constantly u n d er stress, be it
mechanical stress induced by blood flow or acute stress from various "toxins" entering
the blood. Such stresses, along w ith cytokines, induce heat shock proteins in
endothelial cells and probably prevent cell dam age in the vessel w all [Schett et al.,
1995]. Injury to the arteries can result in atherosclerosis, a common and fatal disease in
the w estern w orld [Wick et al., 1995b]. To date, the exact factors that lead to the
initiation of atherosclerosis are unclear, b u t they are thought to be inflammatory in
nature [Wick et al., 1995b]. It has been suggested that the early stages of atherosclerosis
involve an autoim m une response to cpn60, th at is expressed by the stressed
have been isolated from healthy people w ith carotid lesions [Xu et al., 1993]. In in vitro
studies, these antibodies cross-reacted w ith recom binant h um an cpn60 and
mycobacterial cpn60.2 (hsp65) and cpn60 isolated from stressed endothelial cells
[Schett et ah, 1995]. Rabbits im m unized w ith mycobacterial hsp65 showed
inflam m ation at sites know n to be predisposed to lesion form ation [Xu et ah, 1992].
Anti-hsp65 antibodies and hsp65 reactive T cells w ere found in the peripheral blood
and developing lesions of these rabbits. The surprising observation however, came
from tm im m im ized mice which were kept on a high cholesterol diet (cholesterol causes
dam age to blood vessels). These mice had lesions which contained hsp65 reactive T
cells, suggesting that an autoim m une reaction to self cpn60 m ay play a role in the
developm ent of these lesions [Xu et ah, 1992; Wick et ah, 1995a].
Recently, it has been suggested that cpns m ay play a role in m ediating the
inflam m atory response during atherogenesis [Kol et ah, 1999]. Both hum an and
chlam ydial cpn60 stim ulate hum an vascular endothelial cells to produce IL-6 and also
upregulate adhesion molecules in vitro [Kol et ah, 1999]. A similar response to GroEL is
show n by HUVECs, which secrete IL-6, GM-CSF and soluble (s) ICAM-1, sVCAM-1
and sE-selectin while upregulating surface adhesion molecules [Galdiero et ah, 1997].
C pns have also been im plicated in the lysis of stressed endothelial cells [Schett
et ah, 1995]. U nder in vitro conditions, heat shocked endothelial cells were lysed by
anti-hsp65 antibodies isolated from subjects w ith or w ithout carotid lesions. Cell lysis
took place in the presence of complem ent or peripheral blood cells [Schett et ah, 1995].
Similar results were obtained w hen studying the role of Chlamydia pneumoniae
in atherosclerosis [Mayr et ah, 1999]. Here, serum antibodies from patients w ith carotid
lesions contained antibodies that recognized mycobacterial hsp65 (cpn60.2), GroEL,
h u m an cpn60 and chlam ydial cpn60. Antibodies to each m entioned cpn were purified
Chapter 1 Introduction
H ow are these auto-antibodies raised in the first place? Possible suggestions
include infection w ith agents or im m unization w ith dead bacteria that have cpn
molecules hom ologous to hum an cpn60 [Schett et al., 1995]. Sequence similarity
betw een bacterial and hum an cpns makes it possible for antibodies to bacterial cpns to
cross-react and neutralize self-cpn60.
1.6.2.5 C haperonins and autoim m une diseases
A n im m une response to self-cpns has been suggested to be im portant in the
pathogenesis of autoim m une diseases [Lamb and Young, 1990]. Heat-shock proteins,
especially mycobacterial hsp65, are w ell-docum ented in autoim m une diseases such as
arthritis [Gaston et at., 1989; Kaufmann, 1990; Gaston, 1997]. A lthough this protein has
never been show n to induce arthritis on its own, im m unization of rats w ith whole heat-
killed M. tuberculosis induces severe arthritis and also gives rise to T-cells specific for
epitope 180-188 of hsp65 [van Eden et al., 1988]. Van Eden et al (1988) also reported that
im m unization of rats w ith hsp 65, protects the animals from arthritis, that is induced
by im m unization w ith M. tuberculosis. It seems therefore that hsp65, despite being
recognized by T cells during arthritis can actually confer protection in animal models
[van Eden et al., 1988; van Eden, 2000].
The role of cpns in inducing autoim m une diseases was also suggested by Koga
et al, (1989) w ho show ed th at ap-T cells reactive to mycobacterial hsp65, interact w ith
mouse bone m arrow derived macrophages that were stressed w ith IL-4 and
cytomegalovirus. It is thought th at the stress response in these m acrophages involved
upregulation and surface expression of hsp60, w hich stim ulated the mycobacterial
hsp65 specific T cells [Koga et al., 1989]. These and similar observations have led to
suggestions th at in patients w ith rheum atoid arthritis (RA), self-cpn60 is being targeted
by hsp65 antibodies and T cells specific to mycobacterial hsp65. A n independent study