In this solution set, an underline is used to show the last significant digit of numbers. For instance in
x= 2.51693
the 2,5,1, and 6 are all significant. Digits to the right of the underlined digit, the 9 & 3 in the example, are not significant and would be rounded off at the end of calculations. Carrying these extra digits for intermediate values in calculations reduces rounding errors and ensures we get the same answer regardless of the order of arithmetic steps. Numbers without underlines (including final answers) are shown with the proper number of sig figs.
1
Exercise 2.19a pg 86
Given
The standard enthalpy of combustion of cyclopropane is −2091 kJ mol−1 at 25◦C. Additionally, the enthalpy of formation of its isomer propene is +20.42 kJ mol−1.
In terms of given variables, this is written:
∆rH⊖=−2091 kJ mol−1 cyclopropane combustion
∆fH⊖ = +20.42 kJ mol−1 propene enthalpy of formation
Find
Calculate . . .• (a) The enthalpy of formation of cyclopropane
• (b) The enthalpy of isomerization from cyclopropane to propene
Strategy
We begin by constructing the balanced chemical equation for combustion of cyclopropane 2C3H6+ 9O2(g)→6H2O(l) + 6CO2(g) ∆rH⊖= 2× −2091 kJ mol−1
For any such reaction the reaction enthalpy can related to the enthalpy of products and reactants by (Equation 2.32 on pg 68 in your text book)
∆rH⊖ =
∑
Products
νp∆fHp⊖−
∑
Reactants
νr∆fHr⊖
which for this reaction is
∆rH⊖= 6∆fH⊖(H2O(l)) + 6∆fH⊖(CO2(g))
| {z }
products
−2∆fH⊖(C3H6) +−9∆fH⊖(O2(g))
| {z }
reactants
∆fH⊖(C3H6) = 3∆fH⊖(H2O(l)) + 3∆fH⊖(CO2(g))−
1 2∆rH
⊖
= 3× −285.83 kJ mol−1+ 3× −393.51 kJ mol−1−1
2 ×2× −2091 kJ mol
−1
= 52.98 kJ mol−1
With this information we can now calculate the enthalpy of isomerization of cyclopropane to propene. The balanced equation for this process is
C3H6(cyclopropane)→C3H6(propene) ∆rH⊖=?
The enthalpy of this reaction is simply given by
∆rH⊖ = ∆fH⊖(propene)−∆fH⊖(cyclopropane)
= 20.42 kJ mol−1−52.98 = −32.56
Solution
•
The enthalpy of formation of cyclopropane is
∆
fH
⊖(C
3H
6) = 53 kJ mol
−1.
2
Exercise 2.20a pg 86
Given
When a 120 mg sample of naphthalene, C10H8(s), was burned in a bomb calorimeter the temperature rose by 3.05 K.
Additionally, we are to predict the temperature rise for the combustion of 10 mg sample of phenol in the same calorimeter (the enthalpy of combustion of phenol is ∆cH⊖(C6H5OH(s)) =−7061 kJ mol−1).
In terms of given variables, this is written:
mC10H8(s)= 120 mg
∆T= 3.05 K
mC6H5OH(s)= 10 mg
∆cH⊖(C6H5OH(s)) =−7061 kJ mol−1
Find
• Calculate the calorimeter constant.
• By how much will the temperature rise after the phenol sample is burned?
Strategy
To determine the calorimeter constantC we’ll need to determine how much heat was released in this combustion reaction and for this we’ll need to know the combustion enthalpy of naphthalene. We’ll start by constructing the balanced chemical equation for the combustion of naphthalene.
C10H8(s) + 12O2(g)→4H2O(l) + 10CO2(g) ∆rH⊖=?
We can calculate the enthalpy of this reaction from
∆rH⊖ = ∑
Products
νp∆fHp⊖−
∑
Reactants
νr∆fHr⊖
= 4∆fH⊖(H2O(l)) + 10∆fH⊖(CO2(g))−∆fH⊖(C10H8(s)) +−12∆fH⊖(O2(g))
= 4× −285.83 kJ mol−1+ 10× −393.51 kJ mol−1−+78.53 kJ mol−1−12×0 = −5156.950 kJ mol−1
To calculate the heat released in this specific combustion we’ll next need to know how many moles of naphthalene were burned.
n= 120mg C10H8(s) 1g 1 mol
1000mg 128.17g = 9.363×10−
4mol C 10H8(s)
We can now easily calculate the heat producedqpin the combustion of this quantity of moles.
qp = n∆rH⊖
= 9.363×10−4mol× −5156.950 kJmol−1 = −4.883 kJ
The calorimeter absorbed this heat qa =−qp = 4.883 kJ. As we know the temperature change in the calorimeter resulting from the absorption of this heat, we can calculate the calorimeter constantC fromq=C∆T.
C = qa
∆T
= 4.883 kJ 3.05 K = 1.601 kJ K−1
As we now know the calorimeter constant we can go ahead and determine the calorimeter temperature change for burning the phenol sample. The balanced chemical equation for this reaction is
C6H5OH(s) + 7O2(g)→3H2O(l) + 6CO2(g) ∆rH⊖=?
and the enthalpy of this reaction is
∆rH⊖ = ∑
Products
νp∆fHp⊖−
∑
Reactants
νr∆fHr⊖
= 3∆fH⊖(H2O(l)) + 6∆fH⊖(CO2(g))−∆fH⊖(C6H5OH(s)) +−7∆fH⊖(O2(g))
= 3× −285.83 kJ mol−1+ 6× −393.51 kJ mol−1− −165.0 kJ mol−1−7×0 = −3053.55 kJ mol−1
To determine the heat, we’ll need the moles in this combustion.
n= 10mg C6H5OH(s) 1g 1 mol
1000mg 94.11g = 1.06×10
−4mol C 10H8(s)
Now we’ll calculate the heat produced in combustion.
qp = n∆rH⊖
= 1.06×10−4mol× −3053.55 kJ mol−1 = −0.324 kJ
And lastly we’ll determine the temperature change induced in the calorimeter by absorbing this heat.
∆T = qa
C
= 0.324kJ 1.601kJ K−1
Solution
•
The calorimeter constant is
C
= 1
.
6 kJ K
−1.
3
Exercise 2.24a pg 87
Given
For the reaction C2H5OH(l) + 3O2(g)→2CO2(g) + 3H2O(g), ∆rU⊖ =−1373 kJ mol−1 atT = 298 K.
In terms of given variables, this is written:
∆rU⊖ =−1373 kJ mol−1
T = 298 K
Find
Calculate ∆rH⊖ of this reaction.
Strategy
The following equation relates the enthalpy change of a reaction to the internal energy change (Equation 2.21 on pg 58 in your text book).
∆H = ∆U+ ∆ngRT
where ∆ngRT is the change in quantity of gas molecules of the reaction. In analyzing this reaction
C2H5OH(l) + 3O2(g)→2CO2(g) + 3H2O(g)
we see that 5 moles of gas are produced (2 moles of CO2(g) and 3 of H2O(g)) while 3 are consumed (O2(g)). Therefore
∆ng = 2 and ∆rH⊖ is easily calculated as
∆rH⊖ = ∆rU⊖+ ∆ngRT
= −1373×103J mol−1+ 2
mol gas
1 mol reaction×8.314 J
K−1mol−1×298K = −1.36804×106J mol−1
= −1368.04 kJ mol−1
Solution
4
Exercise 2.26a pg 87
Given
Consider the reaction
2NO2(g)→N2O4(g)
atT = 100◦C (T = 373 K) and the information in table 2.8
Find
Calculate the standard reaction enthalpy of this reaction at this temperature.
Strategy
For this problem, we’ll use the fact that enthalpy is a state function to create a series of processes identical to this reaction at the non-standard temperature. By summing summing the change in enthalpy over these steps we’ll arrive at the enthalpy of this reaction at the non-standard temperature.
These processes can be summarized as cooling 2 moles of NO2(g) from T = 373 K to the standard temperature
of T = 298 K. At this standard temperature the 2 moles of NO2(g) can be reacted to form 1 mole of N2O4(g)
and we can calculate the reaction enthalpy at this standard temperature. Lastly, we’ll heat the 1 mole of N2O4(g)
from T = 298 K to T = 373 K. Summing the enthalpy changes over these three processes will give us the enthalpy difference between 2 moles of NO2(g) and 1 mole of N2O4(g) atT = 373 K and this is the enthalpy difference of the
reaction of interest. This is summarized as
2 mol NO2(g) atT = 373 K
cool ∆Hcool
−−−−−−−→ 2 mol NO2(g)atT = 298 K
∆rH⊖(atT= 298 K)
−−−−−−−−−−−−−−→ 1 mol N2O4(g)atT = 298 K
∆Hheat
−−−−−→ 1 mol N2O4(g)atT = 373 K
∆rH⊖(atT = 373 K) = ∆Hcool+ ∆rH⊖(atT = 298 K) + ∆Hheat
The enthalpy of cooling or heating a substance can be determined from the substance’s constant pressure heat capacityCp.
∆H =
∫ Tf
Ti
CpdT
For this problem we’ll assume heat capacities are temperature independent over the temperature range of interest and this simplifies the calculations.
∆H = −
∫ Tf
Ti
CpdT
= Cp
∫ Tf
Ti
dT
= Cp(Tf−Ti) = Cp∆T
Using heat capacities from our textbook we can calculate the enthalpy of heating the 2 moles of NO2(g) and cooling
1 mole of N2O4(g).
∆Hcool = ν×Cp∆T (Cool NO2(g))
= 2×37.20 JK−1mol−1×(298K−373K) = =−5.5800 kJ mol−1
∆Hheat = ν×Cp∆T (Heat N2O4(g))
= 1×77.28 JK−1mol−1×(373K−298K) = = 5.7960 kJ mol−1
(Note that in these last two expressions the reaction coefficients are exact quantities and don’t affect number of significant figures.)
In the original solution sets the temperatures were swapped which changed the signs of these enthalpies.
The enthalpy of the reaction at the standard temperature ofT = 298 can be calculated from enthalpies of formation.
∆rH⊖ = ∑
Products
νp∆fHp⊖−
∑
Reactants
νr∆fHr⊖
= 1×9.16 kJ mol−1−2×33.18 kJ mol−1 = −57.2000 kJ mol−1
Summing these enthalpies gives the reaction enthalpy at the non-standard temperature
∆rH⊖(atT = 373 K) = ∆Hcool+ ∆rH⊖ (atT = 298 K) + ∆Hheat
= −5.5800 kJ mol−1+−57.2000 kJ mol−1+ 5.7960 kJ mol−1 = −56.9840 kJ mol−1
Solution
5
Exercise 2.29a pg 87
Given
We are given the following enthalpies . . .
• Enthalpy of sublimation of Mg(s) ∆subH⊖= +167.2 kJ mol−1
Mg(s)→Mg(g) ∆subH⊖= +167.2 kJ mol−1
• First ionization enthalpy of Mg(g) ∆ion1H⊖= 7.646 eV
Mg(g)→Mg+(g) ∆ion1H⊖ = 7.646 eV
• Second ionization enthalpy of Mg(g) ∆ion2H⊖= 15.035 eV
Mg+(g)→Mg+2(g) ∆ion2H⊖= 15.035 eV
• Dissociation enthalpy of Cl2(g) ∆disH⊖= +241.6 kJ mol−1
Cl2(g)→2Cl(g) ∆disH⊖= +241.6 kJ mol−1
• Electron gain enthalpy of Cl(g) ∆elecH⊖=−3.78 eV
Cl(g)→Cl−(g) ∆elecH⊖=−3.78 eV
• Enthalpy of solution of MgCl2(s) ∆solH⊖=−150.5 kJ mol−1
MgCl2(s)→Mg+2(aq) + 2Cl−(aq) ∆solH⊖ =−150.5 kJ mol−1
• Enthalpy of hydration of Cl−(g) ∆hydH⊖=−383.7 kJ mol−1
Cl−(g)→Cl−(aq) ∆hydH⊖=−383.7 kJ mol−1
Find
Calculate the enthalpy of hydration ∆hydH⊖ of for Mg+2(g).
Strategy
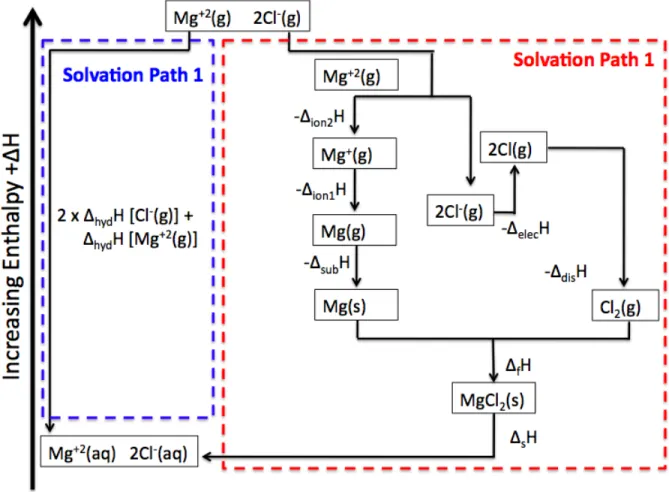
For this problem we need to construct a thermodynamic cycle which includes the unknown process; hydration of Mg+2(g), Mg+2(g) ∆hydH⊖. If we know the enthalpies all other processes in this cycle, then we can solve for the
single unknown enthalpy. Such a cycle is shown is Figure 5 where one mole of Mg+2(g) and two moles of Cl−(g) are solvated through two different pathways. These sum of enthalpies over each of these two paths are thereby equal. For this cycle we found it necessary to include a process that was not given to us by the textbook: the enthalpy of formation of MgCl2(s) from Mg(s) and Cl2(g). We can calculate the enthalpy for this process from its balanced
chemical equation
Mg(s) + Cl2(g)→MgCl2(s)
Figure 1: Thermodynamic cycle diagram
∆rH⊖ = ∑
Products
νp∆fHp⊖− ∑
Reactants
νr∆fHr⊖
= ∆fH⊖(MgCl2(s))−∆fH⊖(Mg(s))−∆fH⊖(Cl2(g))
= −641.32 kJ mol−1−0−0 = −641.32 kJ mol−1
This gives us the enthalpy of the additional process we need:
• The enthalpy of formation of MgCl2(s) ∆fH⊖=−641.32 kJ mol−1
Mg(s) + Cl2(g)→MgCl2(s) ∆fH⊖=−641.32 kJ mol−1
To reiterate, we had to obtain this quantity because it doesn’t appear possible to complete this problem with only the given information. The themodynamic cycle diagram in the figure also illustrates the fact that we can’t complete the cycle unless we link the MgCl2(s) to the corresponding stable elements. The formation reaction is precisely the missing link, and the problem statement didn’t have to provide it because we are able to obtain it using information from the Table 2.8.
If we equate the sum of the enthalpies of the two solvation paths in this thermodynamic cycle we get the following equation:
2×∆hydH⊖(Cl−(g)) + ∆hydH⊖(Mg+2(g)) =
[
−∆ion1H⊖(Mg(g)) +−∆ion2H⊖(Mg+(g)) +−∆subH⊖(Mg(s))
] [
−2×∆elecH⊖(Cl−(g)) +−∆disH⊖(Cl2(g))
]
+∆fH⊖(MgCl2(s)) + ∆solH⊖(MgCl2(s))
This equation can be solved for our unknown ∆hydH⊖(Mg+2(g))
∆hydH⊖(Mg+2(g)) =
[
−∆ion1H⊖(Mg(g)) +−∆ion2H⊖(Mg+(g)) +−∆subH⊖(Mg(s))
] [
−2×∆elecH⊖(Cl−(g)) +−∆disH⊖(Cl2(g))
]
+∆fH⊖(MgCl2(s)) + ∆solH⊖(MgCl2(s)) +
−2×∆hydH⊖(Cl−(g))
= [−737.722 kJ mol−1+−1450.650 kJ mol−1+−167.2 kJ mol−1]
[
−2× −364.71 kJ mol−1+−241.6 kJ mol−1+]+ −641.32 kJ mol−1+−150.5 kJ mol−1+
−2× −383.7 kJ mol−1 = −3346.012 kJ mol−1
where the energies in electron-volts have been converted by the following expression
∆ion1H⊖=
7.646eV 1.60218×10−19
J 1 kJ 6.0221×1023electrons
1eV 1000J 1 mol = 737.722 kJ mol
−1
∆ion2H⊖ =
15.035eV 1.60218×10−19J 1 kJ 6.0221×1023electrons
1eV 1000J 1 mol = 1450.650 kJ mol
−1
∆elecH⊖ = −
3.78eV 1.60218×10−19
J 1 kJ 6.0221×1023electrons
1eV 1000J 1 mol =−364.71 kJ mol
−1
Solution
6
Exercise 2.30a pg 87
Given
When a certain freon used in refrigeration was expanded adiabatically from an initial pressure ofpi = 32 atm and a
Ti= 0◦C to a final pressure ofpf= 1.00 atm, the temperature fell by ∆T = 22 K. In terms of given variables, this is written:
pi= 32 atm
Ti= 0◦C
pf= 1.00 atm
∆T=−22 K
Find
Calculate the Joule-Thomson coefficient,µ, atT = 0◦C assuming it remains constant over this temperature range.
Strategy
The Joule-Thomson coefficient is defined as
µ=
(
∂T ∂p
)
H
If we assumeµis constant over the range of temperature and pressures changes then the derivative can be replaced with finite differences.
µ= ∆T ∆p
Substituting in our temperature and pressure changes gives
µ = 1.00 atm−22 K−32 atm = 0.7097 K atm−1