Clinical review: Neuromonitoring an update
Full text
Figure


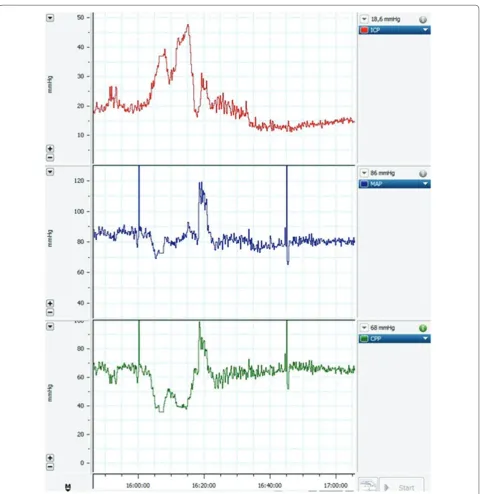
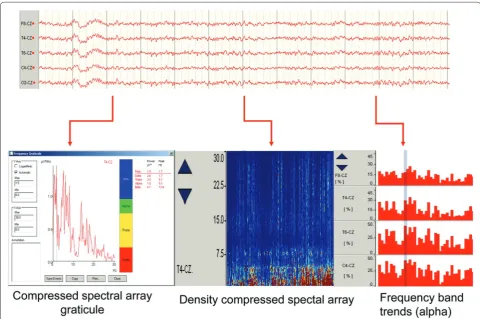
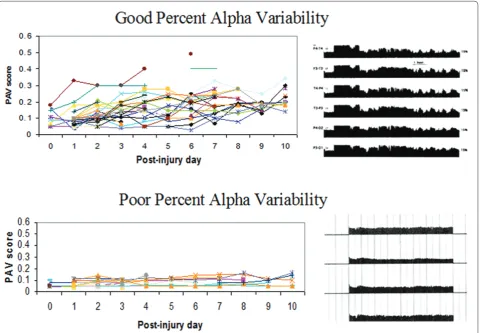
Related documents
The CareTrack Australia study was designed to estab- lish baseline estimates of the appropriateness of care de- livered, at a population level, for 22 common conditions [ 22 ].
A control simulating the coolant flow rate, determines the current flow rate cut of capacitor C, and represents the rate of removal of heat from the reactor by the coolant. It
present coal distribution system, small and moderate sized users contact.. coal brokers and/or marketers with their list of fuel
Thus, a physiotherapy program based on manual therapy may be safe in patients with hemophilia and inhibitor and such therapy may improve joint condition, pain, and joint range of
Conclusions: Posterior decompression and occipitocervical fixation followed by intraoperative vertebroplasty was a safe and valuable palliative method with relatively less invasion
Hewett TE, Myer GD, Ford KR, Heidt RS Jr, Colosimo AJ, McLean SG, van den Bogert AJ, Paterno MV, Succop P: Biomechanical measures of neuromuscular control and valgus loading of the
4) At 4- and 12-month follow-up, changes in the sec- ondary outcomes VAS pain, global disease activity, gen- eric and disease specific functioning and disability (measured by
AO: Arbeitsgemeinschaft für Osteosynthesefragen; CMS: Coleman Methodology Scoring; DI: Deep Infection; KOOS: Knee injury and Osteoarthritis Outcome Score; LOE: Level of Evidence;
