0095-1137/08/$08.00⫹0 doi:10.1128/JCM.01752-07
Copyright © 2008, American Society for Microbiology. All Rights Reserved.
Phylogenetic Backgrounds and Virulence Profiles of Atypical
Enteropathogenic
Escherichia coli
Strains from a
Case-Control Study Using Multilocus Sequence
Typing and DNA Microarray Analysis
䌤
Jan Egil Afset,
1,5* Endre Anderssen,
2Guillaume Bruant, Jose
´e Harel,
3Lothar Wieler,
4and Kåre Bergh
1,5Department of Laboratory Medicine, Children’s and Women’s Health,1and Department of Cancer Research and Molecular Medicine,2
Faculty of Medicine, Norwegian University of Science and Technology, and Department of Medical Microbiology, St. Olavs University Hospital,5Trondheim, Norway; Groupe de Recherche sur les Maladies Infectieuses du Porc,
Faculte´ de Me´decine Ve´te´rinaire, Universite´ de Montre´al, Saint-Hyacinthe, Que´bec J2S 7C2, Canada3; and
Institut fu¨r Mikrobiologie und Tierseuchen, Freie Universita¨t Berlin, P.O. Box 040225, D-10061 Berlin, Germany4
Received 3 September 2007/Returned for modification 26 October 2007/Accepted 30 April 2008
Atypical enteropathogeneticEscherichia coli(EPEC) strains are frequently detected in children with diar-rhea but are also a common finding in healthy children. The aim of this study was to compare the phylogenetic ancestry and virulence characteristics of atypical (eaepositive, stx and bfpA negative) EPEC strains from Norwegian children with (n ⴝ37) or without (nⴝ 19) diarrhea and to search for an association between phylogenetic ancestry and diarrhea. The strains were classified in phylogenetic groups by phylogenetic marker genes and in sequence types (STs) by multilocus sequence typing. Phylogenetic ancestry was compared to virulence characteristics based on DNA microarray analysis. Serotyping and pulsed-field gel electrophoresis (PFGE) were also performed. All four phylogenetic groups, 26 different STs, and 20 different clonal groups were represented among the 56 atypical EPEC strains. The strains were separated into three clusters by overall virulence gene profile; one large cluster with A, B1, and D strains and two clusters with group B2 strains. There was considerable heterogeneity in the PFGE profiles and serotypes, and almost half of the strains were O nontypeable. The efa1/lifA gene, previously shown to be statistically linked with diarrhea in this strain collection (J. E. Afset et al., J. Clin. Microbiol. 44:3703–3711, 2006), was present in 8 of 26 STs. The two phylogenetic groups B1 and D were weakly associated with diarrhea (Pⴝ0.06 andPⴝ0.09, respectively). In contrast, group B2 was isolated most frequently from healthy controls (Pⴝ0.05). In conclusion, the atypical EPEC strains were heterogeneous both phylogenetically and by virulence profile. Phylogenetic ancestry was less useful as a predictor of diarrhea than were specific virulence genes.
Escherichia coli is a commensal of the intestinal tract but also one of the most common causes of diarrhea in humans (34, 40). Several chromosomal and plasmid-borne virulence factors have been linked with the ability of certain E. coli
strains to cause diarrhea. Based on the content of such factors, several differentE. colipathotypes have been defined (30). A key characteristic of two of these pathotypes, enteropathogenic
E. coli(EPEC) and enterohemorrhagicE. coli(EHEC), is a chromosomally located pathogenicity island named the locus of enterocyte effacement (LEE). Genes located on the LEE encode factors enabling the bacterium to adhere intimately to intestinal epithelial cells and to cause attaching-and-effacing (A/E) lesions (36, 37). EPEC is differentiated from EHEC by the possession of the latter of Shiga toxin genes and is further characterized as typical or atypical depending on whether or not it carries the virulence plasmidE. coli adherence factor (EAF) encoding bundle-forming pili (29).
The E. coli species was subdivided into four phylogenetic
groups—A, B1, B2, and D-–based on multilocus enzyme elec-trophoresis (52). It has not been possible to establish reliable ancestral relationships between the phylogenetic groups due either to different speed of evolution among the groups or to frequent recombination (27, 61). Typical EPEC strains from different parts of the world have been shown to belong to four main clonal groups (33). In contrast, it has been argued that atypical EPEC strains may comprise a heterogeneous group of strains from different pathotypes that have acquired the LEE by horizontal transfer or are typical EPEC that have lost the EAF plasmid (58). Some atypical EPEC strains have shown a higher degree of genetic similarity to EHEC (O157:H7) than to typical EPEC (55). It has also been reported that strains classified as atypical EPEC may have very heterogeneous vir-ulence profiles (2, 58).
Whereas typical EPEC is well recognized as a leading cause of severe childhood diarrhea in low-income countries (39), the role of atypical EPEC as a diarrheagenic agent has been con-troversial. Atypical EPEC has been shown to be prevalent both in children with diarrhea and in healthy children (14, 16, 20, 23, 31, 38, 40–44, 47, 50, 57). However, several investigators have reported a statistical association between atypical EPEC and diarrhea (4, 15, 41, 49, 51, 58).
* Corresponding author. Mailing address: Department of Medical Microbiology, St. Olavs Hospital, N-7006 Trondheim, Norway. Phone: (47) 725 73319. Fax: (47) 725 76417. E-mail: [email protected].
䌤Published ahead of print on 7 May 2008.
2280
on May 16, 2020 by guest
http://jcm.asm.org/
In a recent case-control study from Norway, we found a high prevalence of atypical EPEC in children with or without diar-rhea (1). When the strains were analyzed with respect to vir-ulence gene content using DNA microarray and PCR, several genes were significantly associated with diarrhea (2). Among these, the association was highly significant for genes belonging to the pathogenicity island OI-122 (efa1/lifA, nleB, nleE, and
set/ent), and for long polar fimbriae (LPF) when three variants of thelpfAgene were analyzed together. In contrast, theyjaA
gene, which is often used as a phylogenetic marker, was neg-atively associated with diarrhea. This finding indicated a pos-sible association between pathogenic potential of the atypical EPEC strains and their phylogenetic relationships.
Multilocus sequence typing (MLST), by which a set of mul-tiple housekeeping gene loci are compared using sequence analysis, is a powerful tool for long-term epidemiological and phylogenetic analysis (35, 56). In the present study we used MLST to characterize the phylogenetic relationships between the atypical EPEC strains from the above-mentioned case-control study. In addition, we classified the strains in phyloge-netic groups using PCR and compared the phylogephyloge-netic ances-try of the strains with their virulence characteristics based on microarray analysis and with the results of serotyping and pulsed-field gel electrophoresis (PFGE). Finally, we searched for a possible association between phylogenetic ancestry and diarrhea.
MATERIALS AND METHODS
Bacterial strains.Atypical EPEC strains, defined aseae-positiveE. coli
with-outstx1,stx2, andbfpAgenes, were isolated from fecal specimens from children
younger than 5 years old in a case-control study conducted during the period from 2002 to 2003 in the county of Sør-Trøndelag, Norway (1). Thirty-seven of the strains were isolated from 251 children from whom a fecal specimen had been taken due to community-acquired diarrhea, and 19 strains were from 210
healthy children recruited at Maternal and Child Health Centers. Threeeae
-positive strains from the case-control study were classified as typical EPEC (bfpA
positive, two strains) or EHEC (stx2fpositive, one strain) and were therefore not
included in the present investigation. Fifteen strains, which had hybridized with
thestxB1probe in DNA microarray analysis (2), were classified as atypical EPEC
on basis of the following: they did not hybridize with the correspondingstxA1
probe, they were negative in the PCR analysis withstxB1 sequence-specific
primers (5, 10), and they did not produce Shiga toxins (Premier EHEC; Meridian Bioscience) (unpublished results). We therefore concluded that the gene
se-quences detected by thestxB1hybridization probe did not represent a complete
stxBgene. The 56 atypical EPEC isolates were stored at⫺70°C until further
analysis.
Phylogenetic group determination.Each atypical EPEC strain was assigned to
one of theE. coliphylogenetic groups according to the presence or absence of
the geneschuA,yjaA, andtspE4C2as proposed by Clermont et al. (13).
MLST.For each strain the seven housekeeping genesadk,fumC,gyrB,icd,
mdh,purA, andrecAwere amplified and sequenced according to the protocol of
the Escherichia coli MLST database (http://web.mpiib-berlin.mpg.de). After overnight culture, bacterial DNA for PCR was obtained by heat lysis. Amplifi-cation was carried out with primers as previously published (61) in a total volume
of 50l with 50M concentrations (each) of dATP, dCTP, dGTP, and dTTP; 0.5
M concentrations of each primer (MedProbe, Oslo, Norway); 10⫻PCR buffer
(Applied Biosystems, Branchburg, NL); 1.5 mM MgCl2; 1 U of AmpliTaq Gold
polymerase (Applied Biosystems); and 2l of bacterial DNA extract as a
tem-plate. The reaction conditions used were 15 min at 95°C, followed by 30 cycles of 1 min at 95°C, 1 min at the annealing temperature specified for each gene (see
reference 61 or theE. coliMLST website), and a 2-min extension at 72°C,
followed finally by 5 min at 72°C in a thermocycler, either an MJ Research PTC-200 (Bio-Rad, Hercules, CA) or a GeneAmp PCR System 9700 (Applied Biosystems). The PCR products were purified for sequencing with a QIAquick PCR purification kit (Qiagen, Valencia, CA). Both DNA strands were sequenced with the PCR primer set, with primers published by Tartof et al. (54), or with
primers designed in the present study—gyrB(Trh)F, 5⬘-AAGTGATCATGACC
GTTCTG-3⬘; icd(Trh)F, 5⬘-GATGGAATCGGTGTAGATGT-3⬘; icd(Trh)R 5⬘
-GTAGCCCCAGTCTTTAAACG-3⬘; and purA(Trh)R, 5⬘-TGCTTGCAGAGG
AACTCGC-3⬘. Sequencing was performed by using either the CEQ
DTCS-Quick Star kit (Beckman Coulter, Fullerton, CA) or a BigDye terminator cycle sequencing kit (v3.1; Applied Biosystems) with subsequent capillary electro-phoresis, respectively, on a Beckman Coulter CEQ 8800 or an ABI 3130x genetic analyzer according to the protocol of the manufacturers.
Sequence analysis.Raw sequence traces were reviewed by visual inspection using Sequencher software version 4.2 (Gene Code Corp.). Forward and reverse sequences were aligned, and consensus sequences corresponding to the allele
templates were compared to known variants of the corresponding gene at theE.
coliMLST database. Ambiguities in consensus sequences were resolved by
re-sequencing. Each of the seven gene loci was assigned an allele number by
submission of the sequences to theE. coliMLST database. Allelic sequences
previously not reported were given new allele numbers by the curator of the database after independent review of sequence traces. Each isolate was assigned a sequence type (ST) according to its allelic profile.
Phylogenetic analysis.For each bacterial strain the sequences from all seven gene loci were concatenated for phylogenetic analysis. Concatenated sequences were then aligned by using the CLUSTAL W algorithm of the MEGA3 software (32). A rooted neighbor-joining tree was constructed by using a Kimura
two-parameter model of nucleotide substitution and the phylogenetically divergentE.
colistrain Z205 from theE. coliMLST database as an outgroup (61). Tree
stability was assessed by bootstrap analysis with 1,000 iterations. Using SplitsTree 4 software (26), phylogenetic network analysis was done with the neighbor-net
algorithm and untransformed distances (pdistance). The SplitsTreew
recom-bination test was applied to concatenated sequences and for each of the seven gene loci individually to distinguish recombination from recurrent mutation.
The phylogenetic relationships between different STs were analyzed by using the MSTree application of Bionumerics version 4.6 (Applied Maths, Sint-Mar-tens-Latem, Belgium) to identify closely related genotypes. In this analysis atyp-ical EPEC strains are compared based on similarity in allelic profiles. Strains of different STs sharing six of seven alleles were interpreted as belonging to the
same clonal lineage but were assigned to an ST complex by the curator ofE. coli
MLST database only when that lineage included at least three different STs. The phylogenetic diversity of atypical EPEC strains isolated from children with diarrhea and healthy children was compared by using Simpson’s index of
diver-sity:D⫽1⫺[⌺n(n⫺1)/N(N⫺1)], wherenis the number of subjects with
atypical EPEC strains belonging to each ST, andNis the total number of subjects
in each of the two groups (25, 53).
Virulence gene profile.The atypical EPEC strains were compared with respect to virulence gene content based on the previously reported results of DNA microarray experiments and PCR (2). The microarray used in that study, derived
from anE. colivirulence and antimicrobial resistance microarray (8), was
com-posed of 70-mer oligonucleotide probes specific for 182 virulence genes or
markers found in various intestinal and extraintestinalE. colistrains of all known
pathotypes. The oligonucleotide microarray allowed the detection of genetic
variants of theeaegene (17 variants), and three variants each of theespA,espB,
andtir genes, all located on the LEE pathogenicity island. Oligonucleotides
specific for three variants [lpfA(O113),lpfA1, andlpfA(R141)] of the
LPF-encoding genelpfA were also included. The overall virulence profiles of the
atypical EPEC strains were compared by principal-component analysis (R, ver-sion 2.6 [www.r.project.org]) of the 95 virulence genes detected in one or more of the strains (2). The strains were assigned to virulence clusters based on their distribution in the principal component analysis.
PFGE.Macrorestriction analysis (PFGE) of chromosomal DNA was done using XbaI with the following electrophoretic conditions: 14°C, linear ramp of 5
to 60 s over 24 h, 120° switch angle, and a gradient of 6.0 V cm⫺1. Analysis was
done by using Bionumerics software. Similarities of fragments between strains were compared by using a Dice coefficient at 1.0% tolerance and 0.5% optimi-zation, and a dendrogram was constructed with the UPGMA (for unweighted pair-group method with arithmetic averages) clustering method. Significant clus-ters were determined by calculating the cutoff value that produced the highest point-bisectional correlation (Bionumerics manual, version 4.6).
Serotyping.Serotyping of somatic (O) antigens (serogroups O1 to O177) and
flagellar (H) antigens was done by using standard methods (24) at the
Esche-richia,Shigella,Yersinia, andVibrio Reference Unit, Laboratory for Enteric Pathogens at the Health Protection Agency (United Kingdom).
Statistical analyses.Fisher’s exact test was used for the statistical analyses of categorical variables, and the Mann-Whitney U-test was applied to the analysis
of differences between quantitative data which were not normally distributed.P
values⬍0.05 were considered significant.
on May 16, 2020 by guest
http://jcm.asm.org/
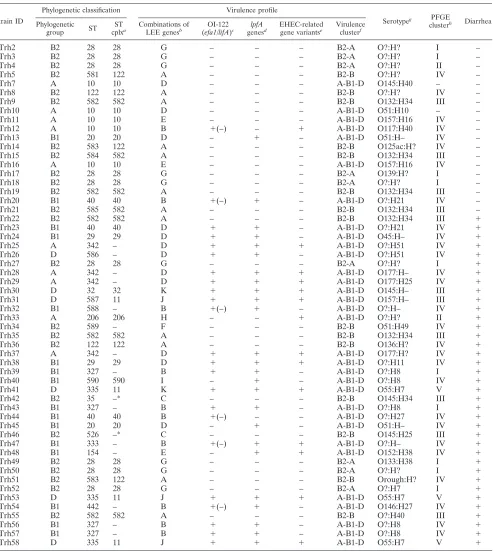
TABLE 1. Comparison of phylogenetic classification, virulence profile, serotype, PFGE clustering, and clinical information for each of the 56 atypical EPEC strains identified in a case-control study among children⬍5 years old in Norway
Strain ID
Phylogenetic classification Virulence profile
Serotypeg PFGE
clusterh Diarrhea
Phylogenetic
group ST
ST
cplxa Combinations of
LEE genesb OI-122
(efa1/lifA)c lpfA
genesd EHEC-related
gene variantse Virulence
clusterf
Trh2 B2 28 28 G – – – B2-A O?:H? I –
Trh3 B2 28 28 G – – – B2-A O?:H? I –
Trh4 B2 28 28 G – – – B2-A O?:H? II –
Trh5 B2 581 122 A – – – B2-B O?:H? IV –
Trh7 A 10 10 D – – – A-B1-D O145:H40 – –
Trh8 B2 122 122 A – – – B2-B O?:H? IV –
Trh9 B2 582 582 A – – – B2-B O132:H34 III –
Trh10 A 10 10 D – – – A-B1-D O51:H10 – –
Trh11 A 10 10 E – – – A-B1-D O157:H16 IV –
Trh12 A 10 10 B ⫹(–) – ⫹ A-B1-D O117:H40 IV –
Trh13 B1 20 20 D – ⫹ – A-B1-D O51:H– IV –
Trh14 B2 583 122 A – – – B2-B O125ac:H? IV –
Trh15 B2 584 582 A – – – B2-B O132:H34 III –
Trh16 A 10 10 E – – – A-B1-D O157:H16 IV –
Trh17 B2 28 28 G – – – B2-A O139:H? I –
Trh18 B2 28 28 G – – – B2-A O?:H? I –
Trh19 B2 582 582 A – – – B2-B O132:H34 III –
Trh20 B1 40 40 B ⫹(–) ⫹ – A-B1-D O?:H21 IV –
Trh21 B2 585 582 A – – – B2-B O132:H34 III –
Trh22 B2 582 582 A – – – B2-B O132:H34 III ⫹
Trh23 B1 40 40 D ⫹ ⫹ – A-B1-D O?:H21 IV ⫹
Trh24 B1 29 29 D ⫹ ⫹ – A-B1-D O45:H– IV ⫹
Trh25 A 342 – D ⫹ ⫹ ⫹ A-B1-D O?:H51 IV ⫹
Trh26 D 586 – D ⫹ ⫹ – A-B1-D O?:H51 IV ⫹
Trh27 B2 28 28 G – – – B2-A O?:H? I ⫹
Trh28 A 342 – D ⫹ ⫹ ⫹ A-B1-D O177:H– IV ⫹
Trh29 A 342 – D ⫹ ⫹ ⫹ A-B1-D O177:H25 IV ⫹
Trh30 D 32 32 K ⫹ ⫹ ⫹ A-B1-D O145:H– III ⫹
Trh31 D 587 11 J ⫹ ⫹ ⫹ A-B1-D O157:H– III ⫹
Trh32 B1 588 – B ⫹(–) ⫹ – A-B1-D O?:H– IV ⫹
Trh33 A 206 206 H – – ⫹ A-B1-D O?:H? II ⫹
Trh34 B2 589 – F – – – B2-B O51:H49 IV ⫹
Trh35 B2 582 582 A – – – B2-B O132:H34 III ⫹
Trh36 B2 122 122 A – – – B2-B O136:H? IV ⫹
Trh37 A 342 – D ⫹ ⫹ ⫹ A-B1-D O177:H? IV ⫹
Trh38 B1 29 29 D ⫹ ⫹ ⫹ A-B1-D O?:H11 IV ⫹
Trh39 B1 327 – B ⫹ ⫹ – A-B1-D O?:H8 I ⫹
Trh40 B1 590 590 I – ⫹ – A-B1-D O?:H8 IV ⫹
Trh41 D 335 11 K ⫹ ⫹ ⫹ A-B1-D O55:H7 V ⫹
Trh42 B2 35 –* C – – – B2-B O145:H34 III ⫹
Trh43 B1 327 – B ⫹ ⫹ – A-B1-D O?:H8 I ⫹
Trh44 B1 40 40 B ⫹(–) – – A-B1-D O?:H27 IV ⫹
Trh45 B1 20 20 D – ⫹ – A-B1-D O51:H– IV ⫹
Trh46 B2 526 –* C – – – B2-B O145:H25 III ⫹
Trh47 B1 333 – B ⫹(–) ⫹ ⫹ A-B1-D O?:H– IV ⫹
Trh48 B1 154 – E – ⫹ ⫹ A-B1-D O152:H38 IV ⫹
Trh49 B2 28 28 G – – – B2-A O133:H38 I ⫹
Trh50 B2 28 28 G – – – B2-A O?:H? I ⫹
Trh51 B2 583 122 A – – – B2-B Orough:H? IV ⫹
Trh52 B2 28 28 G – – – B2-A O?:H7 I ⫹
Trh53 D 335 11 J ⫹ ⫹ ⫹ A-B1-D O55:H7 V ⫹
Trh54 B1 442 – B ⫹(–) ⫹ – A-B1-D O146:H27 IV ⫹
Trh55 B2 582 582 A – – – B2-B O?:H40 III ⫹
Trh56 B1 327 – B ⫹ ⫹ – A-B1-D O?:H8 IV ⫹
Trh57 B1 327 – B ⫹ ⫹ – A-B1-D O?:H8 IV ⫹
Trh58 D 335 11 J ⫹ ⫹ ⫹ A-B1-D O55:H7 V ⫹
a
ST cplx, ST complex, based on the identity in at least six of the seven gene loci analyzed, between strains of at least three different STs. *, STs with identity in six
of seven gene loci but which have not been assigned to an ST complex at theE. coliMLST database.
b
Classified by the combination of the variants of the genesespA,espB,tir, andeaedetected in each strain. See Table 3 for details.
c
Based, in addition to theefa1/lifagene, on the genesnleB,nleE, andset/ent.
d
Based on the gene variantslpfA1,lpfA(O113), andlpfA(R141).
e
Classified by the EHEC plasmid geneskatP,espP,etpD,ehxA, and L7095 and the chromosomalureDgene.
f
Based on principal component analysis of 94 putative virulence genes or gene variants present in one or more of the strains.
g
O?, O serogroup not identified; H?, motile, but H-type not identified.
h
PFGE clusters were defined by the cluster cutoff method of the Bionumerics software. –, Two ST10 strains were not typeable with the method used.
on May 16, 2020 by guest
http://jcm.asm.org/
RESULTS
Phylogenetic group determination. Ten of the atypical EPEC strains belonged to phylogenetic group A, 16 strains belonged to group B1, 24 strains belonged to group B2, and 6 strains belonged to group D when classified according to the scheme proposed by Clermont et al. (13) (Table 1).
MLST analysis. Sequencing of each of the seven MLST gene loci generated acceptable tracings in all 56 atypical EPEC strains tested. Seven new alleles were detected, and altogether 26 different STs were identified (Table 1). Fifteen strains with allele profiles not previously reported were assigned to 10 new STs.
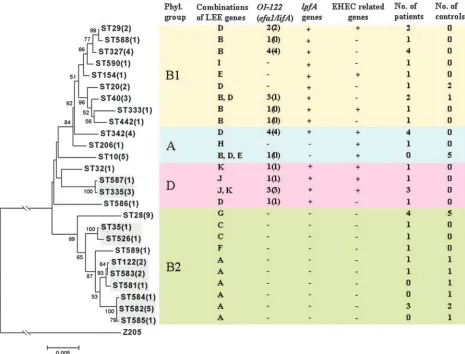
Phylogenetic analysis of concatenated sequences of the seven MLST loci showed that STs within the phylogenetic group B2 were grouped together and were clearly distinct from STs of the three other phylogenetic groups (Fig. 1). SplitsTree analysis revealed several parallel paths indicating phylogenetic incompatibility in the divergence of atypical EPEC clones (Fig. 2). By using thewtest, statistical significant evidence of re-combination was demonstrated (P⫽6.8⫻10⫺6). When each
gene was analyzed separately, evidence for recombination was found for the genesfumC(P⫽0.002) andgyrB(P⫽0.025) but not for the five other genes.
Ten STs had at least six alleles similar to other STs in the study. Eight of these STs belonged to three different ST com-plexes, while two STs (ST35 and ST526) were not assigned to an ST complex due to the lack of a third closely related ST (Table 1 and Fig. 1). Clonal complex 582 (ST582, ST584, and ST585) has not been described previously. Altogether, 20 dif-ferent clonal lineages were represented among the 56 atypical EPEC strains in the study.
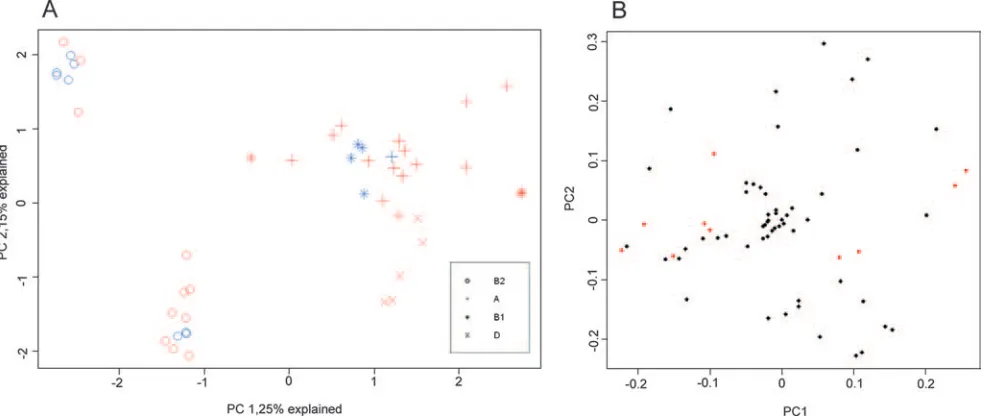
Phylogenetic background compared to virulence character-istics.The atypical EPEC strains were assigned to three dif-ferent clusters by principal-component analysis of the 94 viru-lence genes or markers identified in the study (Fig. 3). Two of the clusters were highly compact, indicating a high degree of similarity in virulence profiles between the strains. Both of these clusters consisted exclusively of phylogenetic group B2 strains and were labeled clusters B2-A and B2-B. The third cluster was much wider, indicating more heterogeneity in
vir-FIG. 1. Phylogenetic relationships between 56 atypical EPEC strains from Norwegian children. A rooted phylogenetic tree was constructed by the neighbor-joining algorithm based on the Kimura two-parameter model of nucleotide substitution. The ST with the number of isolates (in brackets) is given at each branch tip. Bootstrap values greater than 50% based on 1,000 replications are shown at the internal nodes. Clonally related STs are shown by shaded boxes. The classification in phylogenetic groups, distribution of LEE gene variants, the presence of OI-122,lpfA, and EHEC-related genes, and the sources of the strains are shown for each ST. For combinations of LEE gene variants, see Table 3.
on May 16, 2020 by guest
http://jcm.asm.org/
[image:4.585.58.523.68.422.2]ulence genes between these strains. This cluster included all of the phylogenetic group A, B1, and D strains and was accord-ingly labeled cluster A-B1-D. Phylogenetic group B and espe-cially group A strains were widely dispersed within this cluster. Phylogenetic group D strains, in contrast, were located at the periphery and may represent a separate cluster.
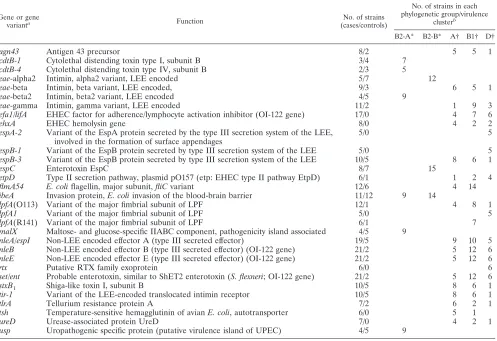
Thirty of the genes or gene variants observed in at least five bacterial strains were restricted to strains of either phyloge-netic group B2 or one or more of the phylogephyloge-netic groups A, B1, and D (Table 2). Among genes present only in phyloge-netic group B2 strains, all but one gene were observed in only one of the B2 virulence clusters, either B2-A or B2-B. In contrast, 18 of the 23 genes detected exclusively in strains belonging to the virulence cluster A-B1-D were present in more than one of the three phylogenetic groups of this cluster. Four genes were found only in strains belonging to phyloge-netic group D, and one gene variant [lpfA(R141)] was detected only in phylogenetic group B1 strains.
The atypical EPEC strains were also analyzed with respect to their total content of the virulence genes and markers ana-lyzed in the study. There were considerable differences in the number of virulence genes between the strains, ranging from 22 of 46 genes per strain (median, 29 genes). Strains belonging to phylogenetic group D contained significantly more virulence genes (median, 37 genes; range, 34 to 42 genes) than strains belonging to the phylogenetic groups A, B1, and B2 (P⫽0.02, 0.01, and 0.001, respectively). The differences between the three other groups were not significant.
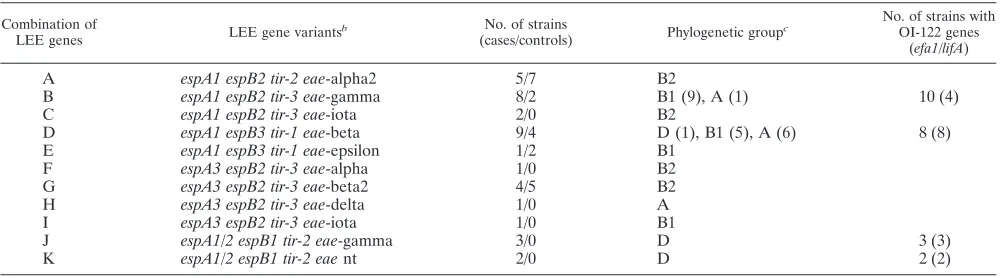
Ten combinations of different variants of the LEE genes
espA, espB, tir, and eae were present among the 56 atypical EPEC strains (Table 3). Each of the four phylogenetic groups, and even some of the STs, included strains with different com-binations of LEE genes (Fig. 1). Some of these comcom-binations were observed only within one phylogenetic group, but two of the most frequent combinations were detected in strains from more than one group. Although most of theeaevariants were associated with only one specific combination of the espA,
[image:5.585.44.283.69.302.2]espB, and tirgene variants, the gamma and the iota variants were linked with two different combinations of the other LEE genes (Table 3).
FIG. 2. Phylogenetic relationships between 56 atypical EPEC strains from Norwegian children are presented as a phylogenetic splits network based on the neighbor-net algorithm using apdistance matrix. STs are indicated at the branch tips. The sources of the strains are listed in parentheses (patients/controls). STs belonging to the same phylogenetic group are enclosed by an ellipse.
FIG. 3. Principal component analysis of the distribution of 94 putative virulence genes or gene variants in 56 atypical EPEC strains isolated from Norwegian children. Scores (A) and loadings (B) for the first two principal components; the phylogenetic group of each strain is indicated by the design of the marker, while the color shows whether the strain was isolated from children with (red) or without (blue) diarrhea. The loadings show genes previously shown to be statistically linked with diarrhea in red (2).
on May 16, 2020 by guest
http://jcm.asm.org/
[image:5.585.47.539.478.687.2]Genes located on the pathogenicity island OI-122 (nleB,
nleE,set/ent, andefa1/lifA) were detected in strains belonging to 12 different STs (Fig. 1). Such genes were present in strains belonging to the three phylogenetic groups A, B1, and D but not group B2. The presence of the OI-122 genes was associated with four different combinations of LEE gene variants (Table 3). Incomplete OI-122s lacking theefa1/lifA gene always cor-responded with one specific combination of LEE gene variants. The three variants of thelpfAgene were detected in 14 differ-ent STs within the phylogenetic groups A, B1, and D (Fig. 1). Genes linked to the EHEC pathotype, the pO157 plasmid geneskatP,espP,etpD,ehxA, and L7095 and the EHEC urease geneureDwere detected in 14 (25%) strains. These genes were present in strains of nine different STs within the phylogenetic groups A, B1, and D (Fig. 1). Eight different combinations between the EHEC-related genes were observed (data not shown). The 15 strains that contained an incomplete stxB1
sequence belonged to seven different STs within the phyloge-netic groups A, B1, and D.
PFGE.When the 56 atypical EPEC strains were analyzed by PFGE, all but two ST10 strains were typeable. Among the 54 typeable strains, the majority displayed unique genotypic
pat-terns (Fig. 4). Except for two strains with identical PFGE restriction patterns isolated from siblings, there was no known epidemiological connection between other strains with closely related PFGE profiles. Five distinct clusters were identified by using the cluster cutoff method, while 42 different clusters identified if a similarity cutoff value of 90% was applied.
Serotypes.An O serogroup was identified in 31 (55.3%) of the 56 atypical EPEC strains (Table 1). The remaining strains were either nontypeable with the O antisera used (24 strains) or rough (1 strain). Four strains belonged to classical EPEC serogroups (39). The other typeable strains belonged to 12 different serogroups. Five O serogroups were detected in more than one strain; O132 (six strains), O145 (four strains), O55 and O157 (three strains each), and O177 (two strains).
Eleven different H-types were detected among the atypical EPEC strains. Most common were the flagellar types H34 (seven strains), H8 (five strains), and H7 (four strains). Seven strains were nonmotile (H⫺), and the H-type was not identi-fied in 14 strains.
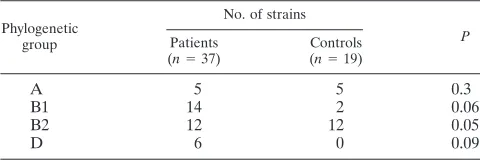
[image:6.585.46.543.82.423.2]Phylogenetic ancestry and diarrhea.An association between phylogenetic decent and diarrhea could indicate the presence of virulence traits linked to phylogenetic ancestry. In the
TABLE 2. Genes or gene variants restricted to one virulence cluster or phylogenetic group in 56 atypical EPEC strains
Gene or gene
varianta Function No. of strains
(cases/controls)
No. of strains in each phylogenetic group/virulence
clusterb
B2-A* B2-B* A† B1† D†
agn43 Antigen 43 precursor 8/2 5 5 1
cdtB-1 Cytolethal distending toxin type I, subunit B 3/4 7 cdtB-4 Cytolethal distending toxin type IV, subunit B 2/3 5
eae-alpha2 Intimin, alpha2 variant, LEE encoded 5/7 12
eae-beta Intimin, beta variant, LEE encoded, 9/3 6 5 1
eae-beta2 Intimin, beta2 variant, LEE encoded 4/5 9
eae-gamma Intimin, gamma variant, LEE encoded 11/2 1 9 3
efa1/lifA EHEC factor for adherence/lymphocyte activation inhibitor (OI-122 gene) 17/0 4 7 6
ehxA EHEC hemolysin gene 8/0 4 2 2
espA-2 Variant of the EspA protein secreted by the type III secretion system of the LEE, involved in the formation of surface appendages
5/0 5
espB-1 Variant of the EspB protein secreted by type III secretion system of the LEE 5/0 5 espB-3 Variant of the EspB protein secreted by type III secretion system of the LEE 10/5 8 6 1
espC Enterotoxin EspC 8/7 15
etpD Type II secretion pathway, plasmid pO157 (etp: EHEC type II pathway EtpD) 6/1 1 2 4 flmA54 E. coliflagellin, major subunit,fliCvariant 12/6 4 14 ibeA Invasion protein,E. coliinvasion of the blood-brain barrier 11/12 9 14
lpfA(O113) Variant of the major fimbrial subunit of LPF 12/1 4 8 1
lpfA1 Variant of the major fimbrial subunit of LPF 5/0 5
lpfA(R141) Variant of the major fimbrial subunit of LPF 6/1 7
malX Maltose- and glucose-specific IIABC component, pathogenicity island associated 4/5 9
nleA/espI Non-LEE encoded effector A (type III secreted effector) 19/5 9 10 5 nleB Non-LEE encoded effector B (type III secreted effector) (OI-122 gene) 21/2 5 12 6 nleE Non-LEE encoded effector E (type III secreted effector) (OI-122 gene) 21/2 5 12 6
rtx Putative RTX family exoprotein 6/0 6
set/ent Probable enterotoxin, similar to ShET2 enterotoxin (S. flexneri; OI-122 gene) 21/2 5 12 6
stxB1 Shiga-like toxin I, subunit B 10/5 8 6 1
tir-1 Variant of the LEE-encoded translocated intimin receptor 10/5 8 6 1
tlrA Tellurium resistance protein A 7/2 6 2 1
tsh Temperature-sensitive hemagglutinin of avianE. coli, autotransporter 6/0 5 1
ureD Urease-associated protein UreD 7/0 4 2 1
usp Uropathogenic specific protein (putative virulence island of UPEC) 4/5 9
a
Only genes present in at least five strains are included in this analysis.
b
*, Strains belonging to phylogenetic group B2 are separated into two clusters (B2-A and B2-B) by principal component analysis of virulence genes (see Fig. 3); †, strains belonging to the phylogenetic groups A, B1, and D are located in the same cluster (A-B1-D) based on principal component analysis of virulence genes (see Fig. 3).
on May 16, 2020 by guest
http://jcm.asm.org/
present study none of the phylogenetic groups A, B1, and D were significantly associated with diarrhea (Table 4). However, a trend toward significance was observed for the groups B1 (P⫽
0.06) and D (P⫽0.09). In contrast, strains belonging to phy-logenetic group B2 were more common in healthy controls than in children with diarrhea. Phylogenetic group A strains were equally common in patients and controls.
Among STs that included three or more strains, three STs were detected only in children with diarrhea, one ST was ob-served only in healthy children, and four STs were present in strains from both healthy and ill children (Fig. 1). The STs that were detected exclusively in strains from children with diarrhea all contained the virulence geneefa1/lifAgene.
There was a higher degree of diversity in phylogenetic an-cestry between strains isolated from children with diarrhea than in strains from healthy children. All but one of the 20 different clonal lineages detected in the study were present among the 37 strains from symptomatic children (Simpson’s diversity index of 0.95). In comparison, no more than six clonal lineages were present among the 19 strains from healthy chil-dren (Simpson’s diversity index of 0.83).
DISCUSSION
In this study we show that atypical EPEC strains from Nor-wegian children with or without diarrhea are very heteroge-neous with respect to phylogenetic ancestry. The 56 strains belonged to all four phylogenetic groups and to 26 different STs within 20 clonal groups (Table 1 and Fig. 1). This finding of phylogenetic heterogeneity is consistent with analyses from the E. coli MLST database, where EPEC strains, probably mainly atypical EPEC (unpublished data), were found within many different STs and clonal complexes (61). In contrast, Lacher et al. have shown that typical EPEC strains are more homogeneous and mainly belong to four clonal groups (33). However, both in the typical EPEC strains in that study and the atypical strains in the present study there was significant evidence of recombination.
Comparison between phylogenetic background and viru-lence characteristics has usually been done only for a limited number of virulence factors (15, 22, 42, 45, 48, 58) or for strains
belonging to specific EPEC serotypes (6). The use of data from DNA microarray experiments in the present study permitted an extensive characterization of the atypical EPEC strains with respect to overall virulence gene content, as well as a compar-ison between virulence profile and phylogenetic ancestry (Fig. 3). The main division between group B2 strains and the three other groups—A, B1, and D—observed in the phylogenetic analysis (Fig. 1) was supported by differences in virulence pro-files (Fig. 3). There was also agreement between the two dif-ferent types of analyses in the separation of phylogenetic group B2 strains in two clusters, while phylogenetic group A and B1 strains were not reliably differentiated by any of the two meth-ods. Group D strains, on the other hand, were narrowly scat-tered at the periphery of this cluster (Fig. 3) and were also shown to contain significantly more virulence genes than strains belonging to the other virulence groups. The link ob-served in the present study between phylogenetic ancestry and virulence profile (Table 2 and Fig. 3) may be explained by the requirement of a specific genetic background for the acquisi-tion of certain virulence factors (17).
One possible reason for poor separation between strains belonging to different groups in the phylogenetic analysis might be the presence of hybrid strains carrying ancestry from more than one source (61). Such hybrid strains were not iden-tified by the PCR method used for the analysis of phylogenetic groups in the present study. However, at least four of the STs identified in the study (ST 28, 32, 154, and 206) were previously observed to contain phylogenetic ancestry from more than one source (61).
[image:7.585.43.543.89.228.2]Typing of LEE genes may be of importance in the charac-terization of A/E pathogens since different variants of these genes have been associated with tropism to different locations in the human intestine (19, 46) and may be used for epidemi-ological characterization of LEE-containing strains. The LEE genesespA,espB,tir, andeaeare all genes that may be exposed to high selection pressure since they encode adhesins and ef-fector proteins that interact directly with the host. In the present study 11 different combinations of these four LEE gene variants were identified. In contrast to previous reports (12, 21), several of theespA,espB,tir, andeaevariants could be
TABLE 3. Distribution of different variants of four LEE genes compared to phylogenetic group and the presence of OI-122 genes in 56 atypical EPEC strains isolated from children⬍5 years old from Norway with or without diarrheaa
Combination of
LEE genes LEE gene variants
b No. of strains
(cases/controls) Phylogenetic group
c No. of strains with
OI-122 genes (efa1/lifA)
A espA1 espB2 tir-2 eae-alpha2 5/7 B2
B espA1 espB2 tir-3 eae-gamma 8/2 B1 (9), A (1) 10 (4) C espA1 espB2 tir-3 eae-iota 2/0 B2
D espA1 espB3 tir-1 eae-beta 9/4 D (1), B1 (5), A (6) 8 (8) E espA1 espB3 tir-1 eae-epsilon 1/2 B1
F espA3 espB2 tir-3 eae-alpha 1/0 B2 G espA3 espB2 tir-3 eae-beta2 4/5 B2 H espA3 espB2 tir-3 eae-delta 1/0 A I espA3 espB2 tir-3 eae-iota 1/0 B1
J espA1/2 espB1 tir-2 eae-gamma 3/0 D 3 (3)
K espA1/2 espB1 tir-2 eaent 2/0 D 2 (2)
aFor hybridization probe sequences, see reference 2.
bFor descriptions of the genes, see Table 2. nt, nontypeable.
cIn cases of more than one group, the number of results for each group is given in parentheses.
on May 16, 2020 by guest
http://jcm.asm.org/
FIG. 4. PFGE cluster analysis compared to phylogenetic ancestry, virulence profile, phylogenetic group, and source of atypical EPEC strains from Norwegian children with or without diarrhea. Clusters identified by the cluster cutoff method (see the text) are visualized by dense lines in the dendrogram. Two strains (Trh7 and Trh10) were not typeable by the PFGE method used. The STs 35 and 526 share six of seven alleles but have not been assigned to a ST complex. See Table 1 for more information.
2287
on May 16, 2020 by guest
observed in combination with more than just one variant of the other LEE genes. This finding is consistent, as recently sug-gested, with horizontal exchange between different strains not only of entire LEE sequences but also of smaller gene ele-ments within the LEE (11). The link between certain combi-nations of LEE gene variants and OI-122 genes shown in the present study (Table 3) may be due to close proximity of the genomic islands in the chromosome of these atypical EPEC strains, a finding similar to what has been shown for O103:H2 EHEC strains (28).
The finding of genes usually linked to the EHEC pathotype in a considerable proportion of the atypical EPEC strains (Ta-ble 1) is consistent with evidence from epidemiological and experimental studies showing that atypical EPEC may convert to, or be a conversion from, the EHEC pathotype through the acquisition or loss ofstxgenes (7, 58, 61). Such a relationship is further supported by the presence of STs belonging to the phylogenetic lineages EHEC1 (ST335 and ST587) and EHEC2 (ST29) (59; http://www.shigatox.net) among the atypical EPEC strains. The variability in the content of plasmid genes between different strains is in agreement with reports of extensive het-erogeneity of large plasmids in STEC (9) and A/EE. coliof animal origin (3).
The main impression from the results of the PFGE analysis is that of extensive heterogeneity between the atypical EPEC strains in the study. Differences in the PFGE banding pattern of several fragments were observed even between strains be-longing to the same clonal lineage or virulence group (Fig. 4). The PFGE results also confirm a pattern of endemic infection among children during the study period and demonstrate, as expected, that the PFGE method has greater discriminatory power than MLST in differentiating between epidemiologically unrelated atypical EPEC strains.
The finding that an O serogroup was identified in less than half of the strains in the study (Table 1) supports the view that O serogrouping is not useful in the diagnosis of atypical EPEC infections, at least with endemic atypical EPEC strains as re-ported here. This view is also supre-ported by the observation that OI-122-positive atypical EPEC strains, which we have previ-ously shown to be significantly associated with diarrhea (2), belonged to many different serogroups or were nontypeable (Table 1).
Phylogenetic ancestry was a less useful indicator of diarrhea-genic potential in this collection of atypical EPEC strains (Ta-ble 4) than specific virulence genes reported previously (2). This is most likely explained by the considerable heterogeneity in virulence factors within each of the phylogenetic groups
(Fig. 3 and Table 2). The OI-122 geneefa1/lifAmost strongly associated with diarrhea in our previous study was present only in some of the strains within the phylogenetic groups A and B1 (Fig. 1). On the other hand, the negative statistical association with diarrhea shown for the phylogenetic group B2 seems to indicate a link between phylogenetic descent and lack of diar-rheagenic potential.
The lack of significant association observed between phylo-genetic groups and diarrhea in the present study may in part be explained by the limited number of strains analyzed. In addi-tion, the high number of STs among the strains made a statis-tical analysis for the relationship with diarrhea for each ST impossible. A considerably larger study than the present will be needed to clarify this issue further. However, the finding that some STs, present in more than two strains, were represented in only one of the groups of children (patients or controls) may indicate a difference in virulence potential between these STs (Fig. 1). Interestingly, the three STs that were detected exclu-sively in strains from children with diarrhea all contained the
efa1/lifAgene.
A limitation of the comparison between phylogeny and vir-ulence genes used in the present study is that only a small part of the genome of each strain was included in the analysis. It is therefore possible that as-yet-unrecognized virulence genes were missed. An alternative could be to do a comparison at the whole-genome level, for instance, by comparative genomic (18) or subtractive (60) hybridization. However, the strength of the present study is the analysis of virulence genes of all patho-types. In contrast, an analysis at the whole-genome level is usually based on hybridization of the test strain against a se-quenced reference strain. In such an analysis, genes which are not present in the genome of the reference strains will not be detected.
In conclusion, we have shown that atypical EPEC strains from Norwegian children with or without diarrhea belonged to all four phylogenetic groups, to 26 different STs, and to 20 different clonal groups. The strains were separated into three clusters by overall virulence gene profile. One large cluster included all phylogenetic group A, B1, and D strains, and two clusters consisted exclusively of strains belonging to group B2. Almost one-third of the virulence genes were detected in only one virulence cluster or phylogenetic group. There was con-siderable heterogeneity in PFGE profiles and serotypes, and almost half of the strains were O nontypeable. Theefa1/lifA
and other OI-122 genes were detected in many different STs, but only within the phylogenetic groups A, B1, and D, and were linked with certain combinations of LEE gene variants. EHEC pathotype-related genes were present in one-fourth of the strains. There was borderline significant association with diarrhea for the phylogenetic groups B1 and D, but phyloge-netic ancestry was less useful as a predictor of diarrhea than the specific virulence genes shown previously.
ACKNOWLEDGMENTS
We thank Kirsti Løseth, Hilde Lysvand, Anne Nor, Sidsel Krokhaug, and Torunn Rønning for excellent technical assistance and Mark Acht-man, curator of the Escherichia coli MLST database, for updated information on ST complexes.
[image:9.585.43.283.99.179.2]J.E.A. was supported by a Ph.D. grant from the Central Norway Regional Health Authority.
TABLE 4. Distribution in phylogenetic groups of 56 atypical EPEC strains from children with or without diarrhea with respect to
the source of the strains
Phylogenetic group
No. of strains
P
Patients
(n⫽37)
Controls
(n⫽19)
A 5 5 0.3
B1 14 2 0.06
B2 12 12 0.05
D 6 0 0.09
on May 16, 2020 by guest
http://jcm.asm.org/
REFERENCES
1.Afset, J. E., L. Bevanger, P. Romundstad, and K. Bergh.2004. Association of
atypical enteropathogenic Escherichia coli(EPEC) with prolonged
diar-rhoea. J. Med. Microbiol.53:1137–1144.
2.Afset, J. E., G. Bruant, R. Brousseau, J. Harel, E. Anderssen, L. Bevanger, and K. Bergh.2006. Identification of virulence genes linked with diarrhea
due to atypical enteropathogenicEscherichia coliby DNA microarray
anal-ysis and PCR. J. Clin. Microbiol.44:3703–3711.
3.Aktan, I., K. A. Sprigings, R. M. La Ragione, L. M. Faulkner, G. A. Paiba, and M. J. Woodward.2004. Characterisation of attaching-effacing Esche-richia coliisolated from animals at slaughter in England and Wales. Vet.
Microbiol.102:43–53.
4.Alikhani, M. Y., A. Mirsalehian, and M. M. Aslani.2006. Detection of
typical and atypical enteropathogenicEscherichia coli(EPEC) in Iranian
children with and without diarrhoea. J. Med. Microbiol.55:1159–1163.
5.Bekal, S., R. Brousseau, L. Masson, G. Prefontaine, J. Fairbrother, and J. Harel.2003. Rapid identification ofEscherichia colipathotypes by virulence
gene detection with DNA microarrays. J. Clin. Microbiol.41:2113–2125.
6.Beutin, L., S. Kaulfuss, S. Herold, E. Oswald, and H. Schmidt.2005. Genetic
analysis of enteropathogenic and enterohemorrhagicEscherichia coli
sero-group O103 strains by molecular typing of virulence and housekeeping genes
and pulsed-field gel electrophoresis. J. Clin. Microbiol.43:1552–1563.
7.Bielaszewska, M., R. Prager, R. Kock, A. Mellmann, W. Zhang, H. Tschape, P. I. Tarr, and H. Karch.2007. Shiga toxin gene loss and transfer in vitro and
in vivo during enterohemorrhagicEscherichia coliO26 infection in humans.
Appl. Environ. Microbiol.73:3144–3150.
8.Bruant, G., C. Maynard, S. Bekal, I. Gaucher, L. Masson, R. Brousseau, and J. Harel.2006. Development and validation of an oligonucleotide microarray for detection of multiple virulence and antimicrobial resistance genes in
Escherichia coli. Appl. Environ. Microbiol.72:3780–3784.
9.Brunder, W., H. Schmidt, M. Frosch, and H. Karch.1999. The large
plas-mids of Shiga-toxin-producingEscherichia coli(STEC) are highly variable
genetic elements. Microbiology145(Pt. 5):1005–1014.
10.Burk, C., R. Dietrich, G. Acar, M. Moravek, M. Bulte, and E. Martlbauer.
2003. Identification and characterization of a new variant of Shiga toxin 1 in
Escherichia coliONT:H19 of bovine origin. J. Clin. Microbiol.41:2106–2112. 11.Castillo, A., L. E. Eguiarte, and V. Souza.2005. A genomic population
genetics analysis of the pathogenic enterocyte effacement island in
Esche-richia coli: the search for the unit of selection. Proc. Natl. Acad. Sci. USA
102:1542–1547.
12.China, B., F. Goffaux, V. Pirson, and J. Mainil.1999. Comparison ofeae,tir,
espA, andespBgenes of bovine and human attaching and effacingEscherichia
coliby multiplex polymerase chain reaction. FEMS Microbiol. Lett.178:177–
182.
13.Clermont, O., S. Bonacorsi, and E. Bingen.2000. Rapid and simple
deter-mination of theEscherichia coliphylogenetic group. Appl. Environ.
Micro-biol.66:4555–4558.
14.Cohen, M. B., J. P. Nataro, D. I. Bernstein, J. Hawkins, N. Roberts, and M. A. Staat.2005. Prevalence of diarrheagenicEscherichia coliin acute
childhood enteritis: a prospective controlled study. J. Pediatr.146:54–61.
15.Dulguer, M. V., S. H. Fabbricotti, S. Y. Bando, C. A. Moreira-Filho, U. Fagundes-Neto, and I. C. Scaletsky.2003. Atypical enteropathogenic Esch-erichia colistrains: phenotypic and genetic profiling reveals a strong
associ-ation between enteroaggregativeE. coliheat-stable enterotoxin and
diar-rhea. J. Infect. Dis.188:1685–1694.
16.Echeverria, P., F. Orskov, I. Orskov, S. Knutton, F. Scheutz, J. E. Brown, and U. Lexomboon.1991. Attaching and effacing enteropathogenic Esche-richia colias a cause of infantile diarrhea in Bangkok. J. Infect. Dis.164:
550–554.
17.Escobar-Paramo, P., O. Clermont, A. B. Blanc-Potard, H. Bui, B. C. Le, and E. Denamur.2004. A specific genetic background is required for acquisition
and expression of virulence factors in Escherichia coli. Mol. Biol. Evol.
21:1085–1094.
18.Feil, E. J.2004. Small change: keeping pace with microevolution. Nat. Rev.
Microbiol.2:483–495.
19.Fitzhenry, R. J., D. J. Pickard, E. L. Hartland, S. Reece, G. Dougan, A. D. Phillips, and G. Frankel.2002. Intimin type influences the site of human
intestinal mucosal colonization by enterohaemorrhagic Escherichia coli
O157:H7. Gut50:180–185.
20.Forestier, C., M. Meyer, S. Favre-Bonte, C. Rich, G. Malpuech, B. C. Le, J. Sirot, B. Joly, and C. C. De.1996. Enteroadherent Escherichia coliand diarrhea in children: a prospective case-control study. J. Clin. Microbiol.
34:2897–2903.
21.Garrido, P., M. Blanco, M. Moreno-Paz, C. Briones, G. Dahbi, J. Blanco, J. Blanco, and V. Parro.2006. STEC-EPEC oligonucleotide microarray: a new tool for typing genetic variants of the LEE pathogenicity island of human
and animal Shiga toxin-producingEscherichia coli(STEC) and
enteropatho-genicE. coli(EPEC) strains. Clin. Chem.52:192–201.
22.Gomes, T. A., K. Irino, D. M. Girao, V. B. Girao, B. E. Guth, T. M. Vaz, F. C. Moreira, S. H. Chinarelli, and M. A. Vieira.2004. Emerging
enteropatho-genicEscherichia colistrains? Emerg. Infect. Dis.10:1851–1855.
23.Gomes, T. A., V. Rassi, K. L. MacDonald, S. R. Ramos, L. R. Trabulsi, M. A. Vieira, B. E. Guth, J. A. Candeias, C. Ivey, and M. R. Toledo.1991. Entero-pathogens associated with acute diarrheal disease in urban infants in Sao
Paulo, Brazil. J. Infect. Dis.164:331–337.
24.Gross, R., and B. Rowe.1985. Serotyping ofEscherichia coli, p. 345–360.In
M. Sussman (ed.), The virulence ofEscherichia coli: reviews and methods.
Cambridge University Press, Cambridge, United Kingdom.
25.Hunter, P. R., and M. A. Gaston.1988. Numerical index of the discrimina-tory ability of typing systems: an application of Simpson’s index of diversity.
J. Clin. Microbiol.26:2465–2466.
26.Huson, D. H., and D. Bryant.2006. Application of phylogenetic networks in
evolutionary studies. Mol. Biol. Evol.23:254–267.
27.Johnson, J. R., K. L. Owens, C. R. Clabots, S. J. Weissman, and S. B. Cannon. 2006. Phylogenetic relationships among clonal groups of
extra-intestinal pathogenicEscherichia colias assessed by multi-locus sequence
analysis. Microbes Infect.8:1702–1713.
28.Jores, J., S. Wagner, L. Rumer, J. Eichberg, C. Laturnus, P. Kirsch, P. Schierack, H. Tschape, and L. H. Wieler.2005. Description of a 111-kb pathogenicity island (PAI) encoding various virulence features in the
entero-hemorrhagicE. coli(EHEC) strain RW1374 (O103:H2) and detection of a
similar PAI in other EHEC strains of serotype O103:H2. Int. J. Med.
Mi-crobiol.294:417–425.
29.Kaper, J. B.1996. Defining EPEC. Rev. Microbiol. Sao Paulo27:130–133. 30.Kaper, J. B., J. P. Nataro, and H. L. Mobley.2004. PathogenicEscherichia
coli. Nat. Rev. Microbiol.2:123–140.
31.Knutton, S., R. Shaw, A. D. Phillips, H. R. Smith, G. A. Willshaw, P. Watson, and E. Price.2001. Phenotypic and genetic analysis of diarrhea-associated
Escherichia coliisolated from children in the United Kingdom. J. Pediatr.
Gastroenterol. Nutr.33:32–40.
32.Kumar, S., K. Tamura, and M. Nei.2004. MEGA3: integrated software for molecular evolutionary genetics analysis and sequence alignment. Brief.
Bioinform.5:150–163.
33.Lacher, D. W., H. Steinsland, T. E. Blank, M. S. Donnenberg, and T. S. Whittam.2007. Molecular evolution of typical enteropathogenicEscherichia
coli: clonal analysis by multilocus sequence typing and virulence gene allelic
profiling. J. Bacteriol.189:342–350.
34.Levine, M. M., and R. Edelman.1984. EnteropathogenicEscherichia coliof classic serotypes associated with infant diarrhea: epidemiology and
patho-genesis. Epidemiol. Rev.6:31–51.
35.Maiden, M. C., J. A. Bygraves, E. Feil, G. Morelli, J. E. Russell, R. Urwin, Q. Zhang, J. Zhou, K. Zurth, D. A. Caugant, I. M. Feavers, M. Achtman, and B. G. Spratt.1998. Multilocus sequence typing: a portable approach to the identification of clones within populations of pathogenic microorganisms.
Proc. Natl. Acad. Sci. USA95:3140–3145.
36.McDaniel, T. K., K. G. Jarvis, M. S. Donnenberg, and J. B. Kaper.1995. A genetic locus of enterocyte effacement conserved among diverse
enterobac-terial pathogens. Proc. Natl. Acad. Sci. USA92:1664–1668.
37.McDaniel, T. K., and J. B. Kaper.1997. A cloned pathogenicity island from
enteropathogenicEscherichia coliconfers the attaching and effacing
pheno-type onE. coliK-12. Mol. Microbiol.23:399–407.
38.Morelli, R., L. Baldassarri, V. Falbo, G. Donelli, and A. Caprioli.1994.
Detection of enteroadherentEscherichia coliassociated with diarrhoea in
Italy. J. Med. Microbiol.41:399–404.
39.Nataro, J. P., and J. B. Kaper.1998. DiarrheagenicEscherichia coli. Clin.
Microbiol. Rev.11:142–201.
40.Nataro, J. P., V. Mai, J. Johnson, W. C. Blackwelder, R. Heimer, S. Tirrell, S. C. Edberg, C. R. Braden, M. J. Glenn, Jr., and J. M. Hirshon.2006.
DiarrheagenicEscherichia coliinfection in Baltimore, Maryland, and New
Haven, Connecticut. Clin. Infect. Dis.43:402–407.
41.Nguyen, T. V., V. P. Le, H. C. Le, K. N. Gia, and A. Weintraub.2005.
Detection and characterization of diarrheagenicEscherichia colifrom young
children in Hanoi, Vietnam. J. Clin. Microbiol.43:755–760.
42.Nunes, E. B., H. O. Saridakis, K. Irino, and J. S. Pelayo.2003. Genotypic
and phenotypic characterization of attaching and effacingEscherichia coli
(AEEC) isolated from children with and without diarrhoea in Londrina,
Brazil. J. Med. Microbiol.52:499–504.
43.Olesen, B., J. Neimann, B. Bottiger, S. Ethelberg, P. Schiellerup, C. Jensen, M. Helms, F. Scheutz, K. E. Olsen, K. Krogfelt, E. Petersen, K. Molbak, and P. Gerner-Smidt.2005. Etiology of diarrhea in young children in Denmark:
a case-control study. J. Clin. Microbiol.43:3636–3641.
44.Orlandi, P. P., G. F. Magalhaes, N. B. Matos, T. Silva, M. Penatti, P. A. Nogueira, and L. H. Silva.2006. Etiology of diarrheal infections in children of Porto Velho (Rondonia, Western Amazon region, Brazil). Braz. J. Med.
Biol. Res.39:507–517.
45.Pelayo, J. S., I. C. Scaletsky, M. Z. Pedroso, V. Sperandio, J. A. Giron, G. Frankel, and L. R. Trabulsi.1999. Virulence properties of atypical EPEC
strains. J. Med. Microbiol.48:41–49.
46.Phillips, A. D., and G. Frankel.2000. Intimin-mediated tissue specificity in
enteropathogenicEscherichia coliinteraction with human intestinal organ
cultures. J. Infect. Dis.181:1496–1500.
47.Regua-Mangia, A. H., T. A. Gomes, M. A. Vieira, J. R. Andrade, K. Irino, and L. M. Teixeira.2004. Frequency and characteristics of diarrhoeagenic
on May 16, 2020 by guest
http://jcm.asm.org/
erichia colistrains isolated from children with and without diarrhoea in Rio
de Janeiro, Brazil. J. Infect.48:161–167.
48.Reid, S. D., C. J. Herbelin, A. C. Bumbaugh, R. K. Selander, and T. S. Whittam.2000. Parallel evolution of virulence in pathogenicEscherichia coli.
Nature406:64–67.
49.Robins-Browne, R. M., A. M. Bordun, M. Tauschek, V. R. nett-Wood, J. Russell, F. Oppedisano, N. A. Lister, K. A. Bettelheim, C. K. Fairley, M. I. Sinclair, and M. E. Hellard.2004.Escherichia coliand community-acquired
gastroenteritis, Melbourne, Australia. Emerg. Infect. Dis.10:1797–1805.
50.Scaletsky, I. C., S. H. Fabbricotti, K. R. Aranda, M. B. Morais, and U. Fagundes-Neto.2002. Comparison of DNA hybridization and PCR assays
for detection of putative pathogenic enteroadherentEscherichia coli. J. Clin.
Microbiol.40:1254–1258.
51.Scaletsky, I. C., M. Z. Pedroso, C. A. Oliva, R. L. Carvalho, M. B. Morais, and U. Fagundes-Neto.1999. A localized adherence-like pattern as a second
pattern of adherence of classic enteropathogenicEscherichia colito HEp-2
cells that is associated with infantile diarrhea. Infect. Immun.67:3410–3415.
52.Selander, R. K., D. A. Caugant, and T. S. Whittam.1987. Genetic structure
and variation in natural populations ofEscherichia coli, p. 1625–1648.In
F. C. Neidhardt, R. Curtiss III, J. L. Ingraham, E. C. C. Lin, K. B. Low, B. Magasanik, W. S. Reznikoff, M. Riley, M. Schaechter, and H. E. Umbarger
(ed.),Escherichia coliandSalmonella: cellular and molecular biology, 2nd
ed. American Society for Microbiology, Washington, DC. 53.Simpson, E. H.1949. Measurement of diversity. Nature163:688. 54.Tartof, S. Y., O. D. Solberg, A. R. Manges, and L. W. Riley.2005. Analysis
of a uropathogenicEscherichia coliclonal group by multilocus sequence
typing. J. Clin. Microbiol.43:5860–5864.
55.Trabulsi, L. R., R. Keller, and T. A. Tardelli Gomes.2002. Typical and
atypical enteropathogenicEscherichia coli. Emerg. Infect. Dis.8:508–513.
56.Urwin, R., and M. C. Maiden.2003. Multi-locus sequence typing: a tool for
global epidemiology. Trends Microbiol.11:479–487.
57.Vernacchio, L., R. M. Vezina, A. A. Mitchell, S. M. Lesko, A. G. Plaut, and D. W. Acheson.2006. Diarrhea in American infants and young children in the community setting: incidence, clinical presentation and microbiology.
Pediatr. Infect. Dis. J.25:2–7.
58.Vieira, M. A., J. R. Andrade, L. R. Trabulsi, A. C. Rosa, A. M. Dias, S. R. Ramos, G. Frankel, and T. A. Gomes.2001. Phenotypic and genotypic
char-acteristics ofEscherichia colistrains of non-enteropathogenicE. coli(EPEC)
serogroups that carry EAE and lack the EPEC adherence factor and Shiga
toxin DNA probe sequences. J. Infect. Dis.183:762–772.
59.Whittam, T. S.1998. Evolution ofEscherichia coliO157:H7 and other Shiga
toxin-producingE. colistrains, p. 195–209.InJ. B. Kaper and A. D. O’Brien
(ed.), Escherichia coli O157:H7 and other Shiga toxin-producingE. coli
strains. ASM Press, Washington, DC.
60.Winstanley, C.2002. Spot the difference: applications of subtractive
hybrid-isation to the study of bacterial pathogens. J. Med. Microbiol.51:459–467.
61.Wirth, T., D. Falush, R. Lan, F. Colles, P. Mensa, L. H. Wieler, H. Karch, P. R. Reeves, M. C. Maiden, H. Ochman, and M. Achtman.2006. Sex and
virulence inEscherichia coli: an evolutionary perspective. Mol. Microbiol.
60:1136–1151.