Acta Cryst.(2003). E59, o395±o396 DOI: 10.1107/S1600536803004306 A. Thiruvalluvaret al. C10H8O2
o395
organic papers
Acta Crystallographica Section E Structure Reports
Online
ISSN 1600-5368
2-Acetylbenzo[
b
]furan
A. Thiruvalluvar,a* S. Silvarani,a
A. Vadivelu,aK. Sithik Aliband
V. R. Venkataramanb
aDepartment of Physics, Rajah Serfoji Government College, Thanjavur 613 005, Tamilnadu, India, andbPost Graduate and Research Department of Chemistry, Jamal Mohamed College, Tiruchirappalli 620 020, India
Correspondence e-mail: [email protected]
Key indicators Single-crystal X-ray study
T= 293 K
Mean(C±C) = 0.003 AÊ Disorder in main residue
Rfactor = 0.036
wRfactor = 0.092
Data-to-parameter ratio = 13.0
For details of how these key indicators were automatically derived from the article, see http://journals.iucr.org/e.
#2003 International Union of Crystallography Printed in Great Britain ± all rights reserved
The benzofuran moiety of the title molecule, C10H8O2, is
planar and forms a dihedral angle of 6.69 (9) with the
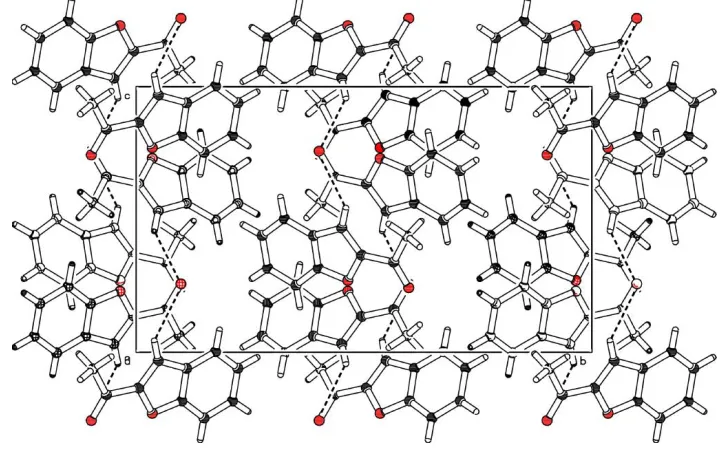
attached acetyl group. In the crystal structure, symmetry-related molecules are linked to form chains by CÐH O intermolecular hydrogen bonds involving the furan H atom and the O atom of the acetyl group. Adjacent chains are interlinked through weak CÐH interactions involving the furan ring.
Comment
A convenient method of preparing 2-acetylbenzofuran, (I), from 2-hydroxybenzaldehyde and chloroacetone in the presence of KOH has been reported (Elliott, 1951). We have obtained (I) using a phase-transfer catalytic method. The present X-ray diffraction study was undertaken to understand the geometry of the benzofuran ring system and the effect of acetyl group substitution at position 2 of the furan ring.
In (I), the benzofuran moiety is planar and the acetyl group is slightly twisted about the C2ÐC21 bond, as seen from the
torsion angles O1ÐC2ÐC21ÐO21 = 5.9 (3) and C3ÐC2Ð
C21ÐC22 = 6.7 (3). The geometry of the benzofuran ring is
comparable to that found in ethyl 3-hydroxybenzo[b ]furan-2-carboxylate (Gould et al., 1998). In the solid state, the
symmetry-related molecules are linked by C3Ð
H3 O21(3
2ÿx, ÿy, ÿ12 +z) hydrogen bonds to form chains
along the c axis. Adjacent chains related by the symmetry operation (ÿ1
2 +x, y,32ÿz) are linked by CÐH hydrogen
bonds involving the furan ring (Table 1), to form double-chain structures.
Experimental
The title compound was synthesized employing a phase-transfer catalytic technique. Salicylaldehyde (6.12 ml, 0.05 mol) and chloro-acetone (4.0 ml, 0.05 mol) were added to benzene (30 ml) and the reaction mixture was magnetically stirred for 3 h with 20% aqueous potassium carbonate (20 ml) solution in the presence of a catalytic amount of tetrabutylammonium hydrogen sulfate (200 mg) as a phase-transfer catalyst. The resulting solid was ®ltered off and dried in air. Recrystallization from 1,4-dioxane afforded the crystals. The yield of the isolated product was 86%.
Crystal data
C10H8O2 Mr= 160.16 Orthorhombic,Pbca a= 8.3865 (13) AÊ
b= 18.273 (4) AÊ
c= 10.652 (2) AÊ
V= 1632.4 (5) AÊ3 Z= 8
Dx= 1.303 Mg mÿ3
MoKradiation Cell parameters from 25
re¯ections
= 10±15 = 0.09 mmÿ1 T= 293 (2) K Block, light brown 0.30.30.3 mm
Data collection
Enraf±Nonius CAD-4 diffractometer
!±2scans
Absorption correction: none 1419 measured re¯ections 1419 independent re¯ections 907 re¯ections withI> 2(I)
max= 25.0 h= 0!9
k= 0!21
l= 0!12
2 standard re¯ections every 100 re¯ections intensity decay: none
Re®nement
Re®nement onF2 R[F2> 2(F2)] = 0.036 wR(F2) = 0.092 S= 1.03 1419 re¯ections 109 parameters
H-atom parameters constrained
w= 1/[2(F
o2) + (0.0348P)2 + 0.2715P]
whereP= (Fo2+ 2Fc2)/3 (/)max< 0.001
max= 0.10 e AÊÿ3
min=ÿ0.12 e AÊÿ3
Table 1
Hydrogen-bonding geometry (AÊ,).
DÐH A DÐH H A D A DÐH A
C3ÐH3 O21i 0.93 2.42 3.190 (2) 140
C7ÐH7 Cg1ii 0.93 2.89 3.431 (2) 119 Symmetry codes: (i)3
2ÿx;ÿy;zÿ12; (ii)xÿ12;y;32ÿz.
The H atoms were ®xed geometrically and were treated as riding on their parent C atoms, with isotropic displacement parameters. The methyl group was found to be disordered over two positions rotated from each other by 60. It was re®ned as an idealized disordered
methyl group.
Data collection: CAD-4 Software (Enraf±Nonius, 1994); cell re®nement:MolEN(Fair, 1990); data reduction:MolEN; program(s) used to solve structure: SIR97 (Altomareet al., 1999); program(s) used to re®ne structure: SHELXL97 (Sheldrick, 1997); molecular graphics: ORTEP-3 (Farrugia, 1997); software used to prepare material for publication:WinGX(Farrugia, 1999).
KS thanks the UGC, India, for partial ®nancial assistance.
References
Altomare, A., Burla, M. C., Camalli, M., Cascarano, G., Giacovazzo, C., Guagliardi, A., Moliterni, A. G. G., Polidori, G. & Spagna, R. (1999).J. Appl. Cryst.32, 115±119.
Elliott, E. D. (1951).J. Am. Chem. Soc.73, 754.
Enraf±Nonius (1994). CAD-4 Software. Enraf±Nonius, Delft, The Nether-lands.
Fair, C. K. (1990).MolEN.Enraf±Nonius, Delft, The Netherlands. Farrugia, L. J. (1997).J. Appl. Cryst.30, 565.
Farrugia, L. J. (1999).J. Appl. Cryst.32, 837±838.
Gould, R. O., Guest, M. F., Joswig, J.-O., Palmer, M. H. & Parsons, S. (1998).
Acta Cryst.C54, 1951±1954.
Sheldrick, G. M. (1997).SHELXL97. University of GoÈttingen, Germany. Spek, A. L. (1990).Acta Cryst.A46, C-34.
Figure 1
The molecular structure of (I), showing 50% probability displacement ellipsoids (Farrugia, 1997).
Figure 2
supporting information
sup-1
Acta Cryst. (2003). E59, o395–o396supporting information
Acta Cryst. (2003). E59, o395–o396 [doi:10.1107/S1600536803004306]
2-Acetylbenzo[
b
]furan
A. Thiruvalluvar, S. Silvarani, A. Vadivelu, K. Sithik Ali and V. R. Venkataraman
S1. Comment
A convenient method of preparing 2-acetylbenzofuran, (I), from 2-hydroxybenzaldehyde and chloroacetone in the
presence of KOH has been reported (Elliot, 1951). We have obtained (I) using a phase-transfer catalytic method. The
present X-ray diffraction study was undertaken to understand the geometry of the benzofuran ring system and the effect
of acetyl group substitution at the second position of the furan ring.
In (I), the benzofuran moiety is planar and the acetyl group is slightly twisted about the C2—C21 bond, as seen from
the torsion angles O1—C2—C21—O21 = 5.9 (3)° and C3—C2—C21—C22 = 6.7 (3)°. The geometry of the benzofuran
ring is comparable to that found in ethyl 3-hydroxybenzo[b]furan-2-carboxylate (Gould et al., 1998). In the solid state,
the symmetry-related molecules are linked by C3—H3···O21(3/2 − x, −y, −1/2 + z) hydrogen bonds to form chains along
the c axis. The adjacent chains related by the symmetry operation (−1/2 + x, y, 3/2 − z) are linked by C—H···π hydrogen
bonds involving the furan ring (Table 1), to form double chain structures.
S2. Experimental
The title compound was synthesized employing a phase-transfer catalytic technique. Salicylaldehyde (6.12 ml, 0.05 mol)
and chloroacetone (4.0 ml, 0.05 mol) were taken in benzene (30 ml) and the reaction mixture was magnetically stirred for
3 h with 20% aqueous potassium carbonate (20 ml) solution in the presence of a catalytic amount of tetrabutylammonium
hydrogen sulfate (200 mg) as a phase-transfer catalyst. The solid obtained was filtered off and dried in air.
Recrystallization from 1,4-dioxane afforded the crystals. The yield of the isolated product was 86%.
S3. Refinement
The H atoms were fixed geometrically and were treated as riding on their parent C atoms, with isotropic displacement
parameters. The methyl group was found to be disordered over two positions rotated from each other by 60°. It was
Figure 1
The molecular structure of (I), showing 50% probability displacement ellipsoids (Farrugia, 1997).
Figure 2
The molecular packing of (I), viewed down the a axis (Spek, 1990).
(I)
Crystal data
C10H8O2 Mr = 160.16
Orthorhombic, Pbca Hall symbol: -P 2ac 2ab a = 8.3865 (13) Å b = 18.273 (4) Å
c = 10.652 (2) Å V = 1632.4 (5) Å3 Z = 8
F(000) = 672 Dx = 1.303 Mg m−3
[image:4.610.123.486.336.561.2]supporting information
sup-3
Acta Cryst. (2003). E59, o395–o396Mo Kα radiation, λ = 0.71073 Å Cell parameters from 25 reflections θ = 10–15°
µ = 0.09 mm−1
T = 293 K
Block, light brown 0.3 × 0.3 × 0.3 mm
Data collection
Enraf-Nonius CAD-4 diffractometer ω–2θ scans
1419 measured reflections 1419 independent reflections 907 reflections with I > 2σ(I) Rint = 0
θmax = 25.0°, θmin = 2.2° h = 0→9
k = 0→21 l = 0→12
2 standard reflections every 100 reflections intensity decay: none
Refinement
Refinement on F2 Least-squares matrix: full R[F2 > 2σ(F2)] = 0.036 wR(F2) = 0.092 S = 1.03 1419 reflections 109 parameters
0 restraints
H-atom parameters constrained w = 1/[σ2(F
o2) + (0.0348P)2 + 0.2715P] where P = (Fo2 + 2Fc2)/3
(Δ/σ)max < 0.001 Δρmax = 0.10 e Å−3 Δρmin = −0.12 e Å−3
Special details
Geometry. All e.s.d.'s (except the e.s.d. in the dihedral angle between two l.s. planes) are estimated using the full covariance matrix. The cell e.s.d.'s are taken into account individually in the estimation of e.s.d.'s in distances, angles and torsion angles; correlations between e.s.d.'s in cell parameters are only used when they are defined by crystal symmetry. An approximate (isotropic) treatment of cell e.s.d.'s is used for estimating e.s.d.'s involving l.s. planes.
Fractional atomic coordinates and isotropic or equivalent isotropic displacement parameters (Å2)
x y z Uiso*/Ueq Occ. (<1)
O1 0.58354 (15) 0.03542 (6) 0.72975 (11) 0.0612 (4) C2 0.6746 (2) 0.00551 (10) 0.63421 (15) 0.0547 (4) C3 0.6747 (2) 0.04877 (10) 0.53242 (16) 0.0575 (5)
H3 0.7277 0.0399 0.4572 0.069*
C4 0.5316 (2) 0.17426 (11) 0.4978 (2) 0.0713 (6)
H4 0.5641 0.1831 0.4157 0.086*
C5 0.4360 (3) 0.22329 (11) 0.5604 (2) 0.0797 (6)
H5 0.4031 0.2656 0.5193 0.096*
C6 0.3873 (2) 0.21127 (11) 0.6835 (2) 0.0774 (6)
H6 0.3236 0.2459 0.7232 0.093*
C7 0.4314 (2) 0.14917 (10) 0.7477 (2) 0.0695 (5)
H7 0.3991 0.1407 0.8299 0.083*
C8 0.5263 (2) 0.10015 (10) 0.68320 (16) 0.0555 (5) C9 0.5787 (2) 0.11078 (10) 0.56100 (16) 0.0555 (5) C21 0.7495 (2) −0.06530 (10) 0.66040 (17) 0.0616 (5) C22 0.8610 (2) −0.09450 (11) 0.56308 (19) 0.0772 (6)
H22A 0.9441 −0.1221 0.6031 0.116* 0.5
H22B 0.9071 −0.0545 0.5172 0.116* 0.5
H22C 0.8035 −0.1256 0.5064 0.116* 0.5
H22E 0.8627 −0.147 0.5673 0.116* 0.5
H22F 0.9663 −0.0759 0.5781 0.116* 0.5
O21 0.72146 (19) −0.09825 (7) 0.75714 (13) 0.0808 (4)
Atomic displacement parameters (Å2)
U11 U22 U33 U12 U13 U23
O1 0.0704 (8) 0.0599 (7) 0.0533 (7) 0.0007 (6) 0.0086 (7) 0.0016 (6) C2 0.0540 (10) 0.0610 (11) 0.0491 (9) −0.0048 (9) 0.0013 (9) −0.0087 (9) C3 0.0522 (10) 0.0720 (11) 0.0482 (10) −0.0095 (10) −0.0001 (9) −0.0030 (10) C4 0.0660 (13) 0.0729 (12) 0.0751 (12) −0.0148 (11) −0.0053 (11) 0.0131 (12) C5 0.0771 (15) 0.0593 (12) 0.1028 (17) −0.0088 (11) −0.0156 (14) 0.0150 (12) C6 0.0656 (13) 0.0623 (13) 0.1043 (17) 0.0025 (10) −0.0035 (13) −0.0101 (12) C7 0.0693 (12) 0.0653 (11) 0.0738 (12) −0.0032 (11) 0.0079 (11) −0.0048 (11) C8 0.0530 (10) 0.0531 (10) 0.0604 (11) −0.0057 (9) −0.0014 (9) 0.0008 (9) C9 0.0507 (10) 0.0603 (11) 0.0553 (10) −0.0124 (9) −0.0058 (9) 0.0043 (9) C21 0.0647 (11) 0.0626 (11) 0.0575 (10) −0.0035 (10) −0.0091 (10) −0.0083 (10) C22 0.0728 (13) 0.0756 (13) 0.0833 (13) 0.0080 (11) 0.0005 (12) −0.0165 (11) O21 0.1054 (12) 0.0714 (8) 0.0657 (8) 0.0089 (8) −0.0034 (8) 0.0052 (8)
Geometric parameters (Å, º)
O1—C8 1.369 (2) C6—H6 0.93
O1—C2 1.3848 (19) C7—C8 1.381 (2)
C2—C3 1.342 (2) C7—H7 0.93
C2—C21 1.465 (3) C8—C9 1.388 (2)
C3—C9 1.423 (2) C21—O21 1.216 (2)
C3—H3 0.93 C21—C22 1.495 (3)
C4—C5 1.374 (3) C22—H22A 0.96
C4—C9 1.398 (2) C22—H22B 0.96
C4—H4 0.93 C22—H22C 0.96
C5—C6 1.391 (3) C22—H22D 0.96
C5—H5 0.93 C22—H22E 0.96
C6—C7 1.376 (3) C22—H22F 0.96
C8—O1—C2 105.55 (13) O21—C21—C2 121.04 (18) C3—C2—O1 111.21 (16) O21—C21—C22 122.12 (19) C3—C2—C21 132.36 (17) C2—C21—C22 116.84 (17)
O1—C2—C21 116.43 (15) C21—C22—H22A 109.5
C2—C3—C9 107.21 (16) C21—C22—H22B 109.5
C2—C3—H3 126.4 H22A—C22—H22B 109.5
C9—C3—H3 126.4 C21—C22—H22C 109.5
C5—C4—C9 118.18 (19) H22A—C22—H22C 109.5
C5—C4—H4 120.9 H22B—C22—H22C 109.5
C9—C4—H4 120.9 C21—C22—H22D 109.5
C4—C5—C6 121.7 (2) H22A—C22—H22D 141.1
C4—C5—H5 119.1 H22B—C22—H22D 56.3
supporting information
sup-5
Acta Cryst. (2003). E59, o395–o396C7—C6—C5 121.3 (2) C21—C22—H22E 109.5
C7—C6—H6 119.3 H22A—C22—H22E 56.3
C5—C6—H6 119.3 H22B—C22—H22E 141.1
C6—C7—C8 116.3 (2) H22C—C22—H22E 56.3
C6—C7—H7 121.9 H22D—C22—H22E 109.5
C8—C7—H7 121.9 C21—C22—H22F 109.5
O1—C8—C7 125.59 (17) H22A—C22—H22F 56.3
O1—C8—C9 110.47 (16) H22B—C22—H22F 56.3
C7—C8—C9 123.94 (18) H22C—C22—H22F 141.1
C8—C9—C4 118.57 (18) H22D—C22—H22F 109.5
C8—C9—C3 105.57 (16) H22E—C22—H22F 109.5
C4—C9—C3 135.86 (18)
C8—O1—C2—C3 −0.20 (19) C7—C8—C9—C4 −0.9 (3) C8—O1—C2—C21 −179.59 (15) O1—C8—C9—C3 −0.49 (18) O1—C2—C3—C9 −0.1 (2) C7—C8—C9—C3 178.76 (17) C21—C2—C3—C9 179.16 (18) C5—C4—C9—C8 0.3 (3) C9—C4—C5—C6 0.5 (3) C5—C4—C9—C3 −179.22 (18) C4—C5—C6—C7 −0.7 (3) C2—C3—C9—C8 0.35 (18) C5—C6—C7—C8 0.2 (3) C2—C3—C9—C4 179.92 (19) C2—O1—C8—C7 −178.81 (17) C3—C2—C21—O21 −173.37 (19) C2—O1—C8—C9 0.43 (18) O1—C2—C21—O21 5.9 (3) C6—C7—C8—O1 179.79 (17) C3—C2—C21—C22 6.7 (3) C6—C7—C8—C9 0.6 (3) O1—C2—C21—C22 −174.04 (14) O1—C8—C9—C4 179.85 (15)
Hydrogen-bond geometry (Å, º)
D—H···A D—H H···A D···A D—H···A
C3—H3···O21i 0.93 2.42 3.190 (2) 140
C7—H7···Cg1ii 0.93 2.89 3.431 (2) 119