organic papers
Acta Cryst.(2005). E61, o941–o943 doi:10.1107/S1600536805006811 Chenet al. C
10H6Br2
o941
Acta Crystallographica Section EStructure Reports Online
ISSN 1600-5368
1,3-Dibromoazulene
Xue-Yi Chen,aJerry R. Dias,a Doug R. Powell,bJ. David Van Horna* and Thomas C.
Sandreczkia
aDepartment of Chemistry, University of
Missouri–Kansas City, 5110 Rockhill Road, Kansas City, MO 64110, USA, andbDepartment
of Chemistry, University of Kansas, Mallott Hall, Room 6044, 1251 Wescoe Hall Drive, Lawrence, KS 66045-7582, USA
Correspondence e-mail: [email protected]
Key indicators
Single-crystal X-ray study
T= 100 K
Mean(C–C) = 0.002 A˚
Rfactor = 0.016
wRfactor = 0.046
Data-to-parameter ratio = 16.1
For details of how these key indicators were automatically derived from the article, see http://journals.iucr.org/e.
#2005 International Union of Crystallography Printed in Great Britain – all rights reserved
In the structure of 1,3-dibromoazulene, C10H6Br2, the planar
molecule sits on a crystallographic mirror plane. The Br atoms are attached to the five-membered ring adjacent to the ring fusion. Head-to-tail charge–charge interactions due to partial positive charges on ring H atoms and partial negative charges within the aromatic ring system attract adjacent 1,3-dibromo-azulene molecules together. This causes them to arrange themselves in a zigzag pattern that allows close packing of the oppositely charged groups.
Comment
Polyazulene and its derivatives are of scientific and practical interest, because they are highly conjugated hydrocarbons that can be made electrically conductive and highly para-magnetic (Wanget al., 2003; Wang, Lai & Han, 2004; Wang, Xu
et al., 2004). One important physical property of a polyazulene that can be tailored is its solubility, which requires appropriate derivatization of the azulene monomer unit. Halogenated azulenes are known to be useful as monomeric building blocks for polyazulene and its derivatives (Wang & Lai, 2003). They are also precursors for the synthesis of dumbbell-shaped alkyl-bridged diazulenyl compounds (Elwahy, 2002). The crystal structures of aromatic halogenated azulenes, however, have not been described in the literature to date. This report of the crystal structure of 1,3-dibromoazulene, (I), and a related report on 1,3-diiodoazulene (Chenet al., 2005), are a portion of our continuing work involving the preparation and analysis of electrically conductive azulene-containing polymers.
Pertinent to this report is the crystal structure of the parent azulene (Robertson et al., 1962), which crystallizes in the monoclinic space group P21/a. Other related structures, also
having monoclinic space groups, include two variously substituted azulenes (Lohr et al., 1984; Wong et al., 1984) which both crystallize in the space groupP21/c, and a fused
bis-azulene (Vogelet al., 1984) found in the space groupP21/n.
This is in contrast with the orthorhombic structure of the dibromoazulene presented here. The related diiodoazulene is
also monoclinic, but is found in the space groupCc(Chenet al., 2005).
A calculation comparing the parent azulene with dibromoazulene [HF/6–31 G(d,p) level, full natural bond orbital analysis; Frischet al., 2003)] indicates that the presence of the Br atoms substantially polarizes the ring current toward the five-membered ring, with significant charge on the C atoms attached to the Br atoms. The calculation also helps to explain the head-to-ring and bromine-to-ring interactions observed in the crystal structure as being of a dipolar (or charge–charge) nature. Finally, the Br atoms have very little calculated charge, which may account for the short intermolecular Br Br distance observed in the solid state.
In the crystal structure of (I), individual molecules lie on a crystallographic mirror plane that passes through atoms C1 and C6, bisecting the fused five- and seven-membered rings. The rings are planar, but stack in an offset herring-bone pattern that apparently maximizes the dipolar interactions between molecules in the same stack. The distance between atom C6 of one molecule and atoms C3 and C30of the ring
fusion bond of a neighboring molecule in the same stack is about 2.76 A˚ (Fig. 2), indicating a strong interaction between the positively charged seven-membered ring and the nega-tively charged five-membered ring. The only significant close contacts between adjacent stacks are intermolecular Br Br interactions [3.549 (2) A˚ ].
Experimental
Compound (I) was obtained by reacting 1.0 equivalent of azulene with 2.1 equivalents ofN-bromosuccinimide in dichloromethane at room temperature overnight (Elwahy, 2002). The solvent was then removed under vacuum below 273 K. Purification was accomplished by column chromatography, using neutral alumina as substrate and hexanes as eluent. Dark-blue blocks were obtained by slow evaporation of the solvent.
Crystal data
C10H6Br2
Mr= 285.97
Orthorhombic,Pnma a= 8.3575 (14) A˚
b= 15.114 (2) A˚
c= 7.1840 (12) A˚
V= 907.4 (2) A˚3
Z= 4
Dx= 2.093 Mg m
3
MoKradiation Cell parameters from 4195
reflections = 2.7–26.0
= 8.87 mm1
T= 100 (2) K Block, blue
0.280.200.20 mm
Data collection
Bruker APEX diffractometer !scans
Absorption correction: multi-scan (SADABS; Bruker, 2002)
Tmin= 0.120,Tmax= 0.170
7311 measured reflections 931 independent reflections
870 reflections withI> 2(I)
Rint= 0.024
max= 26.0
h=10!10
k=18!18
l=8!8
Refinement
Refinement onF2 R[F2> 2(F2)] = 0.016
wR(F2) = 0.046
S= 0.96 931 reflections 58 parameters
H-atom parameters constrained
w= 1/[2
(Fo2) + (0.03P)2
+ 0.6P]
whereP= (Fo2+ 2Fc2)/3
(/)max= 0.001 max= 0.48 e A˚
3 min=0.36 e A˚ 3
Table 1
Selected geometric parameters (A˚ ,).
Br1—C2 1.879 (2)
C1—C2 1.399 (2)
C2—C3 1.396 (2)
C3—C4 1.386 (2)
C3—C3i 1.499 (2)
C4—C5 1.392 (2)
C5—C6 1.395 (2)
C2i
—C1—C2 107.0 (2)
C3—C2—C1 110.9 (2)
C4—C3—C3i
127.6 (1)
Br1—C2—C3—C3i 179.4 (1)
Symmetry codes: (i)x;yþ3 2;z.
organic papers
o942
Chenet al. C [image:2.610.310.568.71.245.2]10H6Br2 Acta Cryst.(2005). E61, o941–o943
Figure 2
[image:2.610.54.278.75.259.2]A view of the packing of (I), showing the zigzag stack of the dibromo-substituted azulene molecules. Charge–charge interactions of parallel [3.24 (1) A˚ ] and herringbone [3.36 (1) A˚] geometry are indicated by dashed lines.
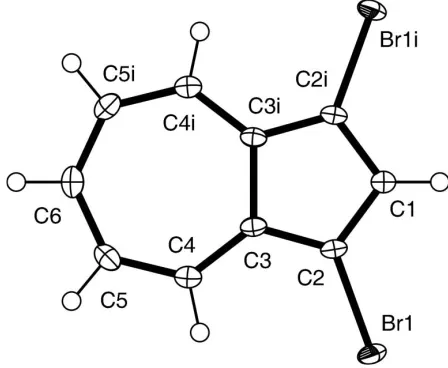
Figure 1
A perspective view of (I) with the atom-numbering scheme [symmetry code: (i)x,y+ 3
All H atoms were included in calculated positions using a riding model, with C—H = 0.95 A˚ and withUiso(H) = 1.2Ueq(C).
Data collection:SMART(Bruker, 1998); cell refinement:SAINT
(Bruker, 1998); data reduction: SAINT; program(s) used to solve structure: SHELXTL (Bruker, 1998); program(s) used to refine structure:SHELXTL; molecular graphics:ORTEP3(Farrugia, 1997); software used to prepare material for publication:SHELXTL.
The authors thank the National Science Foundation (grant No. CHE-0079282) and the University of Kansas for funds to acquire the diffractometer and computers used in this work.
References
Bruker (1998). SMART (Version 5.625), SAINT (Version 6.22) and
SHELXTL(Version 6.12). Bruker AXS, Inc., Madison, Wisconsin, USA. Bruker (2002). SADABS (Version 2.04). Bruker AXS, Inc., Madison,
Wisconsin, USA.
Chen, X.-Y., Dias, J. R., Powell, D. R., Van Horn, J. D. & Sandreczki, T. C. (2005).Acta Cryst.E61, o944–o946.
Elwahy, A. H. M. (2002).Tetrahedron Lett.43, 711–714.
Frisch, M. J., Trucks, G. W., Schlegel, H. B., Scuseria, G. E., Robb, M. A., Cheeseman, J. R., Montgomery, J. A., Vreven, T. Jr, Kudin, K. N., Burant, J. C., Millam, J. M., Iyengar, S. S., Tomasi, J., Barone, V., Mennucci, B.et al.
(2003).GAUSSIAN03. Revision B04. Gaussian Inc., Pittsburgh, Pennsyl-vania, USA.
Farrugia, L. J. (1997).J. Appl. Cryst.30, 565.
Lohr, H.-G., Engel, A., Josel, H.-P., Vogtle, F., Schuh, W. & Puff, H. (1984).J. Org. Chem.49, 1621–1627.
Robertson, J. M., Shearer, H. M. M., Sim, G. A. & Watson, D. G. (1962).Acta Cryst.15, 1–8.
Vogel, E., Wieland, H., Schmalstieg, L. & Lex, J. (1984).Angew. Chem. Int. Ed. Engl.23, 717–719.
Wang, F. & Lai, Y.-H. (2003).Macromolecules,36, 536–538.
Wang, F., Lai, Y.-H. & Han, M.-Y. (2004).Macromolecules,37, 3222–3230. Wang, F., Lai, Y.-H., Kocherginsky, N. M. & Kosteski, Y. Y. (2003).Org. Lett.5,
995–998.
Wang, W., Xu, J., Lai, Y.-H. & Wang, F. (2004).Macromolecules,37, 3546– 3553.
Wong, H. N. C., So, K. P. & Mak, T. C. W. (1984).Z. Kristallogr.169, 117–125.
organic papers
Acta Cryst.(2005). E61, o941–o943 Chenet al. C
supporting information
sup-1
Acta Cryst. (2005). E61, o941–o943
supporting information
Acta Cryst. (2005). E61, o941–o943 [https://doi.org/10.1107/S1600536805006811]
1,3-Dibromoazulene
Xue-Yi Chen, Jerry R. Dias, Doug R. Powell, J. David Van Horn and Thomas C. Sandreczki
1,3-Dibromoazulene
Crystal data C10H6Br2 Mr = 285.97
Orthorhombic, Pnma Hall symbol: -P 2ac 2n a = 8.3575 (14) Å b = 15.114 (2) Å c = 7.1840 (12) Å V = 907.4 (2) Å3 Z = 4
F(000) = 544
Dx = 2.093 Mg m−3
Melting point: 76.5 K
Mo Kα radiation, λ = 0.71073 Å Cell parameters from 4195 reflections θ = 2.7–26.0°
µ = 8.87 mm−1 T = 100 K Block, blue
0.28 × 0.20 × 0.20 mm
Data collection Bruker APEX
diffractometer
Radiation source: fine-focus sealed tube Graphite monochromator
ω scans
Absorption correction: multi-scan (SADABS; Bruker, 2002) Tmin = 0.120, Tmax = 0.170
7311 measured reflections 931 independent reflections 870 reflections with I > 2σ(I) Rint = 0.024
θmax = 26.0°, θmin = 2.7° h = −10→10
k = −18→18 l = −8→8
Refinement Refinement on F2
Least-squares matrix: full R[F2 > 2σ(F2)] = 0.016 wR(F2) = 0.046 S = 0.96 931 reflections 58 parameters 0 restraints
Primary atom site location: structure-invariant direct methods
Secondary atom site location: difference Fourier map
Hydrogen site location: inferred from neighbouring sites
H-atom parameters constrained w = 1/[σ2(F
o2) + (0.03P)2 + 0.6P]
where P = (Fo2 + 2Fc2)/3
(Δ/σ)max = 0.001
Δρmax = 0.48 e Å−3
Δρmin = −0.36 e Å−3
Special details
Geometry. All e.s.d.'s (except the e.s.d. in the dihedral angle between two l.s. planes) are estimated using the full
supporting information
sup-2
Acta Cryst. (2005). E61, o941–o943
Refinement. Refinement of F2 against ALL reflections. The weighted R-factor wR and goodness of fit S are based on F2,
conventional R-factors R are based on F, with F set to zero for negative F2. The threshold expression of F2 > σ(F2) is used
only for calculating R-factors(gt) etc. and is not relevant to the choice of reflections for refinement. R-factors based on F2
are statistically about twice as large as those based on F, and R- factors based on ALL data will be even larger.
Fractional atomic coordinates and isotropic or equivalent isotropic displacement parameters (Å2)
x y z Uiso*/Ueq
Br1 0.46263 (2) 0.558729 (11) 0.79056 (2) 0.02160 (10) C1 0.4714 (3) 0.7500 0.8153 (3) 0.0160 (5) H1 0.5204 0.7500 0.9347 0.019* C2 0.4286 (2) 0.67563 (12) 0.7108 (2) 0.0157 (4) C3 0.36081 (18) 0.70040 (10) 0.5410 (2) 0.0147 (3) C4 0.30704 (19) 0.64441 (11) 0.4015 (2) 0.0169 (3) H4 0.3153 0.5829 0.4272 0.020* C5 0.2426 (2) 0.66626 (12) 0.2288 (2) 0.0202 (4) H5 0.2130 0.6177 0.1522 0.024* C6 0.2156 (3) 0.7500 0.1533 (3) 0.0199 (5) H6 0.1719 0.7500 0.0312 0.024*
Atomic displacement parameters (Å2)
U11 U22 U33 U12 U13 U23
Br1 0.02695 (14) 0.01187 (13) 0.02598 (15) 0.00155 (6) −0.00326 (7) 0.00492 (6) C1 0.0167 (12) 0.0139 (13) 0.0175 (11) 0.000 0.0004 (9) 0.000 C2 0.0171 (8) 0.0100 (8) 0.0200 (9) 0.0008 (7) 0.0018 (6) 0.0017 (6) C3 0.0133 (7) 0.0109 (9) 0.0198 (8) 0.0012 (6) 0.0023 (6) 0.0008 (6) C4 0.0162 (8) 0.0137 (8) 0.0208 (8) −0.0001 (6) 0.0022 (7) −0.0005 (6) C5 0.0181 (8) 0.0201 (10) 0.0223 (8) −0.0016 (8) −0.0012 (7) −0.0066 (7) C6 0.0159 (11) 0.0292 (14) 0.0145 (11) 0.000 −0.0005 (10) 0.000
Geometric parameters (Å, º)
Br1—C2 1.879 (2) C4—C5 1.392 (2) C1—C2 1.399 (2) C4—H4 0.9500 C1—H1 0.9500 C5—C6 1.395 (2) C2—C3 1.396 (2) C5—H5 0.9500 C3—C4 1.386 (2) C6—H6 0.9500 C3—C3i 1.499 (2)
C2i—C1—C2 107.0 (2) C3—C4—C5 128.65 (16)
C2i—C1—H1 126.5 C3—C4—H4 115.7
C2—C1—H1 126.5 C5—C4—H4 115.7 C3—C2—C1 110.9 (2) C4—C5—C6 128.62 (18) C3—C2—Br1 125.45 (13) C4—C5—H5 115.7 C1—C2—Br1 123.59 (13) C6—C5—H5 115.7 C4—C3—C2 126.81 (15) C5i—C6—C5 130.2 (2)
supporting information
sup-3
Acta Cryst. (2005). E61, o941–o943
C2—C3—C3i 105.56 (10) C5—C6—H6 114.9
C2i—C1—C2—C3 0.6 (3) Br1—C2—C3—C3i −179.4 (1)
C2i—C1—C2—Br1 179.63 (7) C2—C3—C4—C5 −178.26 (18)
C1—C2—C3—C4 178.89 (17) C3i—C3—C4—C5 0.9 (2)
Br1—C2—C3—C4 −0.1 (2) C3—C4—C5—C6 −0.2 (3) C1—C2—C3—C3i −0.39 (16) C4—C5—C6—C5i −1.1 (5)