Regulation of primordial germ cell development in the mouse
MASSIMO DE FELICI
Department of Public Health and Cell Biology, University of Rome “Tor Vergata”, Rome, Italy.
ABSTRACT Primordial germ cells (PGCs) are the founders of the gametes. They arise at the earliest stages of embryonic development and migrate to the gonadal ridges, where they differentiate into oogonia/oocytes in the ovary, and prospermatogonia in the testis. The present article is a review of the main studies undertaken by the author with the aim of clarifying the mechanisms underlying the development of primordial germ cells. Methods for the isolation and purification of migratory and post-migratory mouse PGCs devised in the author’s laboratory are first briefly reviewed. Such methods, together with the primary culture of PGCs onto suitable cell feeder layers, have allowed the analysis of important aspects of the control of their development, concerning in particular survival, proliferation and migration of mouse PGCs. Compounds and growth factors affecting PGC numbers in culture have been identified. These include survival anti-apoptotic factors (SCF, LIF) and positive regulators of proliferation (cAMP, PACAPs, RA). Evidence has been provided that the motility of migrating PGCs relies on integrated signals from extracellular matrix molecules and the surrounding somatic cells. Moreover, homotypic PGC-PGC interaction has been evidenced that might play a role in PGC migration and in regulating their development. Several molecules (i.e. integrins, specific types of oligosaccharides, E-cadherin, the tyrosine kinase receptor c-kit) have been found to be expressed on the surface of PGCs and to mediate adhesive interactions of PGCs with the extracellular matrix, somatic cells and neighbouring PGCs.
KEY WORDS:
primordial germ cells, stem cell factor, leukemia inhibitory factor, retinoic acid, cell migration,
apoptosis.
0214-6282/2000/$20.00 © UBC Press
Printed in Spain www.ehu.es/ijdb
*Address correspondence to: Prof. Massimo De Felici. Dipartimento di Sanità Pubblica e Biologia Cellulare, Università di Roma “Tor Vergata”, Via di Tor Vergata 135, 00133 Roma, Italia. TEL: 39 06 7259 6174. FAX: 39 06 7259 6172. e-mail: [email protected]
Abbreviations used in this paper: PGCs, primordial germ cells; APase, alkaline phosphatase; SCF, stem cell factor; LIF, leukemia inhibitory factor; RA, retinoic acid; PACAPs, pituitary adenylyl cyclase activating peptides; ES cells, embryonic stem cells; EG cells, embryonic germ cells; ECM, extracellular matrix; TPA, tetradecanoyl-phorbol-13-acetate; OAG, 1-oleolyl-2-acetyl-sn-glycerol; PKC, protein kinase C; MAPK, mitogen activated protein kinase; cAMP, adenosine 3’5’ cyclic monophosphate; FSH, follicle stimulating hormone; ACTH, adrenocorticotrophin; FRSK, forskolin.
Introduction
Primordial germ cells (PGCs) are the embryonic precursors of the female and male gametes from which all sexually reproducing organisms arise. One of the most fascinating characteristic of these cells is their double nature of highly specialized cells that, at the same time, maintain their differentiation totipotency to pass on to gametes. How this is realized is not known. However, over the last two decades important aspects of PGC development and differentiation have been revealed.
When I first become interested in mammalian PGCs, only morphological studies describing the main steps of PGC develop-ment in some species, including humans, had been performed. Thanks to a high alkaline phosphatase activity, PGCs had been identified in an extraembryonic region near to the yolk sac, very early in embryogenesis, and trace in their migratory route towards the gonadal ridges (for a review, see Eddy et al., 1981). The present essay is a review of the main contributions provided over the last fifteen years by my laboratory to the understanding of the cellular and molecular mechanisms underlying PGC development.
Migratory and post-migratory PGCs can be isolated and
purified from the mouse embryo
ovary and testis, respectively (Fig. 1) (for a review, see De Felici et al., 1992).
The main goal of the early experiments carried out in the Anne McLaren´s laboratory at the MRC Developmental Unit in London was to develop a method for the isolation and purification of mouse PGCs at different developmental stages. We trust that the possi-bility to study PGC behaviour in vitro should allow uncovering some of the mechanisms underlying their development. Using a relatively simple method based on EDTA and mechanical disaggregation of gonadal tissues, we were able to obtain an enriched PGC popula-tion from post-migratory stages (12.5-13.5 days post coitum, dpc, embryos). Moreover, the use of a discontinuous Percoll gradient allowed increasing the purity of such PGC populations up to 80-90% (De Felici and McLaren, 1983). Their very low numbers (from some hundreds to a few thousands per embryo, Tam and Snow, 1981), their migration throughout different tissues and the lack of specific antibodies, rendered difficult the task to isolate PGCs during migratory stages. Later, however, the availability of monoclonal antibodies able to bind to PGC surface molecules (SSEA-1, Fox et al., 1981; EMA-1, Hahnel and Eddy, 1986; TG-1, Donovan et al., 1986), together with the introduction of magnetic cell sorting techniques, allowed us to develop efficient methods for the purification of PGCs from both migratory (10.5-11.5 dpc) and post-migratory (12.5-13.5 dpc) stages (De Felici and Pesce, 1995; Pesce and De Felici, 1995). Fig. 2 shows a schematic representa-tion of the various methods currently used in our laboratory for the isolation and purification of mouse PGCs. Methodological details, advantages and pitfalls of these methods can be found in De Felici (1998a,b).
Why do isolated PGCs not survive in culture?
Our first attempts to culture isolated mouse PGCs had little success. In fact, we observed that germ cells isolated from embry-onic gonads of different ages have different abilities to survive in vitro. In particular, 11.5-12.5 dpc PGCs did not survive at 37°C in any of the several culture conditions employed (De Felici and McLaren, 1983; De Felici and Dolci, 1989). At room temperature, however, about 50% of PGCs survived for 2-3 days and underwent some mitotic proliferation without further differentiation (De Felici and McLaren 1983; Wabik-Sliz and McLaren, 1984). Although until now, culture conditions suitable to allow the survival, proliferation and differentiation of isolated PGCs have not been established, important progress in understanding the reasons for such evident dependence on the surrounding environment has been accom-plished. In our first paper on PGC culture (De Felici and McLaren, 1983), we wrote “ In their normal environment, PGCs do not at any
time exist as an independent tissue, but are always closely asso-ciated with other cells from which they may derive nutrients as well as developmental signals. Such factors may prove critical in controlling the survival of germ cells in vitro and perhaps also their proliferation and development in vivo." Some of these factors have been now identified.
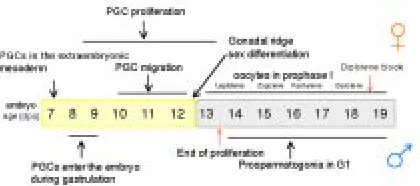
The use of feeder layer culture for PGCs gradually led to improvements in PGC culture and to establish conditions that allowed PGCs not only to survive in vitro for several days, but also to proliferate. Thanks mainly to the use of two in vitro culture systems, PGCs co-cultured on feeder layers of their own gonadal somatic cells or of established cell lines (Fig. 3), survival and/or proliferation factors needed for PGC development were discov-ered. The leukemia inhibitory factor (LIF) was the first growth factor identified in my laboratory as able to significantly increase PGC survival in vitro (De Felici and Dolci, 1991). A second growth factor found to exert a very important action on PGCs was the stem cell factor (SCF). In 1990, several laboratories had identified SCF as a novel growth factor ligand for the tyrosine kinase receptor c-kit encoded by the White (W) locus in hemopoietic cell lines. SCF was found to be encoded by the Steel (Sl) locus and produced from two alternatively spliced mRNAs as a transmembrane or soluble forms (for a review, see Besmer, 1991). Immediately afterwards, Matsui et al. (1990) demonstrated that SCF is produced by the somatic cells surrounding migratory and post-migratory PGCs, and that c-kit is expressed on the surface of PGCs. In 1991, S. Dolci working with P. Donovan’s group at Frederick, showed, simultaneously with other laboratories, that SCF is essential for PGC growth in vitro, and that the PGC life-supporting activity of certain feeder cells Fig. 1. The timing of germ cell development in the mouse embryo. (See
text for details.)
Fig. 2. Schematic representation of methods used for the isolation and purification of mouse PGCs.(A) Mechanical disaggregation of the gonadal tissues after EDTA treatment of the gonadal ridge. (B) PGC purification by Percoll gradient. (C) Immunomagnetic cell sorting by a Mini-MACS apparatus after PGC labeling with TG-1 antibody. (D) An example of PGC purification by Mini-MACS: a) an unsorted cell population obtained by EDTA-trypsin disaggregation of 11.5 dpc gonadal ridges, in which PGCs (arrows) represent less than 10% of the cells, b) after purification, PGC populations with a purity higher than 90% can be obtained. PGCs are stained red by the APase reaction.
A
C
B
is partly due to the production of such a growth factor, in particular of the SCF membrane-bound form (Dolci et al., 1991; Matsui et al., 1991; Godin et al., 1991).
Experiments carried out in my laboratory in Rome confirmed and extended the results about the crucial role that SCF plays in PGC growth. In particular, we showed that recombinant mouse SCF added to PGCs growing on their own gonadal somatic cells, significantly increased the number of PGCs without directly stimu-lating their proliferation (Dolci et al., 1993). Moreover, antibodies against c-kit inhibited PGC survival on STO cell feeder layers (De Felici, unpublished observations). Most importantly, we found that the addition of SCF to the culture medium markedly reduced apoptosis in PGCs during the first hours of culture (Pesce et al., 1993). More recently, we have shown that the effect of SCF on PGC apoptosis is, at least in part, due to a reduction of the expression of the pro-apoptotic gene Bax (De Felici et al., 1999). The finding that PGCs begin to undergo apoptosis a few hours after isolation from the gonadal ridges, finally provided an explana-tion of their rapid degeneraexplana-tion in culture. Clearly in the embryo, PGCs must be prevented to undergo apoptosis by soluble factors and/or direct contact with the surrounding somatic cells. The only transient anti-apoptotic effect of the soluble form of SCF and LIF (Pesce et al., 1993), together with the results reported above concerning the effectiveness of cell feeder layers producing mem-brane bound SCF to support survival/proliferation of PGCs in vitro (Dolci et al., 1991; Matsui et al., 1991; Godin et al., 1991), suggested that direct contact with somatic cells is probably crucial to assure an efficient prevention of PGC apoptosis. Interestingly, we have recently reported that the membrane form of SCF also promotes PGC adhesion to somatic cells (Pesce et al., 1997). This latter finding, together with other experimental evidence discussed in a subsequent section, highlights the importance of adhesive interactions between PGCs and somatic cells to favor PGC sur-vival and proliferation as well their migratory activity. Moreover, this
might be also an elegant way to prevent aberrant PGC migration: PGCs that stray from their migratory pathway are eliminated through apoptosis.
Lastly, it must be pointed out that the susceptibility of PGCs to apoptosis and the effect of SCF on this process, besides clarifying the reason for the degeneration of isolated PGCs in vitro, gives a likely explanation for the germ cell deficiency that characterizes the Sl and W mutations in the mouse (Russel, 1977).
Intracellular pathways involved in PGC proliferation
As reported above, we found that the activation of the c-kit tyrosine kinase receptor does not lead to a direct stimulation of PGC proliferation. In fact, the addition of soluble SCF to the culture medium did not significantly increase the percentage of PGCs incorporating BrdU (Dolci et al., 1993). So far, no growth factors acting via tyrosine kinase receptors have been reported to directly stimulate PGC proliferation. Several studies carried out in my laboratory, showed instead that dbcAMP or cAMP agonists such as forskolin (FRSK) and cholera toxin markedly stimulate PGC proliferation (De Felici et al., 1993). This strongly suggests that cAMP-dependent intracellular pathways are involved in PGC pro-liferation. In line with these results, we found that RP-cAMPS, a selective inhibitor of cAMP-dependent protein kinases, signifi-cantly reduced the effect of FRSK (De Felici et al., 1993). On the other hand, the recent finding (S. Iona and M. De Felici, unpub-lished results) that U0126, a specific potent inhibitor of mitogen-activated protein (MAP) kinases MKK1 and 2, does not counteract the FRSK effect, seems to exclude that activation of MAP kinases is necessary for PGC proliferation. Molecules that may stimulate in vivo the intracellular cAMP increase which appears to be neces-sary to sustain PGC proliferation, have not been identified yet. We have found, however, that, among many peptides known to increase intracellular cAMP via specific receptors (FSH, prostaglandins, ACTH, calcitonin, T3), pituitary adenylyl cyclase activating peptides (PACAPs) are able to stimulate in vitro proliferation of PGCs. In addition, we reported evidence about the presence of these peptides in the fetal gonads (Pesce et al., 1996a).
Fig. 3.(A) Schematic representation of methods used for PGC culture in vitro: 1. Culture of containing cell populations, 2. Culture of PGC-containing cell populations onto pre-formed cell feeder layers, 3. Culture of purified PGCs onto pre-formed STO cell feeder layers. (B) A comparison of 8.5 dpc PGC growth in vitro using methods 1 and 2, showing that the presence of a cell feeder layer is essential for optimal PGC growth; SCs, somatic cells. (C) PGCs cultured onto STO cell feeder layers identified by APase labeling.
Fig. 4.(A) The addition of 5 µM RA and/or 10 µM FRSK to the culture medium increases the number of 8.5, 10.5 and 11.5 dpc PGCs on TM4 cell feeder layers. (B) RA and/or FRSK increase BrdU incorporation by PGCs in culture (percentage of PGCs in S phase). (C) Examples of APase positive colonies formed in culture of 11.5 dpc PGCs on TM4 cells after 5-6 days in the continuous presence of RA and FRSK.
B A
C
A
Another compound that showed to be able to significantly increase the proliferation rate of PGCs was retinoic acid (RA). RA added to the culture medium in the range of 1-5 µM, promoted PGC proliferation on feeder layers of both their own gonadal somatic cells and of TM4 or STO cells (Fig. 4 A,B) (Pesce et al., 1996b). Similar results have been reported by Koshimizu et al. (1995). Although we know that the effect of RA is not mediated by cAMP (R. Canipari and M. De Felici, unpublished results), the molecular pathways involved in its action on PGCs remain to be investigated. Interestingly, we found that FRSK and RA caused a synergistic increase of PGC number (Fig. 4). Moreover, when used in combi-nation these compounds induced in long term culture (6-7 days) of 10.5-11.5 dpc PGCs on TM4 cell feeder layers, the formation of colonies, for the most part APase positive, whose origin (from PGCs or contaminating somatic cells) and identity we were unable to determine (Fig. 4C).
Lastly, the activation of protein kinase C (PKC) does not seem to be involved in PGC proliferation pathways. In fact, the addition to the culture medium of 12-O-tetradecanoyl-phorbol-13-acetate (TPA) or 1-oleolyl-2-acetyl-sn-glycerol (OAG), two potent activa-tors of PKC, did not influence PGC numbers (De Felici and Pesce, 1994).
Taken together these results indicate that, among the key signaling pathways regulating mammalian cell growth, such as receptor protein-tyrosine kinases, MAP kinases, PKC, and cAMP-depend-ent protein kinases, this latter is probably prefercAMP-depend-entially activated during PGC proliferation (Fig. 5).
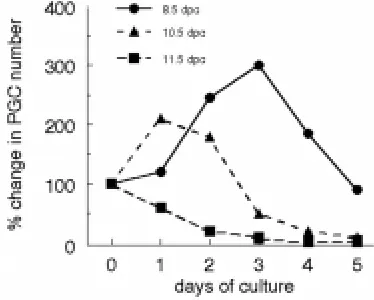
Why do PGCs not properly differentiate in coculture systems? In the developing gonad, PGCs cease mitotic cell division at 13.5 dpc and enter either mitotic arrest in the testis or meiosis in the ovary. Therefore, the period between 12.5 and 13.5 dpc is critical for regulation of PGC proliferation and differentiation. Interestingly, several laboratories including ours, showed that the proliferation behaviour of PGCs in primary culture mimics the growth pattern in vivo. Regardless of the age of the embryo at the time of the explant, PGCs will increase in number in vitro until the time corresponding to about 12-13 dpc in vivo. After this peak, PGC numbers, as measured by their surface APase reaction or by staining with specific antibodies (i.e. SSEA-1, TG-1), unfailingly decline over
several days (Fig. 6). According to these results, it may be speculated that PGC proliferation is an autonomous programmed process. The addition of single compounds or growth factors may increase PGC growth, but does not change the timing of the growth arrest. However, several groups found that a combination of compounds (FRSK, RA) and growth factors (SCF, LIF, bFGF), in certain culture conditions, may lead to prolonged proliferation of a small population of PGCs and their transformation into totipotent ES-like cells, named embryonic germ cells (EG cells) (Matsui et al., 1992; Resnick et al., 1992; Koshimizu et al., 1997). Except under this peculiar culture condition and certainly abnormal situation, two observations argue that in the current coculture systems PGCs die at the end of the proliferation period without undergoing their normal differentiation program in meiotic oocytes or prospermatogonia. First, since meiotic oocytes maintain in vivo detectable APase reaction up to the zygotene-pachytene stage (F. Klinger and M. De Felici, unpublished observations), it is unlikely that differentiating PGCs become undetectable for decline of APase activity. Second, the decline of APase-positive cells in culture is also accompanied by increased frequency of fragmented cells and nuclei, a characteristic of apoptotic cell death. Recently, Richards et al. (1999), using the germ cell nuclear antigen, GCNA1 as a marker of postmigratory PGCs, confirmed the inability of PGCs to differentiate properly in culture beyond the oogonia/ prospermatogonia stage.
Taken together these findings indicate that while factors neces-sary for PGC survival and proliferation are available in the present in vitro culture systems, PGCs in vivo are not normally continuously exposed to such factors, or at a given time, the combined action of these compounds must be modified by a negative regulator/s. In addition, it seems that critical factors allowing the differentiation of PGCs in oocytes/prospermatogonia are lacking in current culture conditions.
Adhesion molecules for PGCs: their expression and func-tions
Between 10 and 13 dpc, PGCs actively migrate from the developing hind gut to the site where the gonad will form. In the process of migration, PGCs move through solid tissues and Fig. 5. A schematic representation of the molecular signals involved in
sustaining PGC survival (SCF, LIF) and in the positive stimulation of their proliferation (cAMP, RA).
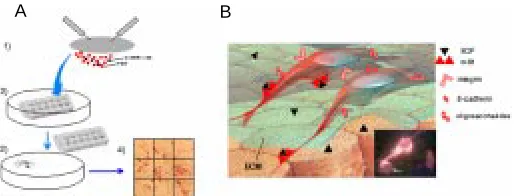
encounter various cell types and extracellular molecules. After arrival into the gonadal ridges, PGCs aggregate into groups of tightly adhering cells and lose their locomotory and proliferation activity. Eventually, PGCs become progressively incorporated into the supporting somatic cells and enter the long process of differen-tiation into the gametes of the adult. Over the years, we have performed a systematic study of adhesion molecules expressed by PGCs at different stages of development, using a combination of techniques. These have included PGC purification, before, during, and after migration (see above), a quantitative adhesion assay (Fig. 7A), and the use of immunohistochemistry (Fig. 7B) and immunoblotting analyses. The results obtained show that PGCs express at the same time integrins able to mediate adhesion to extracellular matrix (ECM) molecules (i.e. fibronectin and laminin) (De Felici and Dolci, 1989; De Felici et al., 1998); cadherins (namely, E-cadherin) that mediate omotypic PGC-PGC aggrega-tion (Di Carlo and De Felici, submitted), and a cytokine-based adhesion system (SCF/c-kit) (Pesce et al., 1997) crucial for their interaction with the surrounding somatic cells. Some indications exist that PGCs might adhere to somatic cell substrates also by specific oligosaccharides (i.e. 3-fucosyl lactosamine) (De Felici and Pesce, 1994), suggesting that selectin-like mediated adhesion systems may also contribute to PGC adhesion during migration. We have been able to demonstrate that some of these adhesion systems are developmentally controlled by changes in their ex-pression and/or functionality, and might play a crucial role in PGC migration and in their ability to survive and proliferate (for a review, see De Felici et al., 1998) (Fig. 7B).
Concluding remarks
Over the last almost two decades my laboratory has contributed to develop methods for isolation, purification and culture mouse PGCs. The use of such methods has allowed obtaining valuable information about mechanisms and factors controlling the
develop-ment of PGCs. We have learnt that throughout the migratory period, PGCs receive signals from the surrounding somatic cells that: 1) secure their survival (at the same time perhaps promoting their apoptotic degeneration in ectopic sites), 2) control their proliferation, and 3) guide them to the developing gonad. At least some of such signals are mediated by adhesive interactions through molecules of distinct adhesion family. PGCs are probably able to modulate their adhesiveness according to different ECM molecules and somatic cells encountered during migration. They interact with ECM components via integrins and with surrounding somatic cells via the SCF/c-kit system as well perhaps via selectin-like molecules. They may even adhere to each other via E-cadherin. It is quite singular that PGCs considered potentially immortal are so ready to undergo apoptosis in the absence of somatic cell support. At the same time, however, they are the only cells of the post-gastrulating embryo that can regain totipotency and immortality when stimulated by the right mix of growth factors. Perhaps such a property is part of the germ cell differentiation program (not yet reproduced in the current in vitro coculture systems) that is normally displayed only following egg fertilization. This is one of the most fascinating secrets of my favorite cell, that I hope to see revealed. The improvement of the culture systems allowing to reproduce in vitro the entire period of PGC develop-ment, including their sex differentiation, the production of immortal-ized PGC cell lines and studies on gene expression in these cells, will probably be of great help to success in this challenge.
Acknowledgments
Most of the results presented in this review were obtained thanks to the precious work of brilliant young scientists who collaborated with me over the years: Susanna Dolci, Maurizio Pesce, Anna Di Carlo, Francesca Klinger and Saveria Iona. I am also grateful to Anne McLaren and Gregorio Siracusa for their precious and constant advice and guide. The research described in this paper was supported by EU BIO-CT96-0183 funds, ASI grant ARS-98-160 and by MURST National Project “Development and Differentiation of Germ Cells”.
References
BESMER, P. (1991). The kit ligand encoded at murine steel locus: a pleiotropic growth factor and differentiation factor. Curr. Op. Cell Biol. 3: 939-946.
DE FELICI, M. (1998a). In vitro culture systems for germ cells from mouse embryo: primordial germ cells and oocytes. In Reproductive Toxicology (Ed. J. Del Mazo). Plenum Press, New York, pp. 41-49.
DE FELICI, M. (1998b). Isolation and culture of germ cells from the mouse embryo. In Cell biology: a laboratory handbook. (Ed. J.E. Celis). Academic Press, New York, pp. 73-85.
DE FELICI, M. and DOLCI, S. (1989). In vitro adhesion of mouse fetal germ cells to extracellular matrix components. Cell Differ. Dev. 26: 87-96.
DE FELICI, M. and DOLCI, S. (1991). Leukemia inhibitory factor sustains the survival of mouse primordial germ cells cultured on TM4 feeder layers. Dev. Biol. 147:
281-284.
DE FELICI, M. and MCLAREN, A. (1983). In vitro culture of mouse primordial germ cells. Exp. Cell Res. 144: 417-427.
DE FELICI, M. and PESCE, M. (1994). Interactions between migratory primordial germ cells and cellular substrates. In: Germline Development (Ed. J. Goodie). Ciba Foundation Symposium, No. 182, pp. 140-153.
DE FELICI, M. and PESCE, M. (1995). Immunoaffinity purification of migratory mouse primordial germ cells. Exp. Cell Res. 216: 277-279.
DE FELICI, M., DI CARLO, A., PESCE, M., IONA, S., FARRACE, M.G. and PIACENTINI, M. (1999). Bcl-2 and Bax regulation of apoptosis in germ cells during prenatal oogenesis in the mouse embryo. Cell Death Dif. 6: 908-915.
A B
DE FELICI, M., DOLCI, S. and PESCE, M. (1992). Cellular and molecular aspects of muose primordial germ cell migration and proliferation in culture. Int. J. Dev. Biol. 36: 205-213.
DE FELICI, M., DOLCI, S. and PESCE, M. (1993). Proliferation of mouse primordial germ cells in vitro: a key role for cAMP. Dev. Biol. 157: 227-280.
DE FELICI, M., PESCE, M., GIUSTINIANI, Q. and DI CARLO, A. (1998). In vitro adhesiveness of mouse primordial germ cells to cellular and extracellular matrix component substata. Micr. Res. Tech. 43: 258-264.
DOLCI, S., PESCE, M. and DE FELICI, M. (1993). Combined action of stem cell factor, leukemia inhibitory factor, and cAMP omn in vitro proliferation of mouse primordial germ cells. Mol. Repr. Dev. 35: 134-139.
DOLCI, S., WILLIAMS, D.E., ERNST, M.K., RESNICK, J.L., BRANNAN, C.I., LOCK, L.F., LYMAN, S.D., BOSWELL, S.H. and DONOVAN, P. (1991). Requirements for mast cell growth factor for primordial germ cell survival in cutlure. Nature 352: 809-811.
DONOVAN, P., SCOTT, D., CAIRNS, A.L., HEASMAN, J. and WYLIE, C. (1986). Migratory and postmigratory muose primordial germ cells behave differently in culture. Cell 44: 831-838.
EDDY, E.M., CLARK, J.M., GONG, D. and FENDERSON, B.A. (1981). Origin and migration of primordial germ cells in mammals. Gamete Res. 4: 333-362.
FOX, N., DAMJANOV, I., MARTINEZ-HERNANDEZ, A., KNOWELS, B.B. and SOLTER, D. (1981). Immunohistochemical localization of the early embryonic antigen (SSEA-1) in postimplantation mouse embryos and fetal and adult tissues. Dev. Biol. 83: 391-398.
GINSBURG, M., SNOW, M.H.L. and McLAREN, A. (1990). Primordial germ cells in the mouse embryo during gastrulation. Development 110: 5231-5238.
GODIN, I., DEED, R., COOKE, J., ZSEBO, K., DEXTER, M. and WYLIE, C.C. (1991). Effects of the steel gene product on mouse primordial germ cells in culture. Nature 352: 807-809.
HAHNEL, A.C. and EDDY, E.M. (1986). Cell surface markers of mouse primordial germ cells defined by tow monoclonal antibodies. Gamete Res. 15: 25-34.
KOSHIMIZU, U., TAGA, T., WATANABE, M., SAITO, M., SHIRAYOSHI, Y., KISHIMITO, T. and NAKATSUJI, N. (1997). Functional requirement of gp-130 mediated signaling for growth and survival of mouse primordial germ cells in vitro and derivation of embryonic germ (EG) cells. Development 122: 1235-1242.
KOSHIMIZU, U., WATANABE, M. and NAKATSUJI, N. (1995). Retinoic acid is potent growth activator of mouse primordial germ cell In vitro. Dev. Biol. 168: 683-685.
MATSUI, Y., TOKSOZ, D., NISHIKAWA, S., NISHIKAWA, S.-I., WILLIAMS, D., ZSEBO, K. and HOGAN, B.L.M. (1991). Effect of steel factor and leukaemia inhibitory factor on murine primordial germ cells in culture. Nature 353: 750-752.
MATSUI, Y., ZSEBO, K. and HOGAN, B.L.M. (1990). Embryonic expression of a haemopoietic growth factor encoded by the Sl locus and the ligand for c-kit. Nature 347: 667-669.
MATSUI, Y., ZSEBO, K. and HOGAN, B.L.M. (1992). Derivation of pluripotential embryonic stem cells from murine primordial germ cells in culture. Cell 70: 841-847.
PESCE, M. and DE FELICI, M. (1995). Purification of muose primordial germ cells by MiniMACS magnetic separation system. Dev. Biol. 170: 722-727.
PESCE, M., CANIPARI, R., FERRI, G.L., SIRACUSA, G. and DE FELICI, M. (1996a). Pituitary adenylate cyclase activating-polypeptide (PACAP) stimulates adenylate cyclase and promotes proliferation of mouse primordial germ cells. Development 122: 215-221.
PESCE, M., DI CARLO, A., CERRITO, M.G. and DE FELICI, M. (1996b). All trans retinoic acid effects on mouse primordial germ cells in culture. Biol. Repr. 54: 411-417.
PESCE, M., DI CARLO, A. and DE FELICI, M. (1997). The c-kit receptor is involved in the adhesion of mouse primordial germ cells to somatic cells in culture. Mech. Dev. 68: 37-44.
PESCE, M., FARACE, MG., PIACENTINI, M., DOLCI, S. and DE FELICI, M. (1993). Stem cell factor and leukemia inhibitory factor promote primordial germ cell survival by suppressing programmed cell death (apoptosis). Development 118: 1089-1094.
RESNICK, J.L., BIXLER, L.S., CHENG, L. and DONOVAN, P.J. (1992). Long term proliferation of mouse primordial germ cells in culture. Nature 359: 550-551.
RICHARDS, A.J., ENDERS, G.C. and RESNICK, J.L. (1999). Differentiation of murine premigratory primordial germ cells in culture. Biol. Reprod. 61: 1146-1151.
RUSSEL, E.S. (1977). Hereditary anemia of the mouse: a review for geneticists. Adv. Genet. 20: 357-459.
TAM, P.P.L. and SNOW, M.H.L. (1981). Proliferation and migration of primordial germ cells during compensatory growth in mouse embryo. J. Embryol. Exp. Morphol. 64: 133-147.

![Effect of Ambient UV B on Stomatal Density, Conductance and Isotope Discrimination in Four Field Grown Soybean [Glycine max (L ) Merr ] Isolines](data:image/gif;base64,R0lGODlhAQABAIAAAP///wAAACH5BAEAAAAALAAAAAABAAEAAAICRAEAOw==)