Severe
Perinatal
Marfan
Syndrome
Donald
M. Gross,
MD, CM,
Luther
K. Robinson,
MD,
Lynne
T. Smith,
PhD,
Nancy
Glass,
MD, Harvey
Rosenberg,
MD, and
Madeleine
Duvic,
MD
From the University of Texas Medical School at Houston, Departments of Pediatrics, Pathology, and Internal Medicine/Dermatology, Houston, and the University of Washington, Department of Biologic Structure, Seattle
ABSTRACT.
The cardiovascular manifestations of theMarfan syndrome in older children and adults have been
well described. Clinical, radiographic, and
echocardi-ographic data regarding three patients with severe
pen-natal Marfan syndrome are descnibed. Two of these pa-tients had the syndrome at birth and died in infancy. The syndrome was diagnosed in the third patient at 6 months of age and the child is still alive at 3 years of age.
The possible relationship among the Marfan syndrome,
Ehiers-Danlos syndrome, and osteogenesis imperfecta is considered. Patients with Marfan syndrome and severe
cardiorespiratory problems early in life tend to have a limited life expectancy. Pediatrics 1989;84:83-89; Marfan
syndrome, cardiorespiratory problem, dermatologic
find-ings.
The Marfan syndrome is a heritable disorder of
connective tissue characterized by ocular,
muscu-loskeletal, cardiovascular and pulmonary abnor-malities.’ Most cases are diagnosed in adolescence
or adulthood because of the clinical appearance,
development of mitral valve prolapse, or sudden
death secondary to aortic rupture. However, a more
severe perinatal clinical appearance has been
de-scribed. Before the availability of
echocardi-ographic diagnosis, a single case of unequivocal
congenital Marfan syndrome was reported in an
infant who died at birth after taking a single
breath.2 In that case, pathologic changes in the
great vessels and cardiac valves were typical of
Marfan syndrome and assumed to be the cause of
death. Other early descriptions include the cases of a 4-month-old infant3 and 5-month-old twins4 who
Received for publication Feb 16, 1988; accepted Apr 26, 1988. Reprint requests to (D.M.G.) University of Texas Medical School at Houston, Dept of Pediatrics, Box 20708, Houston, TX
77225.
PEDIATRICS (ISSN 0031 4005). Copyright © 1989 by the American Academy of Pediatrics.
had congestive heart failure and typical cardiac findings documented at cardiac catheterization.
Since the advent of echocardiography, there have
been sporadic case reports of neonates5’6 and even
premature infants7 with evidence of Marfan
syn-drome. Phornphutkul et al8 described 36 children
with Marfan syndrome, 2 of whom were infants.
Sisk et al9 described the clinical, cardiac, and
ech-ocardiographic features of the Marfan syndrome in
15 children less than 4 years of age. Although
actuarial analysis has shown that mean life expect-ancy for affected men is 40 years and for affected
women 50 years, it seems clear that there is a subset
of patients in whom the disease is diagnosed early in life with severe cardiorespiratory problems and
a limited life expectancy.
Of the three patients recently described in the
literature in whom symptoms of Marfan syndrome
were diagnosed in the neonatal period,57 the age of
death ranged from 3 days to 3 years. Herein we
report two additional patients with severe cardiac
involvement on the first day of life who died of
combined cardiac and respiratory disease at 3
months and 2 years of age, respectively. An addi-tional patient with cardiac involvement at 6 months
of age is still alive, although with progressive car-diac deterioration.
METHODS
M mode and two-dimensional echocardiograms
were performed on Irex 2A (Irex Corporation)
im-aging equipment. Studies were performed in
non-standard patient positions and transducer
onienta-tions when necessitated by chest wall deformity.
Biopsies from skin of the chest wall and lung at
in 35-mm Costar dishes were labeled with
L(2,3,4,5)-3H-proline (Amersham 3.77 BciJmmol) in
0.5%
serum
Dulbecco’s minimum essential mediumwith 100 g/mL L-ascorbate (Sigma).
For light and transmission electron microscopy,
the skin was fixed in half-strength Karnovsky’s
fixative’0 at 4#{176}Cfor at least 24 hours. The tissue
was stained en bloc with 1% 0504, dehydrated through graded alcohols, taken into propylene
ox-ide, and embedded in Epon.” Sections 1-sm thick
were stained according to Richardson et al’2 and
examined by light microscopy using a Zeiss
photo-microscope. Thin sections were collected onto
cop-per grids, stained with 1% phosphotungstic acid,
1%
uranyl acetate, and lead citrate,’3 and viewedwith a Philips 420 scanning transmission electron
microscope.
Labeled procollagens were obtained from media
and pellet by ethanol precipitation and analyzed on
7.5% sodium dodecyl sulfate-polyacrylamide urea gels as described.14 Collagens were obtained by
pep-sin digestion of procollagen chains.’4 Gels were
visualized by autoradiography.
CASE REPORTS
Patient 1
Patient 1 was the product of an uncomplicated full-term pregnancy, labor, and delivery (Table 1). The father
was 26 years of age and there was no family history of connective tissue disease. The baby was a long, thin infant with marked arachnodactyly (Figs 1 to 4), a
de-creased upper to lower segment ratio, skin
hyperextensi-biity, joint contractures of the elbows, wrists, knees, and ankles, and bilateral ectopia lentis. His birth weight was
3.6 kg (75th percentile) and birth length was 58 cm
(>90th percentile). He was in no distress. There was an
apical nonejection click but no murmur or heart failure.
Electrocardiogram and chest radiogram results were
nor-mal. Echocardiographically, mild aortic root dilation was seen and a dysplastic, prolapsing mitral valve. The urine was negative for homocystine.
After discharge from the hospital, the baby had
inter-mittent respiratory distress and chest radiograms results
showed migratory pulmonary consolidation (Fig 1B). At
3 months of age, he had a mitral click and regurgitation
but no heart failure. Results of a second echocardiogram showed marked aortic root dilation, left atnial
enlarge-ment, and marked prolapse of both atnioventnicular
valves (Fig 1C). In spite of maximal medical therapy, progressive respiratory failure developed and the infant required a tracheostomy for home ventilation therapy by
7 months of age. Progressive dilation of the great
yes-sets also developed, leading to tracheal and bronchial
compression. Increasingly frequent and prolonged hos-pital admissions were needed for acute and chronic
res-piratory failure. He died at 21 months of age during an
acute episode of respiratory failure and pneumothorax. At postmortem examination (Table 2), numerous
em-physematous bullae were seen in the lungs, ranging in
size from 0.1 to 5 mm in diameter throughout all lobes.
TABLE 1. Clinical Features of Infants With Marfan Syndrome
Patient No/Sex Age at Diagnosis Family History Skin and Joints Eyes Cardiac Echogram Lung
1/M Newborn Negative Skin and joint hypermobil-ity, joint con-tractures,
an-achnodactyly,
beaked nose, decreased
up-per to lower segment
Ectopia lentis, myopia
Newborn: dilated aor-tic root, mitral valve prolapse
6 mo: atnial
enlarge-ment
11 mo: dilated aorta +
pulmonary artery
Pneumonia, emphysema, atelectasis
2/F Newborn
Mother-con-genital hip
dislocations; father-hy-perextensible joints
Loose skin,
an-achnodactyly, bilateral hip dislocations, hyperextensi-bility Droopy lids, myopia, ec-topia
3 mo: mitral valve pro-lapse, tricuspid valve prolapse, mitral re-gurgitation, tricuspid regurgitation, dilated aortic root, & ventni-des
Bilateral lower lobe infiltrates & heart failure
3/M 6 mo Negative Droopy, loose skin,
arach-nodactyly, joints not
by-perextensible, pectus exca-vatum, right hip disloca-tion, mild nerve hypo-tonia
Ectopia lentis Mitral valve prolapse, mitral regurgitation,
tricuspid valve pro-lapse, tricuspid
re-gurgitation, pul-monic insufficiency, aortic insufficiency,
atnial enlargement,
ventricular hypertro-phy, dilated aortic root
Bilateral lower lobe
c)
V
-‘ -.
.., .
:#{149}#{149}#{149}#{149}#{149}#{149}
, .5’, ,.-
_
Fig 1. A, Patient 1 as neonate. Note redundant skin and
arachnodactyly. B, Anteropostenior chest radiograph of patient 1. Note pulmonary hypeninflation, slightly in-creased heart size, and prominent aortic knob. C, Patient
1, M mode echocardiogram at 3 months of age. Both
mitral and tricuspid valves are seen. Note mid- to late
systolic prolapse of mitral valve and early systolic
pro-lapse of tricuspid valve (arrows). D, Postmortem
photo-graph of patient 2. Note contractures of elbow, and
an-achnodactyly, particularly suggestion of positive thumb
sign (extension of thumb beyond ulnar border when
confined within closed fist). E, Patient 2, anteropostenior chest radiograph at 3 months of age. Note wide superior mediastinum and markedly elongated heart (“Christmas stocking heart”). F, Patient 2, two-dimensional
echocar-diogram in apical four-chamber view showing systolic prolapse of both tricuspid and mitral valves. G, Patient
3 at initial evaluation at 6 months of age. Note pectus excavatum and arachnodactyly. H, Patient 3 at 20
months of age. Chest x-ray film in pulmonary artery
projection. Note marked cardiomegaly with elongation of
heart. There is eventration of left hemidiaphragm and prominence of left atnia appendage. Also note dilated
pulmonary artery branch above left main stem bronchus.
I, Patient 3 at initial evaluation. Two-dimensional
echo-cardiogram in high parasternal oblique projection. Note normal pulmonary artery dimension (1.0 cm) and dilated
aortic root (2.1 cm). Abbreviations: RA, right atrium; LA,
left atrium; TV, tricuspid valve; Ao, aorta; PA, pulmonary artery.
Microscopically, there were markedly distended air
spaces with clubbing of the alveolar septa. Grossly, the
heart had a dilated left atrium and a thick, rubbery, redundant mitral valve that prolapsed into the left atrium. The aortic valve was thickened, opaque, and incompetent. The pulmonary valve was thickened and the sinuses of Valsalva were dilated and thin walled. The tricuspid valve appeared normal.
With light microscopy, the skin showed a thickened
Fig 2. Light microscopy (A) and transmission electron microscopy (B, C) of skin from patient 1. Reticular dermis had increased and densely packed collagen fibers and no apparent increase in elastic fibers (A). Within reticular collagen fiber bundles, fibnil diameters were variable in size and tended to have angular rather than round cross-sectional profiles (B). Elastic fibers were small and irreg-ular (B, C), and frequently appeared to merge with col-lagen fibers (C, arrowheads). Arrow indicates bundle of microfibnils associated with “moth-eaten” elastic fiber. A,
x340; B, x18 900; C, X12 300.
reticular dermis with densely packed collagen fiber
bun-dies and relatively little elastin (Fig 2A). Results of light microscopic examination of the endocardium, including the valves and great vessels, showed similar findings throughout with intimal sclerosis and increased
cellular-ity and blurring of the elastic lamina of the media. There were lakes of material that stained for mucopolysacchar-ide. By electron microscopy of the skin sample, collagen
fibers of the deep reticular dermis were seen to be a mixed population of large and small fibnils and slightly angular
in circumference (Fig 2B). The elastic fibers were
irreg-ular and moderately frayed around the edges (Fig 2B and C).
Analysis of 3H-proline-labeled procollagen chains in
skin appeared normal (Fig 3, lane D) compared with
control skin fibroblasts (Fig 3, lane C). Similarly, collagen
chains a-i and a-2 were not different than control chains (data not shown).
Patient 2
Patient 2 was the product of an uncomplicated full-term pregnancy, labor, and delivery (Table 1). Her birth weight was 4 kg (90th percentile) and birth length was
51 cm (>90th percentile). Because of a family history of
hypertrophic cardiomyopathy in the father (age 37 years),
3A 1
-1
1-PCA1
PNAI
-PCA2
PNA2
I#{248}
.
.-, .
choscopy results showed a normal larynx but partial to complete closure of the trachea and both main stem bronchi during coughing. In spite of vigorous medical therapy, increasing respiratory failure developed and the baby had a cardiorespiratory arrest at 3#{189}months of age from which she could not be resuscitated. A postmortem
A
B
C
D
photograph of the patient is shown Fig 1D to F; chestradiogram and echocardiogram were done shortly before death.
At postmortem examination (Table 2), the lungs were
hypeninflated. Microscopically, the baby had well-aerated alveoli with mild intraalveolar hemorrhage but no
em-physematous changes. By gross examination of the
car-diovascular system, she was shown to have dilation of both atnia, both ventricles, and both great arteries as well
as the coronary arteries near their origins. Both
atnioven-tnicular valves were large and redundant and prolapsed. The semilunar valves were thin and delicate but
redun-Fig 3. 3H-proline-labeled procollagens from cultured
fi-broblasts were separated by sodium dodecyl sulfate-poly-acrylamide gel and radioaudiographed. Lane A, skin fi-broblasts, patient 2; lane B, lung fibroblasts, patient 2;
lane C, skin fibroblasts of control subject; lane D, skin
fibroblasts, patient 1.
Fig 4. Light microscopy (A) and transmission electron microscopy (B, C) of skin from patient 2. At light level,
collagen fibers appeared normal, but elastic fibers were
increased and apparently fragmented (A). Ultrastructure
of dermis confirmed many clusters of small, “moth-eaten”
elastic fibers (B). There was atypical fine, filamentous
material distributed in between collagen fibnils within
dermal collagen fiber bundies (arrowheads) (C). A, x340; B, x18 000; C, x43 200.
of hypertrophic cardiomyopathy. The infant was started on digoxin for congestive cardiomyopathy.
At 2 months of age, results of a dysmorphology evalu-ation revealed droopy eyes, lax skin, arachnodactyly,
hi-lateral hip dislocation, bilateral ectopia lentis, blue
sclerae, and pectus excavatum. According to further
fam-ily history, the mother had also had dislocated hips and
the father was thought to have hyperextensible joints.
The baby had mitral and tricuspid regurgitation without overt heart failure. The chest radiogram showed mild cardiomegaly and the electrocardiogram showed only ST and T wave changes consistent with digoxin effect. The urine was negative for homocystine.
Increasing respiratory failure developed and the infant
was readmitted to the hospital at 3 months of age with
bilateral pneumonia and mild heart failure. Antibiotics,
chest physiotherapy, and hydralazine were started.
Bron-dant.
Microscopically, the baby had focal intimal thickening ofthe great vessels with disruption of medial elastic fibers and lakes of amorphous, metachromatically staining ma-terial. Focal proliferation of the spongiosa was shown in
the atnioventnicular valves with areas of myxoid
degen-eration ofthe fibrous layer. Elastic tissue in the spongiosa
was well preserved.
By light microscopy, the skin showed abundant, small elastic fibers that appeared “fuzzy” with high
magnifica-tion (Fig 4A). By electron microscopy, the skin sample showed clusters of small elastin fibers with irregular
surface contours (Fig 4B). Collagen fibrils of the reticular
dermis had variable diameters with angular rather than
round cross sections (Fig 4B), and there was an additional filamentous material associated with collagen and elastic
fibers (Fig 4C).
Analysis of 3H-proline-labeled procollagen chains in
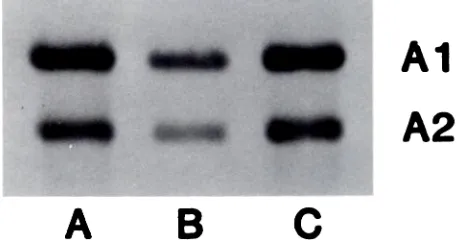
skin (Fig 3, lane A) showed increased amounts of PC a-1 (I), Pc a-2 (I), and a-i and -2 collagen chains relative
to control skin fibroblasts (Fig 3, lane C). The band above
pro a-2, type I, labeled “PNA 1,” the procollagen N-terminus a-2 (I) chain, appears more distinct in the
patient’s skin (lane A) than in the control cells (lane C). By two-dimensional map analysis, this appears to
Al
A2
TABLE 2. Histologic Features of Marfan Syndrome
Patient Lung Heart and Endocardium Valves Skin
No.
1 Emphysema edema Internal sclerosis, increased cellularity, blurring of elastic lamina within media, muco-polysacchanides
Dense collagen in dermis in large & small bundles, decreased elastin
2 Focal hemorrhage L up- Focal thickening of intima, dis- Dense collagen in dermis, large
col-per lobe, hyperplastic rupted elastic fibers, in- lagen bundles, diminished elastin pseudostratified cili- creased mucopolysaccha- or microfibnillar material, dissolu-ated epithelium with rides, atnioventnicular tion of elastin
focal squamous meta- valves-myxoid degenera-plasia tion, focal proliferation of
spongiosa with preserved
elastin
mutant pro a-2, type I chain. In lung fibroblasts, two
bands of approximately 30 kd were seen (Fig 3, lane B)
that were not present in skin. Pepsin-treated samples
showed decreased amounts of a-i, type I and a-2, type I
collagen chains (Fig 5, lane B) relative to control skin (Fig 5, lane A) and to lung (Fig 5, lane C).
Patient 3
Patient 3 was the product of an uncomplicated, full-term pregnancy, labor, and delivery (Table 1). His birth weight was 3.6 kg (75th percentile) and birth length was
55cm (>90th percentile). During the neonatal period and
first 6 months of life, there was a notation of loose skin.
Paternal age was 31 years and there was no family history pointing to the disorder. At 6 months of age, the infant was admitted to the hospital with failure to thrive and a chronic cough. His weight had decreased to the fifth percentile. Prior chest radiogram results had been normal and the baby had not improved with administration of antibiotics.
When he was admitted to the hospital, the infant had little subcutaneous fat, lax skin, arachnodactyly, and hypotonia with no joint contractures or
hyperextensibil-ity. There was a marked pectus excavatum and
disloca-tion of the right hip (Fig 1G). He had bilateral ectopia
lentis, mild hypotonia, and mild myopathy. Nerve
con-duction study results were normal and the urine was negative for homocystine. The baby had a mitral click and regurgitation but no heart failure. The chest radi-ogram showed normal heart size with a wide mediastinum
and bilateral lower lobe atelectasis.
Electrocardiographi-cally, he had left ventricular hypertrophy and left atnial
enlargement, and echocardiographically, he had mitral
and tricuspid prolapse and dilation of the aortic root (Fig 11).
The baby was treated with oral antibiotics and chest physiotherapy, which continued after discharge. He made initial improvement but had increasingly frequent
epi-sodes of cough and fever necessitating antibiotics and chest physiotherapy. A chest x-ray film at 1#{189}years of
age showed marked cardiomegaly with elongation of the heart and a prominent left atnial appendage (Fig 1H) and there was no pulmonary edema. Moderate to severe
mi-tral regurgitation without heart failure persisted and did
not improve with hydralazine treatment. At 4 years of
age his clinical cardiac status had changed little, but left atnial enlargement and left ventricular hypertrophy with
strain were seen on his electrocardiogram. By Doppler
echocardiography, moderate mitral and tricuspid
regur-gitation and mild to moderate pulmonic and aortic regur-gitation, were seen.
DISCUSSION
Although we refer to the Marfan syndrome as if
it were a distinct clinical entity, it seems clear in view of the variety in clinical appearance at a wide
range of ages that the underlying abnormality of
connective tissue must vary greatly, not only in
severity, but also in type. It is important to
recog-nize that no single biochemical abnormality has
been identified as the cause of the Marfan
pheno-type. In fact, it has been proposed that Marfan’s original patient probably had congenital
contrac-tuna! arachnodactyly rather than the condition we
now refer to as the Marfan syndrome.’5’16
Some patients are first diagnosed in the teenage years because of the development of the distinct
body habitus first described by Marfan in 1896.’ It
is of note that this original patient was not known
to have either cardiovascular or ocular
manifesta-tions, now considered to be typical of the Marfan
---
---A
B
C
Fig 5. ‘H-proline-labeled procollagens were digested
with pepsin, and collagen chains were separated by
so-chum dodecyl sulfate-polyacrylamide gel. Lane A, control subject, skin; lane B, patient 2, skin; lane C, patient 2,
syndrome. Others are diagnosed earlier in
child-hood because of the finding of a murmur or valve
dysfunction, myopia or ectopia lentis, or a positive
family history of Marfan syndrome.8’9 Still others
are diagnosed in early infancy or even the neonatal period with musculoskeletal, ocular, cardiovascular,
and pulmonary manifestations.57 These last
pa-tients appear to have the worst prognosis, and it is
with them that this report is concerned.
Ironically, the first report, in 1912, of
cardiovas-cular abnormality in a patient with a Marfanoid
habitus was that of a 2-month-old infant with
car-diomegaly, attacks ofdyspnea, and death.’7 Autopsy
showed dilation of all cardiac chambers and both atrioventricular valves (patient 2). The pulmonary manifestations of the syndrome have been
de-scribed relatively recently’2#{176} and consist mainly
of pneumothorax and emphysematous changes in
the lung that are presumably due to inadequate
support by defective pulmonary connective tissue.
Again, it is clear that not all patients with the
Marfan syndrome have clinically apparent
pulmo-nary involvement.
Patient 1 had the clinical and autopsy findings
typical of the fully developed Marfan syndrome
with musculoskeletal, ocular, cardiovascular, and
pulmonary involvement. It might be reasonable to
speculate that his early demise was hastened by the
mechanical stress of chronic home ventilation on his inadequately supported lungs. However,
Bo-lande and Tucker’s report’8 of the lung findings in
the Marfan syndrome included four infants, 11
weeks to 10 months of age who died with marked
emphysematous changes in one or both lungs. None
of these patients had had chronic mechanical
yen-tilation.
The case of patient 2 is slightly more complex.
During life, she demonstrated many of the typical
findings of Marfan syndrome and died of refractory
cardiorespiratory failure at 3 months of age.
Be-cause the parents gave a history of hyperextensible
joint involvement and congenital hip dislocations,
this patient may represent a compound
heterozy-gote with features of both Ehlers-Danlos type VII
syndrome and Marfan syndrome. The biochemical
studies suggest that this patient may have a defect in one of her a-2, type I collagen chain genes and that her abnormal a-2 collagen chains may be more susceptible to proteolysis with trypsin. Byers et al21
previously described another patient with an
ab-normal a-2, type I collagen chain.
Structural abnormalities in collagen and elastin
have been shown in the dermis of skin biopsies
from patients with inherited disorders of connective tissue including the Marfan syndrome.22 At the
ultrastructural level, there is heterogeneity as well,
and a change in one matrix component may
influ-ence the structure of all others. Abnormal collagen fiber diameter, especially smaller diameter fibers,
has been described with the Marfan syndrome.22
The collagen may be dense with disassociated
bun-dies, angular fibrils, and excessive matrix material that is thought to be mucopolysaccharide. However, the specific abnormality usually associated with the
Marfan syndrome is abnormal elastin fiber with a
“moth-eaten” appearance, which has been found in
skin and aorta.2224
In the skin biopsies of patients 1 and 2, the
histologic features were similar (Table 2). In patient
1,
the dermal collagen was more dense andcom-pacted and there were diminished elastin fibers
relative to normal and to patient 2. In both patients,
the collagen fibnils were variable in size with
an-gular circumference. Elastic fibers were small and
fragmented or frayed around the edges. The biopsy
results of patient 2 showed the presence of fine
matrix material around both collagen fibrils and
elastic fibers. Mucopolysaccharide accumulation
was also seen in the endocardium and aorta of both
patients.
Patient 3 represents a diagnostic and prognostic
problem. Unfortunately, we have been unable to
obtain a skin biopsy for light and electron
micros-copy and collagen biochemical studies. This infant
clearly has had a less stormy course than the other two patients, but nonetheless has been ill for much of his life and is now deteriorating more rapidly. His major difficulty seems to be cardiac, with res-piratory symptoms secondary to compression by
dilated vascular structures.
If all of these children have the Marfan
syn-drome, then the syndrome must have a spectrum
not only of clinical expression (age of onset, cardiac
vs respiratory difficulty and early vs late death), but also of histochemical expression in connective
tissue (normal vs decreased collagen, amount of
disruption of elastin, and amount of acid
mucopo-lysaccharide in vascular endothelium). Although
Marfan described a patient with tall stature,
arach-nodactyly, and joint contractures in his original
report, it is now common to diagnose patients with these features as well as ocular, cardiovascular, and pulmonary manifestations as having the Marfan
syndrome. The underlying biochemical defect
caus-ing the Marfan syndrome is not yet known.
Clini-cally, however, it shares features with the
Ehlers-Danlos syndrome and osteogenesis imperfecta,24
and a single patient with the Marfan syndrome has
been shown to produce an abnormal pro-a-2 type I
collagen chain.2’ However, linkage with type I, II,
and III collagen chain genes has been excluded by
extensive restriction fragment length
polymor-phism linkage studies.25’26 Because the Marfan
structural protein mutation. Sporadic cases occur 15% to 30% of the time and are more severe.9
Osteogenesis imperfecta is heterogeneous at the
clinical level, resulting from molecular
heterogene-ity in mutations affecting both type I collagen chains. The most severe perinatal form (type II) involves mutations affecting triple helix formation
of collagen, especially glycine substitutions.’4 Mild
osteogenesis imperfecta (type IV) involves muta-tions in the a-2, type I collagen chain.27 At the
genetic level, all osteogenesis imperfecta patients
thus far described have different mutations,
al-though linkage studies indicate that either a-i or
a-2 collagen chains are involved.28’29 Our three
pa-tients with infantile Marfan syndrome represent
the most severe form of heterogeneous Marfan
syn-drome. If Marfan syndrome patients were clinically
categorized, this subset would be analogous to
os-teogenesis imperfecta type II of the Sillence classi-fication system.24 Recognition of this severe,
pen-natal subset of the Marfan syndrome by
echocar-diographic and subsequent histologic techniques should permit better definition of this entity at the biochemical level.
ACKNOWLEDGMENTS
This work was supported, in part, by The March of Dimes Basil O’Connor grant 5-501.
The authors thank Yolanda Thomatis for her assist-ance with this manuscript.
REFERENCES
1. Pyeritz RE, McKusick VA. The Marfan syndrome: diagnosis and management. N Engi J Med. 1979;300:772-777
2. Edwards RH. Congenital Marfan syndrome. Birth Defects.
1975;11:329-331
3. Shankar KR, Hultgren MK, Lauer RM, Diehl AM. Lethal tricuspid and mitral regurgitation in Marfan’s syndrome. Am J Cardiol. 1967;20:122-128
4. Hohn AR, Webb HM. Cardiac studies of infant twins with Marfan’s syndrome. Am J Dis Child. 1971;122:526-528 5. Lababidi Z, Monzon C. Early cardiac manifestations of
Marfan’s syndrome in the newborn. Am Heart J. 1981;102:943-.945
6. Rees A, Elbi F, Cook L, Solinger R. Noninvasive documen-tation of cardiovascular involvement in a neonate with Marfan’s syndrome. South Med J. 1982;75:1127-1128
7. Gruber MA, Graham TP, Engel E, Smith C. Marfan syn-drome with contractural arachnodactyly and severe mitral regurgitation in a premature infant. J Pediatr. 1978;93:80-82
8. Phornphutkul C, Rosenthal A, Nadas A. Cardiac
manifes-tations of Marfan syndrome in infancy and childhood.
Cir-culation. 1973;47:587-596
9. Sisk HE, Zahka KG, Pyertiz RE. The Marfan syndrome in early childhood: analysis of 15 patients diagnosed at less than 4 years of age. Am J CardioL 1983;52:353-358
10. Karnovsky MJ. A formaldehyde-glutaraldehyde fixative of high osmolality for use in electron microscopy. J Cell Biol. 1965;27:137A. Abstract
11. Luft JH. Improvements in epoxy resin embedding methods. J Biophys Biochem Cytol 1961;9:409-414
12. Richardson KC, Jarrett L, Finke EH. Embedding in epoxy resins for ultrathin sectioning in electron microscopy. Stain
Technol. 1960;35:313-323
13. Reynolds ES. The use of lead citrate at high pH as an electron opaque stain in electron microscopy. J Cell Biol. 1963;7:208-213
14. Bonadeo J, Byers P. Subtle structural alterations in the chains of type 1 procollagen produce osteogenesis imperfecta type II. Nature. 1985;316:363-366
15. Marfan AB. Un cas de deformation congenitale des quatre membres plus pronounce aux extremites charactenisee par l’allongement des os avec un certain degre d’amincessment.
Bull Mem Soc Med Hop Paris. 1896;13:220-226
16. Hecht F, Beals RK. “New” syndrome of congenital contrac-tural arachnodactyly originally described by Marfan in 1896.
Pediatrics. 1972;49:574-579
17. Salle V. Uber einen Fall von angeborener Abnormalen grosse der extremitaten mit einen an Akromegalie erinnernden Symptomenkomplex. Jahrb Kinderheilk. 1912;75:540-548 18. Bolande RP, Tucker AS. Pulmonary emphysema and other
cardiorespiratory lesions as part of the Marfan abiotrophy. Pediatrics. 1964;33:356-366
19. Lipton RA, Greenwald RA, Seriff NS. Pneumothorax and bilateral honeycombed lung in Marfan syndrome. Am Rev
Respir Dis. 1971;104:924-928
20. Turner JAM, Stanley NN. Fragile lung in the Marfan syn-drome. Thorax. 1976;31:771-775
21. Byers PH, Siegel RC, Peterson KE. Marfan syndrome: abnormal alpha 2 chain in type I collagen. Proc NatI Acad
Sci USA. 1981;78:7745-7749
22. Holbrook KA, Byers PH. Structural abnormalities in the dermal collagen and elastic matrix from the skin of patients with inherited connective tissue disorders. J Invest
Derma-tol.1982;79:7s-16s
23. Perejda AJ, Abraham PA, Cannes WH, et a!. Marfan’s syndrome: structural, biochemical and mechanical studies of the aortic media. J Lab Clin Med. 1985;106:376-383
24. Prockop DJ, Kivirrikko K!. Heritable diseases of collagen.
N EngI J Med. 1984;311:376-386
25. Tsipouras P, Borrensen AL, Bamforth 5, et al. Marfan syndrome: exclusion of genetic linkage to the Col 1A2 gene.
Clin Genet. 1986;30:428-432
26. Ogilvie DJ, Wordsworth BP, Priestley LM, et al. Segregation of all four major fibrillar collagen genes in the Marfan syndrome. Am J Hum Genet. 1987;41:1071-1082
27. Wenstrup RI, Hunter AG, Byers PH. Osteogenesis imper-fecta type IV: evidence of abnormal triple helical structure of type I collagen. Hum Genet. 1986;84:47-53
28. Tsipouras P, Borrensen AL, Dickson LA, et al. Molecular heterogeneity in the mild autosomal dominant forms of osteogenesis imperfecta. Am J Hum Genet.
1984;36:1172-1179