Vit Matuska
A Thesis Submitted for the Degree of PhD at the
University of St. Andrews
2009
Full metadata for this item is available in Research@StAndrews:FullText
at:
http://research-repository.st-andrews.ac.uk/
Please use this identifier to cite or link to this item:
http://hdl.handle.net/10023/828
Ring Compounds
A thesis submitted by
Vit Matuska
In partial fullment for the award of
Doctor of Philosophy of the University of St Andrews
School of Chemistry, University of St Andrews North Haugh, St Andrews, Fife, KY16 9ST
I, Vit Matuska, hereby certify that this thesis, which is approximately 50,000 words in length, has been written by me, that it is the record of work carried out by me and that it has not been submitted in any previous application for a higher degree.
Date: Signature of candidate:
I was admitted as a research student in September, 2004 and as a candidate for the degree of Doctor of Philosophy in May, 2009; the higher study for which this is a record was carried out in the University of St Andrews between 2004 and 2009.
Date: Signature of candidate:
I hereby certify that the candidate has fullled the conditions of the Resolution and Regulations appropriate for the degree of Doctor of Philosophy in the University of St Andrews and that the candidate is qualied to submit this thesis in application for that degree.
Date: Signature of supervisor:
In submitting this thesis to the University of St Andrews we understand that we are giving permission for it to be made available for use in accordance with the regulations of the University Library for the time being in force, subject to any copyright vested in the work not being aected thereby. We also understand that the title and the abstract will be published, and that a copy of the work may be made and supplied to any bona de library or research worker, that my thesis will be electronically accessible for personal or research use unless exempt by award of an embargo as requested below, and that the library has the right to migrate my thesis into new electronic forms as required to ensure continued access to the thesis. We have obtained any third-party copyright permissions that may be required in order to allow such access and migration, or have requested the appropriate embargo below.
The following is an agreed request by candidate and supervisor regarding the electronic publication of this thesis: embargo on both all or part of printed copy and electronic copy for the same xed period of 1 year on the following ground: publication would preclude future publication.
Date: Signature of candidate:
I want to thank my Mum, Dad, Brother and my grandparents for a massive support throughout the studies. Without them I would have not been able to reach this point. I would like to dedicate this thesis to them.
Special thanks to Stu Robertson and Richard Bond, who very patiently introduced me to the british culture and helped me to feel in Scotland like at home. My academic parents Gil Viana and Blerina Kellezi were always ready to cheer me up and for that I thank them a lot. I am indebted to Klára Mí£ková who helped me a lot in the very beginning. I am thankful to my landlords Mr and Mrs Gall, David and Gisele, Tom Lébl and Nathaniel for cosy rooms and to my atmates Mi²o, Alex, Gil, James and Lucia for creating a relaxed atmosphere at home.
The Woollins, Slawin and Kilian groups - thank you folks for a really enjoyable time both in and outside the lab and also for the patience you had with me.
Writing-up of this thesis was easier with LATEX2εand the KOMA-Script bundle. In
ad-dition to the authors I would like to thank Piotr for his help, tips and mainly Konwerter. Paul Waddell checked errors in some parts of this thesis. Grazie Poalo!
I thank Dr. Tomá² Lébl and Melanja Smith for their very kind and instant help whenever needed downstairs in the NMR room. Many thanks to Dr. Joe Crayston (CV), Caroline (mass spec.), Sylvia (microanalysis), Marj, Bobby, Brian, Colin the storesman, Artur and Colin the glassblower.
I am grateful to Dr. Petr Kilián who spent a lot of time bringing me up in the lab, showing me the equipment, its maintenance and explaining new techniques.
A big thank is due to Prof. Alex Slawin who was always ready to measure my crystal structures knowing that they were going to be either S4N4, sulfur, Na2SO4 or they would not diract at all.
A series of 1,3,2,4,5-dithiadiazarsoles with the general formula RAs(S2N2) (R = Me, Et,
iPr, tBu, Ph and Mes) have been prepared and characterised by multinuclear NMR, IR
and Raman spectroscopies and mass spectrometry. The X-ray structures of PhAs(S2N2) and MesAs(S2N2) were determined.
The low temperature X-ray structures of the half-sandwich 5,1,3,2,4-metalladithia-diazoles Cp*M(S2N2) (M = Co, Ir) were determined and Cp*Rh(S2N2) was prepared. All three metalladithiadiazoles were characterised by multinuclear NMR, IR and Raman spectroscopies and mass spectrometry. The X-ray structures of complexes [Cp*RhCp*]Cl, [Cp*Rh(µ-S3N2)(µ-S2O3)RhCp*] and Cp*Ir[S2N2(IrCl2Cp*)] obtained during this work were determined.
The low temperature X-ray structure of Roesky's sulfoxide (S3N2O) is presented to-gether with assignments of its vibrational spectra as suggested by theoretical calculations. The experimental structures of the metalladithiadiazoles and that of Roesky's sulfoxide are compared with calculated geometries.
Declarations 2
Acknowledgements 3
Abstract 4
List of Figures 12
List of Tables 14
Abbreviations 16
General abbreviations . . . 16
NMR spectroscopy abbreviations . . . 17
Vibrational spectroscopy abbreviations . . . 17
I. Introduction
19
1. General considerations 20 1.1. Preparation routes of sulfur-nitrogen heterocycles . . . 201.2. The multiplicity of S−N bonds . . . . 22
1.3. Theoretical calculations . . . 24
1.3.1. Geometry of a molecule . . . 24
1.3.2. Charges and bond orders . . . 25
1.3.3. Aromaticity . . . 25
1.3.3.1. Aromaticity of sulfur-nitrogen compounds . . . 27
1.4. General experimental conditions . . . 28
1.5. Experimental details of the key sulfur-nitrogen reagents syntheses . . . . 29
1.5.1. [S3N2Cl]Cl . . . 29
1.5.2. [S4N3]Cl . . . 30
II. Arsenic-Sulfur-Nitrogen Heterocycles
32
2. Introduction 33
2.1. The toxicity of arsenic . . . 33
2.1.1. Arsenic in the environment . . . 33
2.1.2. Biotransformation of arsenic in nature . . . 34
2.1.3. Arsenic in the human body . . . 35
2.1.3.1. The principle of toxicity of arsenic . . . 35
2.1.3.2. Metabolism of arsenic in the human body . . . 36
2.2. Commercial availability of starting materials . . . 38
2.3. 75As NMR as an analytical tool . . . . 38
2.4. Methods of preparation of common starting materials . . . 39
2.4.1. Arsenic trihalogenides . . . 39
2.4.2. Alkylarsonic acids . . . 40
2.4.3. Arylarsonic acids . . . 41
2.4.3.1. Rosenmund's method . . . 41
2.4.3.2. Bart's reaction . . . 41
2.4.3.3. Modications of Bart's method . . . 46
2.4.3.4. Béchamp's reaction . . . 46
2.4.4. Dialkyl- and diarylarsinic acids . . . 47
2.4.5. Organohalogenoarsines . . . 48
2.4.5.1. From arsonic and arsinic acids . . . 48
2.4.5.2. From AsX3 and organometallic reagents . . . 48
2.4.5.3. Controlled substitution of halogens in AsX3 . . . 50
2.4.5.4. From AsX3 and organosilicon and organotin reagents . . 52
2.4.5.5. From AsX3 by aromatic electrophilic substitution . . . . 54
2.4.5.6. Special methods of RAsX2 preparation . . . 55
2.4.5.7. Halogen exchange reactions of RAsX2 . . . 58
2.5. Arsenic containing main-group ring compounds . . . 59
2.5.1. Organoarsenic heterocycles . . . 59
2.5.1.1. Five-membered heterocycles . . . 59
2.5.1.2. Six-membered heterocycles . . . 60
2.5.2. Arsenic homocycles . . . 61
2.5.2.1. Five-membered arsenic homocycles . . . 61
2.5.2.2. Six-membered arsenic homocycles . . . 62
2.5.2.3. Arsafullerenes . . . 63
2.5.3. Arsenic containing analogues of S4N4 . . . 64
2.5.3.2. Cyclotetraarsazenes . . . 66
2.5.3.3. Cyclotetraarsazanes . . . 67
2.5.3.4. Dithiatetrazadiarsocines . . . 69
2.5.3.5. 1,5,2,4,6,8,3,7-dithiatetrazadiarsocanes . . . 72
2.5.3.6. Other AsSN eight-membered heterocycles . . . 72
2.5.4. Arsenic containing analogues of S2N2 . . . 73
2.5.4.1. As2S2 . . . 73
2.5.4.2. As2N2 . . . 75
2.5.5. Five- and six-membered arsenic-sulfur-nitrogen rings . . . 77
2.5.6. AsEN heterocycles (E = Se, Te) . . . 77
2.5.6.1. Eight-membered As4E4 heterocycles (E = Se, Te) . . . . 77
2.5.6.2. Four-membered As2E2 heterocycles (E = Se, Te) . . . . 78
2.6. Conclusion . . . 79
3. Results and Discussion 80 3.1. Comments on preparative procedures . . . 82
3.1.1. Alkyl/aryldihalogenoarsines . . . 82
3.1.2. 5-alkyl/aryl-1,3λ4δ2,2,4,5-dithiadiazarsoles . . . . 87
3.2. NMR spectroscopy . . . 89
3.2.1. 1H and 13C NMR data . . . . 89
3.2.2. 14N NMR data . . . . 92
3.3. Mass spectrometry . . . 94
3.4. IR and Raman spectroscopy . . . 95
3.4.1. The assignments and their discussion . . . 96
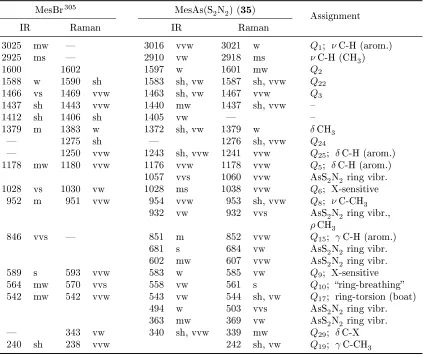
3.5. X-ray structures . . . 105
3.5.1. PhAs(S2N2) . . . 105
3.5.2. MesAs(S2N2) . . . 108
3.6. Cyclic voltammetry . . . 109
3.7. Reactivity of PhAs(S2N2) . . . 110
3.8. Conclusion . . . 112
4. Experimental 114 4.1. Preparation of AsCl3 . . . 114
4.2. Preparation of CH3AsI2 . . . 115
4.3. Preparation of EtAsI2 . . . 115
4.4. Preparation of iPrAsCl 2 . . . 117
4.5. Preparation of tBuAsCl 2 . . . 117
4.7. Preparation of mixture of mesityldihalogenoarsines . . . 119
4.8. Preparation of p-phenylenediarsonic acid by Bart's reaction . . . 120
4.9. Reaction of MeAsI2 with [nBu 2Sn(S2N2)]2 . . . 121
4.10. Reaction of EtAsI2 with [nBu 2Sn(S2N2)]2 . . . 122
4.11. Reaction ofiPrAsCl 2 with [nBu2Sn(S2N2)]2 . . . 123
4.12. Reaction oftBuAsCl 2 with [nBu2Sn(S2N2)]2 . . . 124
4.13. Reaction of PhAsCl2 with [nBu 2Sn(S2N2)]2 . . . 125
4.14. Reaction of mesityldihalogenoarsines with [nBu 2Sn(S2N2)]2 . . . 126
4.15. Reaction of PhAs(S2N2) with Pt(COD)Cl2 . . . 127
4.16. Methylation of PhAs(S2N2) with CH3I . . . 127
4.17. Reaction of PhAs(S2N2) with [Mo(CO)4(piperidine)2] . . . 128
4.18. Reaction of PhAs(S2N2) with Cp2Ti(CO)2 . . . 128
4.19. Reaction of PhAs(S2N2) with Cp*Co(CO)2 . . . 128
4.20. Reaction of PhAs(S2N2) with sulfur . . . 129
III. Transition Metal Sulfur-Nitrogen Ring Compounds
130
5. Introduction 131 5.1. Four-membered rings . . . 1315.2. Five-membered rings . . . 134
5.2.1. M(S2N2) complexes containing the (S2N2)2 anion . . . 134
5.2.1.1. Preparation from S4N4 or S2N2 . . . 134
5.2.1.2. Preparation from other SN rings . . . 135
5.2.1.3. Transmetallation reactions . . . 137
5.2.1.4. Deprotonation of M(S2N2H) complexes . . . 137
5.2.1.5. Preparation from acyclic SN compounds . . . 138
5.2.2. M(S2N2) complexes containing the (NSSN) moiety . . . 138
5.2.3. M(S2N2H) ring complexes . . . 139
5.2.3.1. Preparation from S4N4 . . . 139
5.2.3.2. Protonation of M(S2N2) complexes . . . 140
5.2.4. M(S3N) ring complexes . . . 140
5.2.4.1. Preparation from [S7N] . . . 140
5.2.4.2. Desulfuration of [S4N] . . . 141
5.2.4.3. Preparation from other SN compounds . . . 141
5.2.5. Structure of the ve-membered MSN rings . . . 141
5.3. Six-membered rings . . . 142
5.3.2. M(S2N3) ring complexes bearing the (S2N3) anion . . . 143
5.3.3. M(S2N3) ring complexes bearing the (S2N3)3 anion . . . 143
5.3.4. M(S2N3) ring complexes bearing the [N2S2N(SO2NH2)]2 anion . 144 5.4. Seven-membered rings . . . 145
5.5. Eight-membered rings . . . 145
5.5.1. Complexes of the (S3N4)2 anion . . . 145
5.5.2. Complexes of the (S4N3) anion . . . 146
5.6. Nine-membered rings . . . 146
5.6.1. Monocyclic nine-membered rings . . . 146
5.6.2. Insertion of a metal centre in S4N4 . . . 146
5.7. Half-sandwich M(S2N2) complexes . . . 148
5.8. Conclusion . . . 148
6. Results and discussion 149 6.1. Syntheses of the complexes . . . 149
6.1.1. Cp*Co(S2N2) . . . 149
6.1.2. Cp*Rh(S2N2) . . . 150
6.1.3. Cp*Ir(S2N2) . . . 152
6.2. The X-ray structures . . . 152
6.2.1. Cp*M(S2N2) (M = Co, Rh, Ir) . . . 152
6.2.1.1. The inuence of M in CpM(S2N2) and Cp*M(S2N2) . . . 156
6.2.2. Cp*Rh(µ−S3N2)(µ−S2O3)RhCp* and [Cp*RhCp*]Cl . . . 158
6.2.3. Cp*Ir[S2N2(IrCl2Cp*)] . . . 160
6.3. NMR spectroscopy . . . 163
6.3.1. 14N NMR spectroscopy . . . 164
6.4. Mass spectrometry . . . 165
6.5. IR and Raman spectroscopy . . . 165
6.6. Protonation of Cp*Co(S2N2) . . . 165
6.7. Conclusion . . . 166
7. Experimental 168 7.1. Preparation of [Cp*RhCl2]2 . . . 168
7.2. Preparation of Cp*Co(CO)2 . . . 169
7.3. Preparation of Cp*Co(S2N2) . . . 169
7.4. Cp*Rh(S2N2) . . . 170
7.5. [Cp*RhCp*]Cl . . . 171
7.6. [Cp*Rh(µ-S3N2)(µ-S2O3)RhCp*] . . . 171
7.8. Cp*Ir(S2N2) . . . 173
7.9. Cp*Ir[S2N2(IrCl2)]Cp* . . . 173
7.10. Protonation of Cp*Co(S2N2) with HBF4 . . . 174
7.11. Protonation of Cp*Co(S2N2) with HCl . . . 175
7.12. Computational details . . . 175
IV. Roesky's Sulfoxide, S
3N
2O
176
8. Introduction 177 8.1. Synthesis of Roesky's sulfoxide . . . 1778.2. Structure and reactivity . . . 179
8.3. Conclusion . . . 181
9. Results and Discussion 182 9.1. The crystal structure . . . 182
9.1.1. Aromaticity of Roesky's sulfoxide . . . 184
9.2. Vibrational spectra . . . 184
9.3. 14N NMR spectroscopy . . . 185
9.4. Cyclic voltammetry . . . 185
9.5. Reactivity of Roesky's sulfoxide . . . 185
9.6. Conclusion . . . 188
10.Experimental 189 10.1. Preparation of S3N2O . . . 189
10.2. Reaction of S3N2O with Cp*Co(CO)2 . . . 189
10.3. Reaction of S3N2O with Cp2Ti(CO)2 . . . 190
10.4. Reaction of S3N2O and [(Ph3P)Au(CH3CN)][ClO4] . . . 190
10.5. Protonation of S3N2O with HBF4 . . . 191
10.6. Reaction of S3N2O with Woollins' Reagent . . . 191
10.7. Reaction of (S2N2)CO with Woollins' Reagent . . . 192
10.8. Computational details . . . 192
Conclusion 193
Further work 195
Appendices
211
1.1. Selected S-N compounds with S-N bond lengths . . . 24
2.1. Arsenobetaine, arsanilic acid and roxarsone . . . 34
2.2. The structures of syn- and anti- arenediazoates . . . 43
2.3. Ball and stick model of the anion [As@Ni12@As20]3 . . . . 64
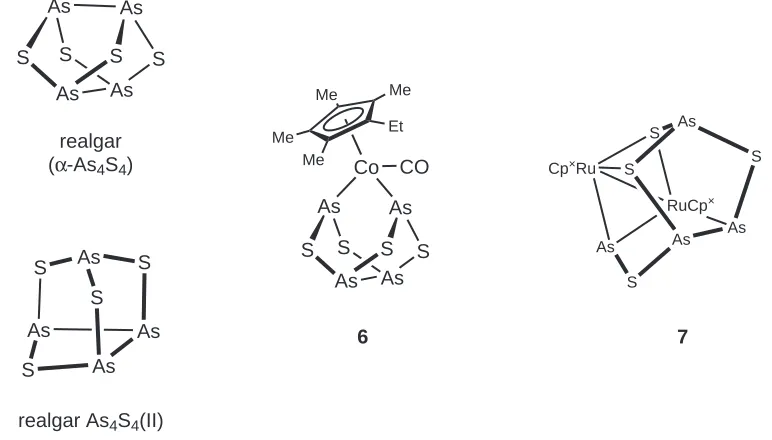
2.4. The structures of realgar (α-As4S4), realgar As4S4(II) and their metal complexes . . . 66
2.5. A schematic demonstration of the structural dierence between S4N4 and dithiatetrazadiarsocines . . . 70
2.6. Possible conformations of the As2S2 ring . . . 73
2.7. A part cut out from the polymeric anion [NiAs4S8]2n n . . . 74
2.8. Mesomeric structures of bis(1,3,2-dithiarsolylium) dication . . . 75
3.1. 1,4-bis(dichloroarsino)benzene, p-phenylenediarsonic acid and Atoxyl . . 86
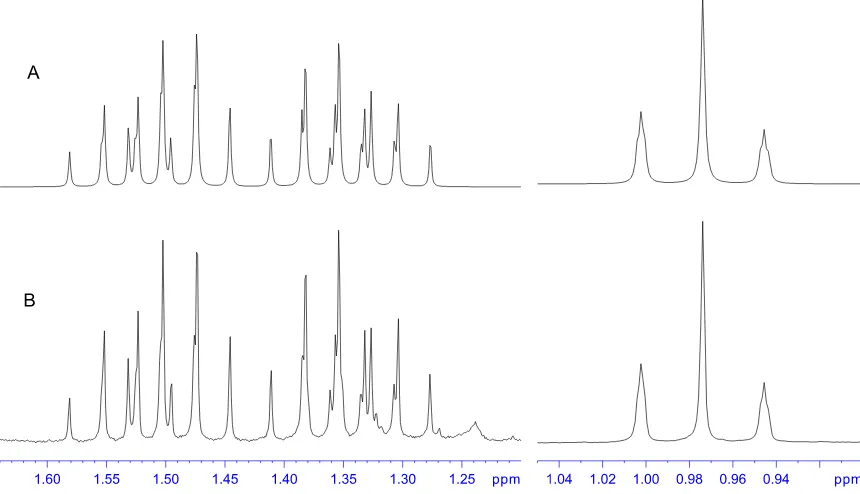
3.2. Simulated and recorded 1H NMR spectrum of EtAs(S 2N2) . . . 91
3.3. A general presentation of conformers for EtAsI2, iPrAsCl 2, EtAs(S2N2) and iPrAs(S 2N2) . . . 96
3.4. The X-ray structure of PhAs(S2N2) . . . 106
3.5. The stacking of PhAs(S2N2) molecules in the crystal . . . 107
3.6. The X-ray structure of MesAs(S2N2) . . . 108
3.7. Cyclic voltammogram of PhAs(S2N2) . . . 110
5.1. Possible coordination modes of the N2S and NS2 ligands onto a metal centre . . . 131
5.2. The structure of [Cl2Re(S2N3)(Cl3ReSN3)]2 2 . . . 144
5.3. The structures of complexes [(PPh3)(CO)IrCl(f ac-S4N4)] and [PPh4][Cl3Pt(f ac-S4N4)] . . . 147
5.4. The structures of complexes [(PPhMe2)PtCl2(mer-S4N4)] and [(PPhMe2)PtCl2(f ac-S4N4)] . . . 147
5.5. Numbering of the CpM(S2N2) and Cp*M(S2N2) complexes . . . 148
6.2. The X-ray structure of Cp*Ir(S2N2) . . . 156
6.3. The X-ray structure of Cp*Rh(µ−S3N2)(µ−S2O3)RhCp* . . . 158
6.4. A Newman projection of Cp*Rh(µ−S3N2)(µ−S2O3)RhCp* . . . 160
6.5. The X-ray structure of [Cp*RhCp*]Cl·H2O . . . 160
6.6. The X-ray structure of Cp*Ir[S2N2(IrCl2Cp*)]·nBu 2SnCl2 . . . 161
6.7. Cp*Ir[S2N2(IrCl2Cp*)] and [Cp*Ir[S2N2(IrCl(PPh3)Cp*)]][PF6] . . . 163
9.1. The crystal structure of S3N2O . . . 183
9.2. The packing of S3N2O molecules in the crystal . . . 183
9.3. Cyclic voltammogram of S3N2O . . . 186
3.1. Melting and boiling points of Me3SiX andnBu
2SnX2 . . . 81
3.2. 1H NMR data of RAsX 2 . . . 90
3.3. 1H NMR data of RAs(S 2N2) . . . 91
3.4. 13C NMR data of RAsX 2 . . . 92
3.5. 13C NMR data of RAs(S 2N2) . . . 93
3.6. 14N NMR data of RAs(S 2N2) . . . 93
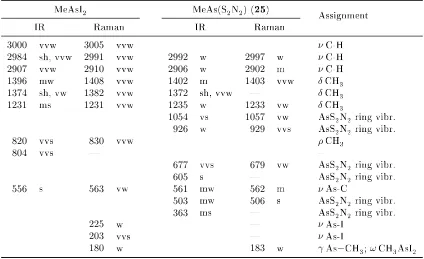
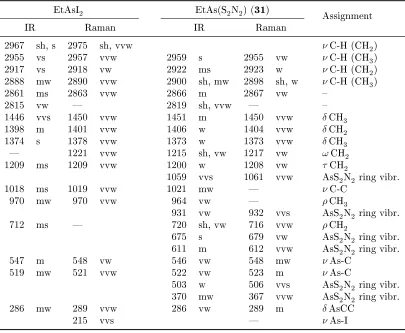
3.7. Selected IR and Raman wavenumbers of MeAsI2 and MeAs(S2N2) . . . . 98
3.8. Selected IR and Raman wavenumbers of EtAsI2 and EtAs(S2N2) . . . 99
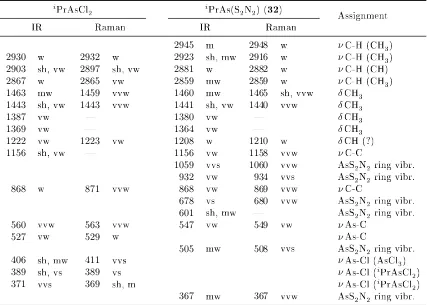
3.9. Selected IR and Raman wavenumbers ofiPrAsCl 2 and iPrAs(S2N2) . . . 100
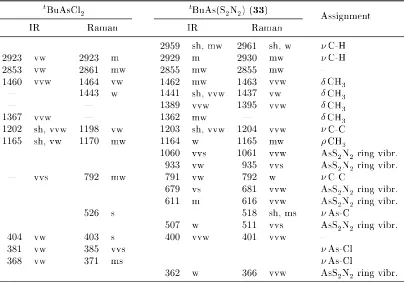
3.10. Selected IR and Raman wavenumbers oftBuAsCl 2 and tBuAs(S2N2) . . . 101
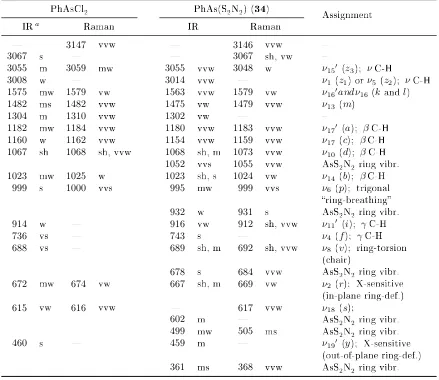
3.11. Selected IR and Raman wavenumbers of PhAsCl2 and PhAs(S2N2) . . . 103
3.12. Selected IR and Raman wavenumbers of MesBr and MesAs(S2N2) . . . . 105
3.13. Selected bond lengths and angles of PhAs(S2N2) . . . 106
3.14. Selected bond lengths and angles of MesAs(S2N2) . . . 109
3.15. Experimental and simulated voltammetric data for PhAs(S2N2) . . . 110
6.1. Selected bond lengths and angles of CpCo(S2N2), Cp*Co(S2N2) and Cp*Ir(S2N2) . . . 154
6.2. Selected calculated geometrical data for CpM(S2N2) and Cp*M(S2N2) . . 154
6.3. Calculated Wiberg bond orders for the most important bonds in CpM(S2N2) and Cp*M(S2N2) . . . 157
6.4. Selected bond lengths and angles for Cp*Rh(µ−S3N2)(µ−S2O3)RhCp* . 159 6.5. Selected bond lengths and angles for [Cp*RhCp*]Cl·H2O . . . 160
6.6. Selected bond lengths and angles for Cp*Ir(S2N2), Cp*Ir[S2N2(IrCl2Cp*)] and [Cp*Ir[S2N2(IrCl(PPh3)Cp*)]][PF6] . . . 162
6.7. 1H NMR data of compounds 47, 49, 51, 52, 53 and 54 . . . 163
6.8. 13C NMR data of compounds 47, 49, 51, 52, 53 and 54 . . . 164
9.1. The bond lengths and angles for S3N2O . . . 183
9.2. Selected IR and Raman wavenumbers of S3N2O . . . 184
A.1. Crystal data and structure renement for PhAs(S2N2) . . . 212
A.2. Crystal data and structure renement for MesAs(S2N2) . . . 213
A.3. Crystal data and structure renement for Cp*Co(S2N2) . . . 214
A.4. Crystal data and structure renement for Cp*Ir(S2N2) . . . 215
A.5. Crystal data and structure renement for [Cp*Rh(µ−S3N2)(µ−SSO3)RhCp*]·CH2Cl2 . . . 216
A.6. Crystal data and structure renement for [Cp*RhCp*]Cl·H2O . . . 217
A.7. Crystal data and structure renement for Cp*Ir[S2N2(IrCl2Cp*)]·nBu 2SnCl2 . . . 218
General abbreviations
◦C degrees Celsius
Å Ångström, 1×10−10m
Ac acetyl
approx. approximately
b.p. boiling point
nBu but-1-yl
tBu tertiary butyl, 2-methylprop-2-yl
calc. calculated
CI chemical ionisation
COD cycloocta-1,5-diene
conc. concentrated
Cp cyclopentadienyl (C5H5)
Cp* 1,2,3,4,5-pentamethylcyclopentadienyl (C5Me5)
Cp0 methylcyclopentadienyl (C
5H4Me)
Cp× 1-ethyl-2,3,4,5-tetramethylcyclopentadienyl (C
5Me4Et)
Cy cyclohexyl
DBN 1,5-diazabicyclo[4.3.0]non-5-ene
DBU 1,8-diazabicyclo[5.4.0]undec-7-ene
decomp. decomposition
DME 1,2-dimethoxyethane
DMF N,N -dimethylformamide
DMSO dimethylsulfoxide
EI electron impact
ES electrospray
Et ethyl
Fc ferrocenyl
HOMO Highest Occupied Molecular Orbital
IR infrared
LCAO Linear Combination of Atomic Orbitals
LUMO Lowest Unoccupied Molecular Orbital
m.p. melting point
Me methyl
Mes mesityl, 2,4,6-trimethylphenyl
min. minute, minutes
MS mass spectrometry
NBS N -bromosuccinimide
NMR Nuclear Magnetic Resonance
Ph phenyl
Phth phthalimide
iPr iso-propyl, prop-2-yl
r.t. room temperature
toil oil bath temperature
THF tetrahydrofuran
TOF Time Of Flight
XRD X-Ray Diraction
NMR spectroscopy abbreviations
d doublet
dd doublet of doublet
m multiplet
qqrt quartet of quartet
qrt quartet
quart. quarternary (written as a subscript to an atom, e.g. Cquart.)
s singlet
t triplet
Vibrational spectroscopy abbreviations
m medium
ms medium-strong
s strong
sh shoulder
v very
w weak
β bending (in-plane)
γ bending (out-of-plane)
δ deformation
ν stretch
νnumber a fundamental frequency; also used for labelling vibrational mode corresponding to that frequency
ρ rocking
τ twist
ω wag
This thesis concentrates on the preparation and characterisation of ve-membered sulfur-nitrogen ring compounds with the emphasis on ve-membered heterocycles. Since par-ticular projects involved in this thesis deal with elements from dierent areas of the Periodic System, it seemed better to compose the thesis along these lines.
The rst part describes the syntheses of the key sulfur-nitrogen reagents used through-out the research period as well as denitions of basic terms in computational chemistry, which are used in the sebsequent parts. The focus of the second part is on 1,3,2,4,5-dithia-diazarsoles with the general formula RAs(S2N2). The third part deals with half-sandwich 5,1,3,2,4-metalladithiadiazoles of the Group 9 metals (Co, Rh, Ir) and in the last part the structure and reactivity of Roesky's sulfoxide (S3N2O) is discussed.
Every part has its own introduction, discussion of results and experimental section. The overall contribution described in the thesis is summarised in a nal conclusion, after which a short account on future work follows. References are listed at the end of the thesis followed by appendices.
1.1. Preparation routes of sulfur-nitrogen
heterocycles
General synthetic routes leading to sulfur-nitrogen heterocycles have been thoroughly reviewed.13 The most useful are the cyclocondensation reactions which start with cheap
and commercially available acyclic materials and nish with ring compounds. These ring compounds can be further converted into other ring compounds with suitable reagents.1,3
A number of methods will be described in the introductions of particular chapters. The routes leading to the most frequent suur-nitrogen reagents used during this work are described here.
[S3N2Cl]Cl (eq. 1.1).4
2 NH4Cl + 4 S2Cl2 + S + 8 HCl + 5 S
reflux N
S N
S S+ Cl
Cl
-(1.1)
Although unpleasant and despite relatively low yields, this synthesis can be conveniently scaled-up to provide good amounts of the material. Interestingly, [S3N2Cl]Cl has not been used in many syntheses other than conversions to other purely SN rings. One such example is the preparation of [S4N3]Cl (eq. 1.2).4
N
S N S S+ Cl
Cl- + S2Cl2
3 2 + 3 SCl2
CCl4
reflux
N
S N S
S N S
Cl
-(1.2)
The bright yellow crystalline compound is also air-sensitive but can be stored for long time periods with only marginal decomposition. The conversion from [S3N2Cl]Cl to [S4N3]Cl is nearly quantitative.
[S4N3]Cl became extremely popular over the years because it is a safe source of the [S2N2]2 anion, which is the building block in a large number of metal-sulfur-nitrogen
complexes (see Part III of this thesis). S4N3Cl reacts with liquid ammonia to form a complex mixture, which was thoroughly studied by 14N NMR spectroscopy.5 The major
product was shown to be the [S3N3] anion (eq. 1.3), while [S
2N2]2 was not detected.
However, experiments preceding the NMR study conrmed the formation of metalla-dithiadiazoles (PR3)2M(S2N2) (M = Pt, Pd) from Na[S3N3] (eq. 5.10, page 136)6
in-dicating that [S2N2]2 could be formed from [S
3N3]. The role of the metal centre in
the transformation of [S3N3] was not investigated but it was suggested that [S 3N3]
disproportionates to give S4N4 and [S2N2]2 (eq. 1.4).6
7 [S4N3]+ + 6 NH3 9 [S3N3]- + H2S + 16 H+ (1.3)
4 [S3N3]- 2 S4N4 + 2 [S2N2]2- (1.4)
Based on the results from previous works7 it is very probable that the disproportionation
of [S3N3] occurs also in the liquid ammonia reaction mixture. The generated [S 2N2]2
One class of metalladithiadiazoles, namely the tin analogues dithiadiazastannoles (or stannadithiadiazoles) became versatile reagents in SN synthesis. [nBu
2Sn(S2N2)]2
repre-sents a good example of a versatile tin reagent prepared from nBu
2SnCl2 and [S4N3]Cl
in liquid ammonia (eq. 1.5).8
18 nBu2SnCl2 + 28 S4N3Cl + 92 NH3 +
NH3 (l)
+ 18 S4N4 + 4 NH4SH + 64 NH4Cl Sn
N Sn N S
N S S N S
nBu nBu
nBu nBu
9
(1.5)
The procedure is relatively time consuming (23 days) but this is mainly due to long purication period (Soxhlet extraction). The time is reasonable with respect to the quantities of the tin reagent obtained.
Both the liquid ammonia method and the use of the tin reagent [nBu
2Sn(S2N2)]2 have
been frequently employed during this work. The experimental details of the syntheses of the above SN reagents are described at the end of this introductory part.
1.2. The multiplicity of S
−
N bonds
It is often important to know whether a chemical bond is single, double or triple. This can be formally decided on the basis of a bond order, which can be calculated according to the well known formula
b = n−n∗
2 ,
where b is the bond order, n is the number of electrons in bonding orbitals and n∗ is
the number of electrons in antibonding orbitals.9 Cystallographic methods showed that
triple bonds are shorter than double, which are shorter than single bonds. Thus, it is possible to make an assessment of bond multiplicities of a novel compound on the basis of its X-ray structure. Organic molecules are excellent examples, in which the types of bonds correspond very well with their lengths. Is this also the case for sulfur-nitrogen compounds?
Pauling's original covalent radii of singly bound nitrogen (0.70 Å) and other rst row
elements were corrected by Schomaker and Stevenson (0.74 Å), who also provided the covalent radii for doubly and triply bound main group elements.10 Pauling included
recommended to use them only for the second period elements.11 Thus, the covalent radii
for singly, doubly and triply bound nitrogen are 0.74, 0.62 and 0.55 Å, respectively, and those of sulfur are 1.04, 0.94 and 0.87 Å, respectively.∗
For the calculation of the length of a single bond between atoms A and B, Pauling suggested to use Schomaker and Stevenson's equation
D(A−B)=rA+rB−c|xA−xB|,
where rA and rB are Pauling's and - for the second period elements - Schomaker and Stevenson's covalent radii of the two elements,xAandxB are Pauling's electronegativities of the two elements and c is a correction coecient. Pauling suggested the coecient to be 0.08 Å for any bond involving one or two atoms from the second period of the
Periodic System. This is the case for a S−N single bond, which should be 1.74 Å long.
The theoretical S−−N bond length calculated by the addition of the covalent radii for doubly bound nitrogen and sulfur11 is 1.56 Å and the S−−−N length is 1.42 Å.
The reality is not so straightforward. Classication of S−N bonds on the basis of their length is tricky, as the intervals for the lengths of particular bond types are very wide. For example the experimental values of a S−N single bond range from 1.74 to 1.61 Å (Fig. 1.1).14,15 The experimental values of a S−−N bond lie in the interval 1.561.46 Å
(in INSNI and [S−−N−−S]+, respectively)16,17 and the S−−−N bond lengths were found from 1.46 to 1.42 Å (in NSPh3 and NSF3, respectively).1820
The overlap between the double and triple bond length interval is striking. On one hand it was emphasised that the valence description of [S−−N−−S]+ most probably does
not correspond to reality and that the S−−N bonds in the cation have a signicant triple character.17Nevertheless, the form [S−−N−−S]+is still commonly used. On the other hand
it was shown, that the length of a S−−N bond depends on the charge of a S−N molecule, i.e. the total number of electrons within the molecule. For example S4N4 contains nearly equal S−N bonds with the length of 1.63 Å (mean value).21 A withdrawal of one or two
electrons from the antibonding HOMO orbitals results in a dramatic structural change and shortening of the bonds to 1.55 Å observed in the radical cation [S4N4]·+ and in
the planar forms of the [S4N4]2+ cation, respectively.22,23 If similar bond shortening by
0.08 Å is to be expected upon withdrawing electrons from the hypothetical thio-analogue
∗Two recent studies brought a list of new single bond covalent radii for nearly all the elements. While
the summary by Cordero et al.12is based on a vast amount of experimental values published in the
Cambridge Structural Database, Pyykkö and Atsumi brought a more consistent set of radii based on experimental or calculated data of several model molecules, which were subsequently optimised by least-squares t.13 For nitrogen, both Cordero et al. and Pyykkö and Atsumi published a covalent
radius of 0.71 Å. The value is very close to the Pauling's original value but since Pauling himself adopted the corrected value of 0.74 Å, 0.74 Å will be used throughout this thesis.
I N
S N
I N
S
N
S N
S N S
O O
1.61 Å
1.56 Å
1.55 Å
1.58 Å
1.60 Å 1.56 Å
(a) (b) (c)
(1.57 Å) (1.53 Å) 1.74 Å
HN
S
S S
S NH S S
1.69 Å
Fig. 1.1. Selected S-N compounds with S-N bond lengths: (a) S6(NH)2 (1,4-isomer); (b) S4N4O2; (c) INSNI; the bond lengths in parentheses belong to another INSNI molecule in the unit cell.
of nitrogen dioxide radical (·NS2), then Pauling's S−−N bond length (1.56 Å) will change to 1.48 Å, which is very close to the experimental value in the [S−−N−−S]+ cation.17 A
signicant triple character then must be put in question. According to Parsons and Passmore, the formal bond order in [S−−N−−S]+ is indeed 2.24
Overall the determination whether a S−N bond is single or double is precarious. Sim-ilarly problematic is the commonly used term a length between a single and a double S−N bond for molecules where π-electrons delocalisation is possible.
1.3. Theoretical calculations
Research in chemical synthesis has been for some time strongly supported by the use of computing techniques. Nowadays, there are only a few synthetic chemists who would refuse to benet from predictions of structure or electron arrangements within molecules of their interest. Since some compounds discussed in this thesis were thoroughly analysed by computational methods, a brief section is included about how structure, bonding and aromaticity can be described by the results obtained from theoretical calculations.
1.3.1. Geometry of a molecule
The structure of a molecule is given by a set of atoms arranged in certain manner in three-dimensional space. A particular set-up of atoms will be stable only if the total energy of this set-up will be low. A correlation between energy of a particle and its position in a three-dimensional space is mathematically described by the Schrödinger equation. Once a mathematical description of a problem exists, calculations can be started to solve the problem.
Schrödinger equation for a multiatomic system, i.e. the energies of atomic orbitals.9
Be-cause the Schrödinger equation cannot be precisely solved for more than a two-particle system, the equation used is simplied by several approximations. These approximations lead to inaccurate orbital energy values and therefore it is necessary to apply post-ab initio methods (e.g. DFT). The post-ab initio methods elaborate the ab initio results by taking into account circumstances neglected or averaged by the previously used ap-proximations and as a result lower orbital energy values are obtained.25
In the second step the ab initio geometry is subjected to molecular mechanics cal-culations (force eld calcal-culations), which assess how distorted are particular structural parameters of the ab initio structure - i.e. bond lengths, bond angles, dihedral angles and non-bonding interactions - from ideal values given by agreed standards.25The higher
the distorsion, the higher energy contribution and the higher the overall energy of the ab initio geometry. The overall energy must correspond to a minimum on a Potential Energy Surface (PES) if the ab initio structure is to be considered as stable. During these molecular mechanics calculations the vibrational frequencies are also calculated.25,26
1.3.2. Charges and bond orders
The atomic charges and bond orders are calculated by partitioning the total electron density in the regions of the atoms. For both the atomic charge calculation and the bond order calculation several concepts have been developed. The principles are complex.26
1.3.3. Aromaticity
Aromaticity deserves to be regarded as something special chemistry has been endowed with. One cannot forget the rst lectures in organic chemistry where planar ring struc-ture, fully conjugated system of multiple bonds and 4n+2 π-electrons were described as the basic criteria of aromaticity.27 However, the concept of aromaticity has deeper roots.
For complicated systems a more general approach must be opted for and that is where aromaticity starts to be even more interesting. The three known criteria for aromaticity need to be generalised and modied to give a set of ve new ones:28,29
• (4n+2) π-electrons
• Structural criteria
• Energetic criteria
• Electronic criteria
The origin of the Hückel's rule, (4n+2) π-electrons, is in quantum mechanics and the rule is a condition of aromaticity valid for both organic and inorganic compounds.30The
term structural criteria refers predominantly to planarity and bond length equalisa-tion in aromatic molecules. Thanks to the delocalisaequalisa-tion of π-electrons, the experimen-tally found bond lenghts in aromatic molecules do not have values of localised single or double bonds but bear an average value between the two.28 The delocalisation
energy represents another possible aromaticity criterion28 since the delocalisation of
the π-electrons brings lowering of the energy of aromatic systems in comparison with
their non-delocalised analogues.27 Electronic criteria refer to the description of the
delocalised π-electronic system by electron population analysis.31 The typically high
chemical shift of aromatic protons in 1H NMR spectrum27,32,33 points at the magnetic
properties of aromatic compounds which are of big signicance, especially in the area of theoretical calculations as is described in the next paragraph.
It was shown that none of the ve criteria can serve as an indicator of aromaticity on its own.28 The desire to dene a single quantity which would be directly connected with
aromaticity became stronger and in 1968 a quantity called Diamagnetic Susceptibility Exaltation (Λ) was introduced as a unique criterion of aromaticity.34 This quantity is
dened as the dierence in magnetic susceptibilities between systems with delocalised and non-delocalised π-electrons:
Λ =χM −χM0 (1.6)
Since magnetic susceptibility is directly linked to the delocalised π-electrons in aromatic compounds,35,36 diamagnetic susceptibility exaltation represents the best instrument of
aromaticity description.28,37 Unfortunately, despite being a single quantity describing
aromaticity, diamagnetic susceptibility exaltation is not convenient for semi-empirical theoretical calculations, as its value depends strongly on the ring size (area) and requires suitable calibration standards.38Instead, it was suggested to calculate absolute magnetic
shieldings at the centres of aromatic rings. The obtained values were named Nucleus-Independent Chemical Shifts (NICS).38 The precision of this method was later enhanced
when functional groups attached to the parent aromatic hydrocarbons were taken into consideration. Dierent functional groups have dierent magnetic environments which inuence the overall environment within the resulting molecule. Therefore it was found more exact to calculate NICS at 1 Å above the ring plane.39 For non-planar molecules
least-squares plane through the ring must be calculated and in addition the use of NICS at 1 Å below the plane was proposed.40 NICS is then denoted as NICS(1), NICS(0) and
NICS(-1) for NICS 1 Å above, at the centre and 1 Å below the plane, respectively.39,40The
1.3.3.1. Aromaticity of sulfur-nitrogen compounds
The Hückel's rule was originally postulated for benzene, i.e. a molecule with six equi-valent atoms forming a ring with delocalised π-electrons. Inorganic analogues of benzene (e.g. borazine) are usually formed by dierent atoms. Dierent atoms bring inequiva-lent electronic environments to the molecule and therefore the solution of the quantum mechanical problem could oer results signicantly dierent to those obtained for ben-zene.30 This could make Hückel's rule impossible to use for inorganic rings. However,
since the semi-emprirical parameters used in the quantum mechanical calculations are very similar for certain heteroatoms, the Hückel's rule may be applied to some inor-ganic compounds.30 In the case of binary sulfur-nitrogen rings, the Hückel's rule can be
applied. The number of π-electrons can be easily counted according to the method of Banister:41,42 every sulfur (6 valence electrons) and nitrogen (5 valence electrons) atom
uses 2 electrons for the connection with their neighbours by σ bonds and every atom
in the ring keeps 2 more electrons as a lone pair. Thus, sulfur has 2 and nitrogen 1 electron spare which can be used for π bonding. If the sum of the spare electrons within a SN cycle equals a (4n+2) number, then a high probability arises that the compound is aromatic to some extent.
Additional assurance about aromaticity can be obtained by calculating Topological Resonance Energy (TRE)†, an analogue to delocalisation energy. If TRE >+0.01β, a
compound is aromatic, if +0.01β > TRE > −0.01β, a compound is nonaromatic and TRE values lower than −0.01β indicate antiaromaticity.30,43 It was shown that (4n+2)
SN rings give TRE values in the aromatic range. Also, the preference of some (4n+2) SN rings to adopt nonplanar geometry was explained on the basis of the TRE values: nonplanar rings gave signicantly lower TRE values than the planar ones.43
Also in case of inorganic rings, the calculation of NICS became very popular among theoretical chemists and is frequently used as an instrument of aromaticity determina-tion.31,40,4446
Except for the extremely useful tool forπ-electron counting, Banister also revealed that the content of π-electrons in the SN rings is higher than in the aromatic hydrocarbons of the same ring size.41 The explanation for this comes from the quantum mechanical
(Hückel and Extended Hückel) calculations which showed that the energies of sulfur and nitrogen atomic orbitals contain lower coulombic repulsion contributions than those of carbon.43 The energy of the molecular orbitals formed by the LCAO is thus lower than
in aromatic hydrocarbons.1,30,47 In addition, the gap between HOMO and LUMO was
found to be smaller than in aromatic hydrocarbons.1As a result, the excessiveπ-electrons
occupy nonbonding or antibonding π orbitals which are more accessible.1,30,47 Since the
number of electrons in bonding and antibonding orbitals determines the magnitude of a bond order,9 the occupation of antibonding orbitals in SN aromatics brings lowering of
the bond order. This is then reected in higher reactivity and lower thermal stability of SN aromatics in comparison with aromatic hydrocarbons.1,47
1.4. General experimental conditions
The following general experimental details apply for all the following parts of the thesis. Unless otherwise stated, all reactions were carried out in an oxygen-free nitrogen at-mosphere using standard Schlenk and syringe techniques. The term high vacuum cor-responds to the pressure of 0.3 Torr.
Dry solvents were used from the Solvent purication system MB-SPS-800 (MBraun GmbH). Less common solvents were dried, puried and stored according to common procedures.48
1H, 13C and 31P NMR spectra were recorded on a Jeol GSX spectrometer at the
fre-quencies of 270.2, 67.9 and 109.4 MHz, respectively. Chemical shifts δ were referenced to external tetramethylsilane and phosphoric acid, respectively. 14N NMR spectra were
recorded on a Bruker Avance II 400 spectrometer at 28.9 MHz withδreferenced to exter-nal liquid ammonia. All 13C and 31P NMR spectra are proton-decoupled; for practical
reasons 13C and 31P descriptions will be used instead of 13C{1H} and 31P{1H},
respec-tively.
Mass spectrometry was performed by the University of St Andrews Mass Spectrometry Service and elemental analyses were performed by the St Andrews University School of Chemistry Service.
Cyclic voltammetry was performed using an EcoChemieµAutolab apparatus controlled by GPES 4.2 software. Three-electrode system consisted of a glass-embedded platinum disc working electrode (area = 0.008 cm2), platinum wire counter electrode and Ag/AgCl
reference electrode. Typically, 5 mm solutions of the analyte in dry CH3CN were placed in an electrochemical cell of 10 ml capacity and were provided with 1.0 mmol of supporting electrolyte ([nBu
4N][PF6]). A small amount (0.2 mg) of solid ferrocene was added as
internal standard. The voltammograms were calibrated for the half-wave potential of the ferrocene/ferrocenium couple E1/2 = 0 V. Digital simulation of cyclic voltammograms was performed using the DigiSim software.
Single crystal X-ray structure data were collected on Rigaku MM007 confocal op-tics/Saturn or Mercury CCD diractometers using Mo-Kα radiation (confocal optic,
λ = 0.710 73 Å), and corrected for absorption. The structures were solved by direct methods and rened by full-matrix least-squares methods on F2 values of all data. Re-nements were performed using SHELXTL (Version 6.1, Bruker-AXS, Madison WI, USA, 2001).
Powder X-ray diraction patterns were recorded on a Stoe STADI/P diractometer using CuKα1 radiation.
1.5. Experimental details of the key sulfur-nitrogen
reagents syntheses
1.5.1. [S
3N
2Cl]Cl
4All joints were coated with teon sleeves or teon tape and slightly greased. Where teon coating could not be used, the joints were properly greased.
[NH4]Cl (300 g, 5.61 mol) and sulfur owers (60.0 g, 0.230 mol) were placed in a 1 litre
ask. S2Cl2 (300 ml, 506 g, 3.75 mol) was quickly added, the mixture was mixed a little bit with a glass rod and the ask was covered with a lid. The lid was tted with an air condenser (3.5 cm outer diameter, 75 cm long) and the top of the condenser was tted with a drying outlet (anhydrous CaSO4). The top of the lid was insulated with glass wool and aluminium foil and the mixture was gently reuxed. The heating was adjusted so that the upper level of the condensing S2Cl2 lies just within the bottom joint of the air condenser (150152◦C). The reaction is nished usually within 15
20 hours with the lower part of the condenser being covered with a layer of orange/brown crystals of [S3N2Cl]Cl. After cooling down to ambient temperature, the air condenser was transferred quickly on air onto an empty, predried and weighed 500 ml Schlenk ask with a nitrogen gas ow. The drying tube was removed, the top of the condenser was plugged with a well greased stopper and the apparatus was evacuated for 30 minutes to dry the product. The nitrogen gas was reintroduced and the dry product was scraped o the condenser walls with a spatula attached to a long metal rod. Yield 73 g (40 %, based on S2Cl2).
IR data: 1406 (mw), 1164 (mw), 1127 (vw), 1013 (ms), 960 (sh), 942 (vs), 705 (vs), 677 (sh), 581 (ms), 562 (sh), 472 (m), 458 (m), 432 (mw) cm−1.
1.5.2. [S
4N
3]Cl
4All joints were coated with teon sleeves and slightly greased. In a 250 ml ask equipped with a stirring bar, dry CCl4 (50 ml) was cooled to -10 to -20◦C (acetone/dry ice) and
under stirring S2Cl2 (50 ml, 84.4 g, 0.630 mol) was added in one portion. The solution was cooled to -20◦C (acetone/dry ice) and [S
3N2Cl]Cl (14.0 g, 0.072 mol) was added in
one portion. The mixture was gently reuxed (toil = 96◦C) for 5 hours. Still warm, the mixture was ltered through a sinter and the canary yellow product was washed twice with 20 ml and 10 ml CCl4 respectively. The product was dried under high vacuum for 1 hour. Yield 7.0 g (71 %).
IR data: 1262 (vw), 1165 (vs), 1130 (sh), 1001 (vs), 936 (sh), 794 (vw), 719 (vw), 683 (s), 639 (mw), 608 (m), 589 (sh), 565 (s), 527 (sh), 469 (vs), 454 (s) cm−1.
Raman data: 1173 (w), 1004 (w), 607 (mw), 568 (m), 447 (vs), 250 (s), 210 (ms) cm−1.
1.5.3. [
nBu
2
Sn(S
2N
2)]
28Ammonia (200 ml) was condensed at −78◦C (acetone/dry ice) in a predried 1 litre ask
equipped with a stirring bar and [S4N3]Cl (7.0 g, 0.034 mol) was added in three portions. The dark red mixture was stirred for 30 minutes at −78◦C after which time nBu
2SnCl2
(20.0 g, 0.066 mol) was added in small portions over the period of 5 minutes. After a
while the mixture thickened and changed colour from dark red to brown/orange. The mixture was stirred at −78◦C for 2.5 hours, then the cooling bath was removed and
the mixture was allowed to warm up to room temperature overnight with the ammonia being evaporated. After the evaporation the mixture was evacuated for an hour to remove residual ammonia. The ask was then transferred to a glove box together with a Soxhlet apparatus. The crude solid was carefully scraped o the walls and was placed in a Soxhlet thimble, the thimble was placed into the apparatus and the apparatus was closed with stoppers. In this way protected crude [nBu
2Sn(S2N2)]2 was moved back to
a fume cupboard where the Soxhlet apparatus was attached to a 1 litre ask containing dry and degassed petroleum ether (650 ml). The Soxhlet apparatus was insulated with glass wool and aluminium foil and to the top a water condenser with an oil-bubbler was attached. The crude solid was extracted for 8.5 hours (toil = 7880◦C), at that point the extracts were nearly colourless. The extract was allowed to cool to room temperature, its volume was reduced to approx. 1
2 and the concentrate was placed to freezer overnight, where bright yellow precipitate separated. This was ltered o, washed twice with cold (−40◦C) degassed petroleum ether, dried under high vacuum for 1 hour and stored under
argon.
in the same way as the rst crop. The second crop showed less satisfactory elemental analyses. Overall yield 14.4 g (67 %).
Arsenic-Sulfur-Nitrogen Heterocycles
Abstract
The chemistry of arsenic oers a lot of adventure. Generations of chemists have been investigating the chemical behaviour of arsenic in the hope that the amount of knowledge will at least match that of arsenic's lighter neighbours in Group 15, phosphorus and nitrogen. It seems correct to say that these expectations have only been fullled partly. Without any deep considerations three main reasons can be named which have hindered the chemical research of arsenic: toxicity, lack of commercially available starting materials and low applicability of arsenic NMR.
2.1. The toxicity of arsenic
Arsenic and its compounds are very often referred to as strong poisons and carcinogenic substances. The use and synthesis of arsenic compounds forms a substantial part of this thesis. To better understand where is the cause of arsenic's toxicity, a short literature survey is included in the following brief section.
2.1.1. Arsenic in the environment
Arsenic is the 20th most abundant element in the Earth's crust and is found in nature mainly in suldic ores.49 Its spread into the environment can occur by natural means
such as volcanic activity, wind erosion or the biological activity of microorganisms. The human population contributes considerably to the overall arsenic content in the environ-ment through industrial activities such as mining or smelting, but also by burning fossil fuels or by the use of arsenic-based pesticides.49 On the other hand, the possibilities of
2.1.2. Biotransformation of arsenic in nature
Arsenic is present everywhere in the environment and all organisms are unwillingly ex-posed to it. Fortunately, every organism has a metabolic pathway in which its body is detoxied. For example fungi and molds methylate arsenicals and release the correspond-ing alkyl(aryl)arsines as the nal metabolic products.49 It was also found that fungi in
rotting wood, previously treated with arsenic-based preservatives, are able to transform these arsenicals into trimethylarsine.50,51
The attitude of plants towards arsenicals is diverse. While some plants succumb to the eects of on purpose invented weed killers, other plants have an extraordinary tolerance to arsenic and can take up and concentrate arsenic from the environment (e.g. water hyacinth).52 The high arsenic content in the ash of some plants (e.g. Douglas Fir) was
even interpreted as a biogeochemical indicator of the presence of gold veins buried in the subsoil.53
Fish and other water fauna metabolise arsenicals to arsenobetaine (Fig. 2.1). This
As+
O
-O H3C
H3C
H3C
NH2 As O OHOH
OH As O OHOH
NO2
3 2
1
Fig. 2.1. Arsenobetaine (1), arsanilic acid (2) and roxarsone (3)49
highly methylated organoarsenic compound is believed to be the nal product of the arsenic metabolism in sh. Analyses have shown that the amount of arsenic in marine sh is greater than in the freshwater sh.49,54 The possible poisoning of people by the arsenic
contained in sh and seafood is of no concern, since arsenobetaine is not metabolised in the human body and is excreted unchanged.55
The arsenic tolerance of terrestrial animals is low. Much is known about the arsenic metabolism of mice, rats and rabbits as they are commonly used for toxicity testing. Ar-senic in their bodies undergoes methylation and is excreted in urine.56,57Similar
metabo-lites were observed in the urine of dogs, pigs and cows.58Poultry and pigs show increased
2.1.3. Arsenic in the human body
The fact that arsenic is toxic to humans is strongly backed up by a large volume of medical, toxicological and biochemical studies. It is predominantly the inorganic forms of arsenic to which people are nowadays exposed. The main routes of exposure are in-halation and ingestion. Similarly to other toxic substances, the consequences of exposure to arsenic depend on the scale of the dose and on the period during which the dose was taken up.
Acute poisoning with huge doses of arsenic leads to a cardiovascular collapse and heavy brain damage with death coming within hours.60
Smaller doses cause acute arsenic poisoning with slower progress. The rst responses come from the gastrointestinal tract and take hours to days. They are followed by cardiovascular system shock which takes from minutes to hours and already causes no small amount of deaths. The poisoning proceeds further with multiple organ failures (kidney failure, liver enlargement, lung oedem) and also bone marrow damage. Within a week, symptoms on the skin and brain start to develop. Typical for the skin is an increase of pigmentation, hyperkeratoses and dermatitis. Brain aection is distinguished by perplexity, convulsions or prolonged coma.60
Even if acute arsenic poisoning is successfully warded o, the nal consquences in-cluding tumors and cancers may appear. These consequences are tricky because they always develop with a latency period and with no respect to the amount of arsenic one was exposed to.60
Chronic arsenic poisoning develops during a prolonged exposure to arsenic. In the past, arsenic was frequently ingested as a medicine in the form of Fowler's solution (i.e. 1 % solution of potassium arsenate prepared by dissolving As2O3 in aqueous KHCO3).61,62
Nowadays it is predominantly people living in areas where drinking water is taken from wells sunk in arsenic-rich subsoils who suer from chronic arsenic poisoning. The most well known areas are e.g. Bangladesh, the West Bengal state in India, several regions in Argentina and Chile and the southwest coast of Taiwan.61 The consequences of chronic
arsenic poisoning are tumors and cancers of various tissues, dermatitis, gangrenes (Black Foot disease) and other complications.61,63
2.1.3.1. The principle of toxicity of arsenic
There are nowadays two theories explaining the reason why arsenic is toxic to people. The rst and commonly accepted theory is based on the anity of arsenic to sulfur. In the human body, arsenic in both oxidation states AsIII and AsVreacts with the free thiol
groups of proteins, enzymes and cofactors.56,64 This process was successfully simulated.
2.12.3.64
RxAsO(OH)3-x + 2 R'SH RxAs(SR')2(OH)3-x + H2O (2.1)
RxAs(SR')2(OH)3-x RxAs(OH)3-x + R'SSR' (2.2)
RxAs(OH)3-x + (3-x) R'SH RxAs(SR')3-x + (3-x) H2O (2.3)
The stability of the products is variable. While aliphatic As−S compounds are not so stable, As−S heterocycles are characterised by their increased stability. As expected, the ve- and six membered rings are the most stable.65 The cofactors of the fundamental
enzymes in cell metabolism provide the source of vicinal SH groups which react with arsenic to form stable heterocycles. The cofactor and thus the activity of the enzyme then become blocked, subsequent biochemical reactions are disrupted, cell metabolism collapses and the cell dies.66
The second theory is based on the fact that inorganic AsIII compounds stimulate the
production of H2O2 in the human body.67 In the presence of Fe2+, H
2O2 may dissociate
in free hydroxyl radicals according to the Fenton reaction (eq. 2.4).68
Fe2+ + H2O2 Fe
3+ + OH + OH
-(2.4)
The radicals oxidise proteins and enzymes, as a consequence of which the cell metabolism collapses causing death of the cell.66,69 This theory is currently under investigation but
represents a strong challenge to the previously described thiol group theory.
Arsenic in the oxidation state +5 has another toxic eect. Due to its similarity to phosphate ion, arsenate can enter the ATP formation process and can replace phosphate. The resulting compounds are unstable, hydrolyse spontaneously and thus cannot serve as energy reservoirs.56,70
2.1.3.2. Metabolism of arsenic in the human body
Arsenic from all sorts of sources gets to the blood by which it is distributed through the whole body. Arsenicals are however quickly eliminated from blood and are redis-tributed into the organs. The organs most aected with arsenic poisoning are the liver, kidneys, bowel and spleen.60 Also well known is the accumulation of arsenic in hair
and nails, a feature frequently utilised in forensics. Hair and nails are skin derivatives composed of keratins, i.e. structural proteins with high ratio of aminoacids containing thiol groups. Arsenic carried to the hair by blood interacts with these thiol groups and remains stored.71,72
The metabolism of inorganic arsenicals takes place in the kidney.60Arsenites are
to methylarsonous acid and methylated again to dimethylarsinic acid. Inorganic arse-nates must be rst reduced to arsenites so that they can undergo methylation. The methylated products are then excreted in urine. Scheme 2.1 summarises the metabolical process.55,56,64
[AsVO4]3- [AsIIIO3]3- [MeAsVO3]
2-[MeAsIIIO2]2- [Me2AsVO2]- [Me2AsIIIO]
-Me3AsVO Me3AsIII
2e
-2e
-2e
-2e -Me+
Me+
Me+
2e
-Me+
Scheme 2.1 Metabolism of inorganic arsenate in human body64
During the oxidative methylation, a methyl group from a donor is enzymatically trans-ferred onto the AsIII centre. The following reduction is also an enzymatic process and
the source of electrons are thiols which at the same time are oxidised to disuldes in accordance with equations 2.1 and 2.2.64,73,74
It was found that soon after the arsenic intake, the unmetabolised inorganic forms are excreted but after 16 hours it is already dimethylarsinic acid as the main metabo-lite. Also, during chronic arsenic poisoning the main metabolite is dimethylarsinic acid whereas for acute arsenic poisoning the excretion of methylarsonic acid is dominant. This is caused by the inhibition of the methylation by AsIII.75 Despite the possibility of
reducing dimethylarsinic acid to dimethylarsinous acid and continue the sequence up to trimethylarsine as suggested by Scheme 2.1, no trimethylated species have been detected in human urine.55,57 An interesting recent discovery is that methylarsonous acid, an
2.2. Commercial availability of starting materials
∗The toxicity of arsenic and its compounds (especially inorganic) represents probably the toughest obstacle from both practical and organisational points of view. In the past arsenicals were used as chemical weapons and nowadays the risk of their misuse still persists, this time imposed by terrorist organisations. This forces commercial suppliers to introduce strict policies both for the production and distribution. On the industrial level, the companies producing arsenicals must have special governmental licences which must be regularly renewed for no small fees. Preparations of major amounts of arsenic compounds are subject to strict evidence and limits within both the production, stock holdings and dispatching. Waste disposal is strictly monitored and therefore there must be investment in technologies which can make the waste arsenic-free. The overall high nancial demand on the production of arsenicals adds a signicant amount to the prices of the nal products which are then dicult to sell (e.g. 250 g of AsCl3 for over ¿400). Only major companies can aord to produce arsenicals and the range of products is usually very narrow. Especially universities nd this inconvenient. Simple starting materials need to be prepared which is not necessarily dicult but it can be time consuming and it slows down the progress of the research.
The cheapest available material is arsenic trioxide, As2O3, which can be purchased in large quantities. It is, however, not useful for synthesis since it is not soluble in common organic solvents and it is not particularly reactive at ambient temperatures. Arsonic and arsinic acids are usually poorly available with only phenylarsonic (C6H5As(O)(OH)2), arsanilic (Fig. 2.1 on page 34) and dimethylarsinic (Me2As(O)(OH)) acids being the reg-ular items in the stocks. Arsenic(III) chalcogenides As2E3 (E = Se, Te) can be purchased but they nd application as semiconducting materials rather than starting materials in chemical synthesis.76,77 Arsenic trihalides AsF
3, AsCl3 and AsI3 are available but the
ra-tio quantity:price is not convenient. Mono- and dialkyl/aryl substituted halogenoarsines are not commercially produced due to their possible warfare use. Triphenylarsine is the only trisubstituted arsine available.
2.3.
75As NMR as an analytical tool
The rapid development in phosphorus chemistry was signicantly helped by the magnetic properties of the element. 31P NMR enabled virtually immediate product or mixture
characterisation. Arsenic, similarly to phosphorus, is monoisotopic (75As) but its
nu-∗The commercial availability data were taken from the internet catalogues of Sigma-Aldrich
clear spin of 3
2 and relatively high quadrupolar moment result in broad lines in spectra and make 75As NMR impossible to use for characterisation of other than symmetrical
molecules.78,79
2.4. Methods of preparation of common starting
materials
The term common starting materials relates to simple inorganic and organoarsenic compounds, which were described in vast majority of the studied literature as starting materials for particular syntheses. They can be sorted in 4 classes:
• Arsenic trihalogenides
• Arsonic acids
• Arsinic acids
• Organohalogenoarsines
2.4.1. Arsenic trihalogenides
The most convenient laboratory preparations of arsenic trihalogenides on a medium scale use arsenic trioxide and follow similar procedures. AsF3 and AsI3 are prepared by the treatment of As2O3with suitable halogenating agent (CaF2, KI) in concentrated acid:80,81
As2O3 + 3 CaF2 + 3 H2SO4 2 AsF3 + 3 CaSO4 + 3 H2O (2.5)
As2O3 + 6 KI + 6 HCl 2 AsI3 + 6 KCl + 3 H2O (2.6)
The preparation of AsF3 is carried out in a combined apparatus where a still head with a water condenser is attached to the reaction ask from the beginning. The mix-ture requires gentle heating in a water bath. The reaction starts at 30◦C and at 65◦C
the colourless product distills to the collection ask. AsF3 can be puried from small amounts of HF by the addition of dried NaF which is usually placed in the collection ask. Subsequent distillation to a metal storage cylinder aords suciently pure AsF3 as a colourless, air-sensitive liquid.80,82,83
Solid AsI3 precipitates when mixture of As2O3 and conc. HCl is carefully added to an aqueous solution of KI (eq. 2.6).81AsI
3 is ltered o and puried by recrystallisation from
CS281 or diethyl ether.84 AsI