M A J O R A R T I C L E
Mycobacterium tuberculosis
Complex DNA
from an Extinct Bison Dated 17,000 Years
before the Present
Bruce M. Rothschild,1,2,3,4Larry D. Martin,4Galit Lev,5Helen Bercovier,5Gila Kahila Bar-Gal,5Charles Greenblatt,8 Helen Donoghue,5Mark Spigelman,5and David Brittain6
1Arthritis Center of Northeast Ohio, Youngstown,2Department of Internal Medicine, Northeastern Ohio Universities College of Medicine, Rootstown, Ohio;3The Carnegie Museum, Pittsburgh;4University of Kansas Museum of Natural History, Lawrence, Kansas;5Department of Bacteriology, Royal Free Hospital and University College London, London;6Veterinary Sciences Division, Department of Agriculture and Rural Development, Belfast; and8Kuvin Center for the Study of Infectious and Tropical Diseases and Ancient DNA, Hadassah Medical School, Hebrew University, Jerusalem
In order to assess the presence of tuberculosis in Pleistocene bison and the origin of tuberculosis in North America, 2 separate DNA extractions were performed by 2 separate laboratories on samples from the metacarpal of an extinct long-horned bison that was radiocarbon dated at17,870230 years before present and that had pathological changes suggestive of tuberculosis. Polymerase chain reaction amplification isolated fragments of tuberculosis DNA, which were sequenced, and on which spoligotyping was also performed to help determine its relationship to the various members of the Mycobacterium tuberculosiscomplex. Extensive precautions against contamination with modernM. tuberculosiscomplex DNA were employed, including analysis of pa-leontologic and modern specimens in 2 geographically separate laboratories.
Recognition of tuberculosis-compatible pathology in bones of North American Pleistocene bovids has sug-gested that tuberculosis was present from an early date on that continent. This is now confirmed by the results of DNA sequencing for a bone sample from a bison dated to 17,000 years before the present (BP). The pres-ence of similar pathology in fossil bighorn sheep and musk ox suggests thatMycobacterium tuberculosis com-plex organisms were widespread in bovids that immi-grated to North America over the Bering Strait con-nection and widespread during the late Pleistocene. It suggests Holarctic distribution and that bovids were the likely vector and reservoir for dispersion of what be-came known as the white plague.
Received 9 November 2000; electronically published 5 July 2001.
Financial support: The Center for the Study of Emerging Diseases, Jerusalem.
Reprints or correspondence: Dr. Bruce M. Rothschild, Arthritis Center of Northeast Ohio, 5500 Market St., Youngstown, OH 44512 ([email protected]).
Clinical Infectious Diseases 2001; 33:305–11
2001 by the Infectious Diseases Society of America. All rights reserved. 1058-4838/2001/3303-0006$03.00
Clarification of the evolutionary history of infectious diseases affords great opportunity for understanding the factors that have created current epidemiological patterns. Study of pathognomonic lesions in fossils has allowed progress in this endeavor [1–3], but for many lesions a specific etiology cannot be determined. PCR and application of DNA spoligotyping and sequencing techniques [4–8] allow that limitation to be tran-scended, but the very sensitivity of PCR raises the ad-ditional challenge of contamination [7, 8]. Contami-nation of ancient by modern DNA is now sufficiently recognizable (and usually preventable) that confident diagnosis of ancient DNA is possible [7, 8].
of M. tuberculosis, Mycobacterium bovis, Mycobacterium afri-canum, andMycobacterium microti[13]. The term “M. tuber-culosis complex” distinguishes this group of organisms from saprophytic mycobacteria (e.g., soil organisms) and atypical mycobacteria such asMycobacterium avium–intracellulare[14]. Additional analysis is required to distinguish among the members of theM. tuberculosiscomplex. Spoligotyping clearly distinguishes modern M. tuberculosis from modern M. bovis
and from non–M. tuberculosiscomplex mycobacteria [15]. Documentation of M. tuberculosis complex in humans on both sides of the Atlantic Ocean who lived 13 millennia ago
[2, 16–18] has led to a great deal of speculation about its origins. Such work suggests that at least some of the previously reported presumptive diagnoses [16–30] were accurate. The presence of
M. tuberculosiscomplex on both sides of the Atlantic at such early dates suggests that it either arrived with early settlers in North America or was present in North America when they arrived. The latter hypothesis is supported by suggestive evi-dence in the fossils of North American bovids [31].
One of the classic findings in tuberculosis is the undermined subchondral articular surface [3, 32]. That finding, which is relatively specific for the diagnosis of tuberculosis, is common in fossil bones from the Late Pleistocene in North America [31].
This study involved the use of molecular DNA techniques to identify the presence ofM. tuberculosiscomplex in a skeletal specimen with lesions suggestive of tuberculosis from Natural Trap Cave (Wyoming). That unique site was an unavoidable hazard on a game trail, providing an unbiased sampling of the many passing species. Identification of the DNA fragments was done by spoligotyping and direct DNA sequence determination. Analysis was conducted at 2 separate laboratories (Department of Bacteriology, Royal Free Hospital and University College, London, and Department of Parasitology, Hebrew University, Jerusalem) to ensure the reproducibility of the findings and eliminate the possibility of contamination.
Our results provide the first definitive diagnosis of M. tu-berculosis complex in a fossil. They suggest that bovids were the vectors that transported the primordial organism that caused what we today call the white plague: tuberculosis.
METHODS
Sample source. Bones were recovered in Wyoming from the Natural Trap Cave, a chamber nearly 30 m deep with a 4-m opening. The cave lies across an ancient game trail that leads to and from low-altitude pastures. For 1100,000 years it has
trapped animals such as bears, wolves, foxes, wolverines, mar-tens, cheetahs, lions, horses, mammoths, camels, sheep, musk oxen, and bison [33]. The depth of the cave makes falls
in-variably fatal and also keeps the temperature nearly constant at 4C–5C.
During a survey of all of the approximately 40,000 bones recovered from Natural Trap Cave, a classic but relatively rare skeletal expression of granulomatous infection, involving un-dermining of subchondral surfaces, was noted in bovids (i.e., bighorn sheep, extinct musk ox, and bison). These lesions were absent in the very large horse and American pronghorn an-telope samples from that site [31]. Radiological analysis revealed peri-erosive osteopenia (decrease in the bone density around the involved area). The most likely etiologic agent of this pa-leopathology wasM. tuberculosis.
Samples and controls. Two separate samples were taken from the undermined articular surface region of a metacarpal of an extinct long-horned bison(Bisoncf.antiquus)that was enclosed in sediments dated by radiocarbon analysis at years BP. Areas distinct from the pathological 17,870230
lesion were also examined. Fossil controls consisted of lesion-free bones fromCanisspecies andEquusspecies.
DNA extraction. Two separate DNA extractions were in-dependently analyzed by 2 geographically separate laboratories. Cell lysis with guanidinium thiocyanate and DNA capture onto silica was used on nondecalcified samples in the Jerusalem lab-oratory and on decalcified samples in the London lablab-oratory [34–36]. Stringent precautions were taken at both laboratories to avoid cross-contamination. DNA extraction and PCR were performed with standard precautions, including the use of ultraviolet-irradiated safety cabinets, ultrapure reagents, and sterile disposables under full isolation procedures. Control sam-ples that contained no bone material and bone material from nonlesional regions and from unaffected species were used to assess contamination during the extraction and amplification processes.
DNA amplification. Different PCR systems were used in the 2 laboratories. The Jerusalem laboratory used primers from the ribosomal protein S12 [5, 6] to amplify a 204-bp DNA fragment: forward 5-TCGTCGGGACAAGATCAGTAAG-3 (position 33–54) and reverse 5 -ATCGAGTGCTCCTGCAGG-TTG-3(position 216–236). In addition, a primer based on the insertion sequence IS6110 [37] was also used to amplify a region of 245 bp: forward 5-CGTGAGGGCATCGAGGTGGC-3 (po-sition 633–652) and reverse 5 -GCGACGTAGGCGTCGGTGA-CAAA-3(primer position 880–858).
Amplification was performed in a solution of 20 mMof Tris-HCL (pH, 7.5), 40 mMof NaCl, 2 mMof sodium phosphate, 0.1 mM of EDTA, 1 mMof dithiothreitol stabilizers (Sigma-Aldrich), 50% (v/v) glycerol, 1.5 mMof MgCl2, 40 pMof each
primer, 0.2 mMeach of nuclear triphosphates (MBI, Fermen-tas), and 1.25 U of Platinum Taq DNA polymerase (GibcoBRL Life Technologies). Amplification consisted of initial denatur-ation at 94C for 2 min, followed by 39 cycles of 94C for 30 s, 61C for 45 s, 1 min at 72C, and, finally, 10 min at 72C.
DNA sequence analysis. The Jerusalem laboratory per-formed direct sequencing using the Termo-Sequenase kit (Amersham), followed by agarose gel electrophoresis of the PCR product. The band was cut from Nu-Sieve low-melting-point agar and used as such in the labeling mix. The London laboratory removed the selected band from the gel and ex-tracted it using a MERmaid Spin Kit (10–200 nucleotides; An-achem). The purified DNA was then fluorescence-labeled with the ABI PRISM Dye Terminator Cycle sequencing kit with AmpliTaq DNA Polymerase, FS (PE Applied Biosystems). The sequence was analyzed in an ABI PRISM 310 automated se-quencer (PE Applied Biosystems).
Spoligotyping. To determine the species ofMycobacterium
within the M. tuberculosiscomplex, the spoligotyping system (Isogen) was applied in the Jerusalem laboratory. “Spoligotyp-ing” stands for “spacer-oligos.” The process is based on PCR amplification of a series of 43 nonrepetitive short spacer se-quences (of between 36 and 41 bp), located between small repeats (DRs) in the DR locus of the Mycobacteriumgenome [39]. This system differentiates between modernM. bovisand
M. tuberculosis,as demonstrated by the array of the oligonu-cleotide sequences attached to a membrane [15]. The spacers are arranged on the blot in the order in which they appear in the H37Rv DR sequence (spacers 1–19, 22–32, and 37–43) and
the sequence of M. bovis BCG (spacers 20, 21, and 33–36). Subsequent sequencing of the DR regions of otherM. tuber-culosis, M. bovis, M. microti,andMycobacterium canettiisolates has elucidated 65 novel spacer sequences (26 fromM. canetti). The order of spacers in all isolates is very well conserved, apart from a few duplications of spacers [5, 6, 40]. One primer is biotinylated, and the amplified products are hybridized under stringent conditions to the membrane furnished in the Isogen kit. The final readout is made exquisitely sensitive by the bind-ing of streptavidin–horseradish peroxidase to the biotinylated hybridized spacers, followed by use of a chemiluminescent de-tection system (Amersham).
Similar spoligotyping results for different DNA extractions can imply an accurate result based on the spacer regions of the genome or the spacer regions of a consistent pattern of frag-mentation, perhaps because the molecule’s stability varies at different sites along the DNA strand [38]. The applicability of
this technique to ancient specimens has been established by Taylor et al. [38].
RESULTS
DNA recovery and sequencing. PCR amplification enabled recovery ofM. tuberculosiscomplex at both laboratories. Se-quencing revealed that the PCR product that was obtained belonged to a species in theM. tuberculosiscomplex. With use of primers for the S12 ribosomal gene, the isolated gel bands yielded a perfect match for a 163-bp fragment of theM. tu-berculosis complex (of a possible 204 bp). IS6110 sequencing revealed a base-pair substitution difference. The London lab-oratory 92-bp segment had a 12-bp section missing (822–832 of X17348) in the forward strand, but the flanking sequences of 35 bp and 31 bp were identical to those in the database. A sequence for the reverse strand was obtained from bases 782–853, including the missing region on the positive strand. The total length sequenced was 92 bp, which included the total length between primers IS3 and IS4. Base discrepancy was spe-cifically noted in the Jerusalem laboratory at 798 (T for C). However, the sequence obtained in London agreed with that in the database.
Extraction control samples and PCR control samples at both laboratories yielded no tuberculosis DNA. For all the other animal bone samples and specimens from the affected bone at locations away from the lesions, multiple extractions and PCR sequencing analyses found no evidence of tuberculosis DNA.
Spoligotyping. Spoligotyping revealed that the Pleistocene bison lesions containedM. tuberculosiscomplex segments not associated with modernM. bovisorM. microti[37]. The pres-ence of spacers 39, 40, 41, and 43 eliminates the possibility that the DNA is from modernM. bovis[9, 10] or from the related BCG [9, 10], and thus eliminates the possibility that the sample was contaminated by DNA from a BCG-vaccinated individual. On the basis of identification of the spoligotype pattern as a 43-digit binary number, the pattern can be assigned descriptive nomenclature: taking a negative as 0 and positive as 1 and dividing the pattern into 6 blocks of 7 or 8 digits results in 6 blocks containing spacers 1–7, 8–14, 15–21, 22–28, 29–36, and 37–43. The binary number in each block can then be converted to the hexidecimal base. The result is a code of6⫻2 digits [41]. The “hex code” for the bison pattern is thus 7F-6E-7E-7F-F8-7D.
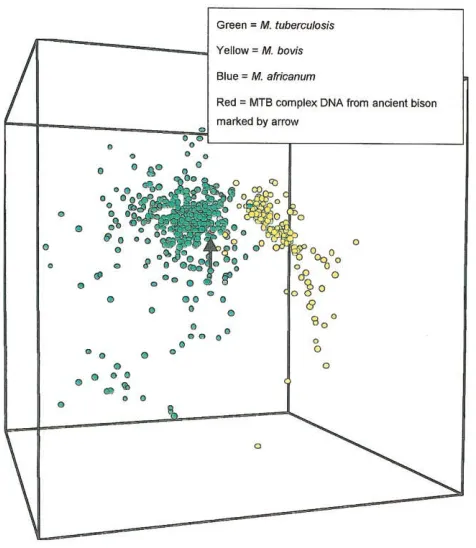
cal-Figure 1. Pseudo–3-dimensional representation of discriminant
anal-ysis of 634Mycobacterium bovis, Mycobacterium tuberculosis,and
My-cobacterium africanumspoligotype patterns from a combined database of modern patterns collated by the National Institute of Public Health and Environment (RIVM), Utrecht, The Netherlands, and the Veterinary
Sciences Division, Belfast. MTB,M. tuberculosiscomplex.
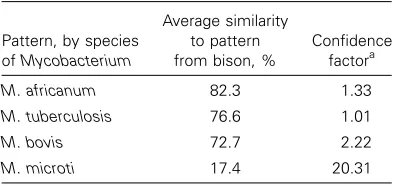
Table 1. Average similarity of bison spoligotype pat-tern to a library of spoligotype patpat-terns, divided into units based on species.
Pattern, by species ofMycobacterium
Average similarity to pattern from bison, %
Confidence factora
M. africanum 82.3 1.33
M. tuberculosis 76.6 1.01
M. bovis 72.7 2.22
M. microti 17.4 20.31
a
Calculated by comparing the average similarity between the bison pattern and the library unit’s entries and the average sim-ilarities of the library unit’s entries with each other, thus taking account of the heterogeneity of the library units.
culate similarities by means of the Dice coefficient. The bison pattern was found to be most similar to pattern ST203 (hex code 7F-7F-7E-7F-F0-7F), with a similarity value of 93.2%. This pattern has been obtained from 2M. tuberculosisisolates.
The pattern from the bison sample was then compared with a library of species-defined units. The highest average similarity to the bison pattern was obtained from theM. africanumunit (82.3%), followed by M. tuberculosis (76.6%) and M. bovis
(72.7%; table 1). Therefore, the bison pattern fits well within the M. africanumand M. tuberculosis units. Further study of the bison spoligotype pattern by discriminant analysis shows that the bison pattern plots more closely to theM. tuberculosis
group than to the M. bovisand M. africanum groups (figure 1). Principal-components analysis of the spoligotype patterns in the database, excluding those spacers that were negative for the bison pattern, shows that the bison pattern continues to group withM. tuberculosisandM. africanumrather than with
M. bovis(figure 2). This demonstrates that grouping the bison pattern withM. tuberculosisandM. africanumis not related to any difficulties in obtaining a full spoligotype pattern.
DISCUSSION
Clarification of the evolutionary history of infectious diseases is advanced by comparison of DNA in ancient and modern specimens. Our understanding of the antiquity of tuberculosis was previously based upon presumptive, macroscopic pathol-ogy–based diagnosis of skeletal remains [16–30]. The work of Allison et al. [19], Cockburn et al. [42], Garcia-Frias [43], and Zimmerman [20] allowed histologic confirmation of Myco-bacterium-related infection [18].
In the current study, DNA analysis of a 17,000-year-old skel-etal specimen from Natural Trap Cave confirms the diagnosis ofM. tuberculosiscomplex infection. IS6110 DNA fingerprint-ing is now the standard method for molecular analysis of tu-berculosis epidemiology, because this sequence is apparently restricted to species that belong to theM. tuberculosiscomplex
[39]. The results of our study further specified the particular species of this 4-member complex that is responsible for disease in bison.
M. bovisand relationships. It is clear from the presence of spacer elements 39, 40, 41, and 43 that the bison DNA is not modernM. bovisDNA. However,M. bovis, M. tuberculosis, M. africanum, and M. microti probably evolved from an M. tuberculosis precursor ∼15,000–20,000 years BP [44], so the bison sample may be representative of the precursor or of group 1 species shortly after speciation.
Information from theM. tuberculosisgenome sequence has very recently been used to construct a DNA microarray, in which almost every open-reading frame is displayed, allowing a global analysis of genetic differences betweenM. tuberculosis, M. bovis,and BCG substrains. Multilocus sequencing [44] had implied that there was a high degree of genetic conservation betweenM. bovisandM. tuberculosis. The new findings suggest that genetic diversity within theM. tuberculosiscomplex may be much greater than previously supposed and that gene de-letion, rather than point mutation, may be a key source of genetic variation [9, 10].
M. microti. Van Soolingen et al. [37] demonstrated that
Figure 2. Pseudo–3-dimensional representation of
principle-compo-nents analysis of 634Mycobacterium bovis, Mycobacterium tuberculosis,
andMycobacterium africanumspoligotype patterns from a combined da-tabase of modern patterns collated by the National Institute of Public Health and Environment (RIVM), Utrecht, The Netherlands, and the Vet-erinary Sciences Division, Belfast. Analysis excludes spacers absent from spoligotyping of DNA from ancient bison (i.e., spacers 8, 10, 25–37, and
43). MTB,M. tuberculosiscomplex.
may be of limited value for application to ancient DNA because of taphonomic changes, the short direct-repeat regions ofM. microti identified by spoligotyping allow its recognition and elimination as a causative organism for tuberculosis in Pleis-tocene bison. AlthoughM. microtiis not known to affect bovids, it does affect mice. Isolation ofM. tuberculosiscomplex DNA from the lesion only and not from unaffected species and bones suggests that contamination of the site by mice is unlikely. Results of spoligotyping totally eliminate the possibility of such contamination.
M. africanum. M. africanumwas originally described by Castets et al. [45]. It is responsible for 60% of cases of tuber-culosis in Africa. Hass and des Prez [34] refer toM. africanum
as intermediate betweenM. tuberculosis and M. bovis. Spoli-gotyping data support this. The possibly close relationship be-tween the isolated organism andM. africanumis therefore per-haps not surprising. If the oldest identified organism (that described in this study) is more closely related toM. africanum
than to other members of theM. tuberculosiscomplex, it would seem reasonable to consider this organism to be the primordial
form of the organisms today recognized asM. tuberculosisand
M. bovis.
M. tuberculosiscomplex. The Eisenach/Taylor primers are specific for theM. tuberculosiscomplex. Larger IS6110 primers and those for S12 are not. Hence, any product with the nested PCR is specific, as further documented by sequencing studies. It is intriguing that the spoligotyping pattern of the bison isolate (as determined by comparison with the extensive col-lection of spoligotyping patterns in data banks) is closer to the pattern of modernM. africanumand M. tuberculosis than to the patterns of the other members of theM. tuberculosis com-plex. The patterns were compared to a database containing several hundred different patterns obtained fromM. tuberculosis
isolates,1170M. bovispatterns, 34M. microtipatterns, and 6 M. africanumpatterns. The pattern obtained from the bison material most closely matches with anM. tuberculosispattern (figure 2). However, the bison pattern fits most closely with the mean of theM. africanumpatterns as a group. The bison isolate, however, cannot be assigned to a particular species of theM. tuberculosiscomplex on the basis of spoligotyping data. Assignment of this isolate to any one of the species in theM. tuberculosiscomplex must be done with reservations, because speciation of the M. tuberculosis complex may not yet have occurred by 17,000 years BP. The M. tuberculosiscomplex is very homogeneous at the DNA level and may have speciated from a progenitor organism only very recently [44].
Caveats. The slight base-pair difference found at one lab-oratory between the ancient DNA from the bison specimen and modern sequences requires confirmation. It should be noted that there have been no reports of base changes in the 92-bp sequence of IS6110, although other tested samples have been considerably less ancient.
Base-pair differences do, however, provide an additional proof that contamination is not a factor. Kolman and Tuross [7] have criticized previous work because the investigators found ancient DNA that had sequences identical to those of modern DNA. They state that “the finding of identical se-quences in an archaeological specimen and the control DNA sample precludes convincing proof that ancient DNA was ex-tracted and analyzed” [7, p. 16]. The lack of a perfect match between the DNA extracted from the ancient bison and the DNA from contemporary organisms not only documents ab-sence of contamination, but the character of that DNA suggests that it may represent the primordial organisms that developed into the organisms that are today used as positive controls.
CONCLUSION
be-cause the temperature has remained nearly constant and rel-atively low, and the climate is semiarid with little water circulation.M. tuberculosiscomplex bacteria have hydrophobic cell walls, rich in long-chain mycolic acids. Location within bones provides further protection of DNA from environmental taphonomic loss [46].
Because this documentation is of tuberculosis occurring in the Pleistocene long before domestication of cattle, it suggests a different scenario for the spread of tuberculosis than is usually conceived [4, 21]. The presence of characteristic lesions in sheep
(Ovis canadensis catclawensis)and extinct musk ox(Bootherium bombifrons)[31] and in bison—now confirmed as tubercular in origin—indicates that the disease was widespread throughout the Pleistocene. It is suggested that tuberculosis in bovids had a Holarctic pattern and that it reached North America at least 20,000 years BP. Absence of tubercular lesions in pronghorn
Antilocapra americanumandEquusspecies suggests that it was absent in North America before the immigration of the ad-vanced northern bovids.
Analysis of additional specimens that have tuberculosis le-sions is now indicated to elucidate the evolution of tuberculosis. It is likely that the bison isolate contains some “novel” spoli-gotyping spacers—and perhaps spacers as yet not identi-fied—between the spacers that appeared on the blot for our specimen. The paleontologic record provides an excellent geo-graphic and chronological database for understanding disease origins and spread, especially for diseases with significant os-seous impact.
Acknowledgments
The support of The Center for the Study of Emerging Dis-eases, Jerusalem, is gratefully acknowledged. Dr. Dick van Sool-ingen’s cogent input is acknowledged with appreciation.
References
1. Greenblatt CL, ed. Digging for pathogens: ancient emerging diseases: their evolutionary, anthropological and archeological context. Jerusa-lem: Balaban Publishers,1998.
2. Martin LD, Rothschild BM. Earth history and the evolution of sickness. In: Greenblatt CL, ed. Digging for pathogens: ancient emerging dis-eases: their evolutionary, anthropological and archeological context. Jerusalem: Balaban Publishers,1998:15–46.
3. Rothschild BM, Martin LD. Paleopathology: disease in the fossil record. London: CRC Press,1999.
4. Arriaza BT, et al. Pre-Columbian tuberculosis in northern Chile: mo-lecular and skeletal evidence. Am J Phys Anthropol1995; 98:37–45. 5. Cole ST, Bosch R, Parkhill J, et al. Deciphering the biology of
Myco-bacterium tuberculosis from the complete genome sequence. Nature 1998; 393:537–44.
6. Finken M, Kirschner P, Meier A, Wrede A, Bottger EC. Molecular basis of streptomycin resistance inMycobacterium tuberculosis:alterations of the ribosomal protein S12 gene and point mutation within a functional S16 ribosomal RNA pseudoknot. Mol Microbiol1993; 9:1239–46.
7. Kolman CJ, Tuross N. Ancient DNA analysis of human populations. Am J Phys Anthropol2000; 111:5–23.
8. Kolman CJ, Centurion-Lara A, Lukehart SA, et al. Identification of
Treponema pallidum subspecies pallidum in a 200-year-old skeletal specimen. J Infect Dis1999; 180:2060–3.
9. Behr MA, Wilson MA, Gill WP, et al. Comparative genomics of BCG vaccines by whole-genome DNA microarray. Science1999; 284:1520–3. 10. Dye C, Scheele S, Dolin P, et al. Global burden of tuberculosis: esti-mated incidence, prevalence, and mortality by country. JAMA1999; 282:677–86.
11. Musial CE, Tice LS, Stockman L, Roberts GD. Identification of my-cobacteria from culture using the Gen-Probe rapid diagnostic system for Mycobacterium avium complex and Mycobacterium tuberculosis
complex. J Clin Microbiol1988; 26:2120–3.
12. van der Heijden IM, Wilbrink B, Schouls LM, van Embden JD, Breed-veld FC, Tak PP. Detection of mycobacteria in joint samples from patients with arthritis using a genus-specific polymerase chain reaction and sequence analysis. Rheumatology1999; 38:547–53.
13. Frothingham R, Strickland PL, Bretzel G, Ramaswamy S, Musser JM, Williams DL. Phenotypic and genotypic characterization of Mycobac-terium africanumisolates from West Africa. J Clin Microbiol1999; 37: 1921–6.
14. Eisenach KD, Cave MD, Bates JH, Crawford JT. Polymerase chain reaction amplification of a repetitive DNA sequence specific for My-cobacterium tuberculosis. J Infect Dis1990; 161:977–81.
15. Cousins DV, Williams SN, Dawson DJ. Tuberculosis due to Mycobac-terium bovis in the Australian population: DNA typing of isolates, 1970–1994. Int J Tuberc Lung Dis1999; 3:722–31.
16. Verano JW, Ubelaker DH. Disease and demography in the Americas. Washington, DC: Smithsonian Institution Press,1992.
17. Widmer L, Perzigian AJ. The ecology and etiology of skeletal lesions in late prehistoric populations from eastern North America. North-western Univ Archeol Prog Sci Papers1981; 5:99–113.
18. Salo WL, Aufderheide AC, Buikstra J, Holcomb TA. Identification of
Mycobacterium tuberculosisDNA in a pre-Columbian Peruvian mum-my. Proc Natl Acad Sci USA1994; 91:2091–4.
19. Allison MJ, Mendoza D, Pezzia A. Documentation of a case of tuber-culosis in pre-Columbian America. Am Rev Respir Dis 1973; 107: 985–991.
20. Zimmerman MR. Pulmonary and osseous tuberculosis in an Egyptian mummy. Bull N Y Acad Med1979; 55:604–8.
21. Buikstra JE. The Caribou Eskimo: general and specific disease. Am J Phys Anthropol1976; 45:351–68.
22. Aceves-Avila FJ, Baez-Molgado S, Medina F, Fraga A. Paleopathology in osseous remains from the XVI century: a survey of rheumatic dis-eases. J Rheumatol (in press).
23. Aufderheide AC, Rodriguez-Martin C. The Cambridge encyclopedia of human paleopathology. Cambridge, UK: Cambridge University Press,1998.
24. Formicola V. X-linked hypophosphatemic rickets: a probable Upper Paleolithic case. Am J Phys Anthropol1995; 98:403–9.
25. Morse D. Tuberculosis. In: Brothwell D, Sandison AT, eds. Diseases in antiquity. Springfield, IL: Charles C. Thomas,1967:249–71. 26. Palfi G. The osteo-archaeological evidence of vertebral tuberculosis in
the 8th century. Acta Biologica Szeged1991; 37:101–5.
27. Sager P, Schalimtzek M, Moller-Christensen V. A case of spondylitis tuberculosa in the Danish Neolithic age. Danish Med Bull1972; 19: 176–80.
28. Strouhal E. Vertebral tuberculosis in ancient Egypt and Nubia. In: Ortner DJ, Aufderheide AC, eds. Human paleopathology: current syn-theses and future options. Washington, DC: Smithsonian Institute Press,1991:181–94.
29. Wells C. Bones, bodies and diseases. London: Thames and Hudson, 1964.
30. Bartels P. Tuberkulose (Wirbelkaries) in der jungeren Steinzeit. Arch Anthropol1907; 6:243–55.
sample from Natural Trap Cave (late Pleistocene). J Vert Paleontol l989;9:31A.
32. Resnick D, Niwayama G. Diagnosis of bone and joint disorders. 2d ed. Philadelphia: Saunders,1988.
33. Wang X, Martin LD. Natural Trap Cave. Natl Geographic Soc Res Rep 1993; 9:422.
34. Hass DW, des Prez RM. Mycobacterium tuberculosis. In: Mandell GL, Bennett JE, Dolin R, eds. Principles and practice of infectious diseases. 4th ed. New York: Churchill Livingstone,1995:2213–43.
35. Boom R, Sol A, Salimans M, Jansen C, Wertheim-van Dillen P, van der Noordaa J. Rapid and simple method for purification of nucleic acid. J Clin Microbiol1990; 28:495–503.
36. Donoghue HD, Spigelman M, Zias J, Gernaey-Child AM, Minnikin DE. Demonstration ofMycobacterium tuberculosiscomplex DNA in calcified pleura from remains 1400 years old. Lett Appl Microbiol 1998; 27:265–9.
37. van Soolingen D, van der Zanden AG, de Haas PE, et al. Diagnosis of
Mycobacterium microtiinfections among humans by using novel ge-netic markers. J Clin Microbiol1998; 36:1840–5.
38. Taylor GM, Goyal M, Legge AJ, Shaw RJ, Young D. Genotyping analysis ofMycobacterium tuberculosisfrom medieval human remains. Micro-biology1999; 145:899–904.
39. Niemann S, Richter E, Rusch-Gerdes S. Stability ofMycobacterium
tuberculosisIS6110 restriction fragment length polymorphism patterns and spoligotypes determined by analyzing serial isolated from patients with drug-resistant tuberculosis. J Clin Microbiol1999; 37:409–12. 40. van Embden JD, van Gorkom T, Kremer T, Jansen R, van der Zeijst
BA, Schoulds LM. Genetic variation and evolutionary origin of the direct repeat locus ofMycobacterium tuberculosiscomplex bacteria. J Bacteriol (2000); 182:2393–401.
41. Dale JW, Brittain D, Cataldi AA, et al. Spacer oligonucleotide typing of bacteria of theMycobacterium tuberculosiscomplex: recommenda-tions for standardised nomenclature. Int J Tuberc Lung Dis2001; 5: 216–9.
42. Cockburn A, Cockburn E, Reyman TA. Mummies, disease and ancient cultures. Cambridge, UK: Cambridge University Press,1998. 43. Garcia-Frias J. La tuberculosis en los antiguos Peruanos. Actualidad
Medicina Peruana1940; 5:274–91.
44. Streevatsan S, Pan X, Stockbauer KE, et al. Restricted structural gene polymorphism in the Mycobacterium tuberculosiscomplex indicates evolutionarily recent global dissemination. Proc Natl Acad Sci USA 1997; 94:9869–74.
45. Castets M, Boisvert H, Grumbach F, Brunel M, Rist N. Tuberculosis bacilli of the African type. Rev Tuberc Pneumonol1968; 32:179–84. 46. Eglinton G, Logan GA. Molecular preservation. Philos Trans R Soc