Proc. Natl.Acad. Sci. USA
Vol. 90, pp. 11623-11627,December 1993 CellBiology
The mdm-2 gene
is induced
in response to
UV
light
in a
p53-dependent manner
(cellcycle/transcription)MARY ELLEN
PERRY, JACQUES PIETTE*,
JEROME A.ZAWADZKI,
DIANEHARVEYt,
ANDARNOLD J.LEVINEt
Department of Molecular Biology, Princeton University, Princeton, NJ 08544-1014
ContributedbyArnold J. Levine, August31, 1993
ABSTRACT Irradiation ofmammalian cellswithUVlight results in a dose-dependent accumulation of the p53 tumor-suppressor gene product that is evident within 2 hr. UV treatmentcauses a dramatic increase inp53-specific transcrip-tional transactivation
activity
and an increase in expression of thep53-responsive gene mdm-2. UV-stimulated mdm-2 expres-sion is notdirectycorrelated with the level of p53 protein in a cell becausemdm-2inductionisdelayed at highUVdoses even though p53 levels rise almost immediately. Cells lacking p53 protein do not respond to UV by increasing their expression of mdm-2. The delayed induction of mdm-2 at high UV doses suggests that, inadditionto p53 proteinlevels,other factors contribute to theregulation of mdm-2 expressionfollowing UV treatment.The time of induction of mdm-2 incellstreated with UV light correlates with recovery ofnormal rates of DNA synthesis, presumably after DNA repair. These data indicate a possible role for mdm-2 in cell cycle progression.Mammalian cells respond to irradiation with UV light by transiently decreasingboth RNA and DNAsynthesis and by inducing expression of several genes whose products are thought to haveprotective effects against DNA damage (1). Theregulation of these UV response genes appears to be mediated by several transcription factors which function after UV exposure (2). The p53 tumor-suppressor gene productisatranscription factor(3, 4) that also appears to be involved in the response to UVlight.Thep53 proteinlevels increase due to thestabilizationof this protein in both murine (5, 6)and human(7, 8)cellstreated with UVlight. Although therole ofp53in the response to UVlighthas not beenfully characterized,this tumor-suppressorproteinhasbeen shown to act as a cell cycle checkpoint in the response to y irradiation(9, 10). fyirradiation induces bothaG1and a G2 phase-specific cell cycle block, andexpression of wild-type
p53
isnecessaryfor theG1butnottheG2block. Thespecific DNA-binding activity ofp53 isincreased after yirradiation and aDNAdamage-inducible, growth arrest-specific gene, GADD45,hasbeenshown tocontainap53responseelement (10).Characterization of the role of
p53
in the response to'y
irradiationled to thehypothesisthatp53actsas acell cycle checkpoint, causing a delay in the G1 phase of the cycle during which damage is thought to be repaired (9, 10). It seemedlikelythat
p53
mightalso act as acheckpointin the responseof cellstoUV exposure. Inaddition,Zhanetal.(8) haverecentlyshown thatp53
transcriptionaltransactivation activity is increased in human cells exposed to UVlight.
Possible targetsfor
p53
transcriptionaltransactivation activ-ity in the UV response are the GADD45 gene, which wasisolatedas aUV response gene(11),and themdm-2 gene.
p53
and mdm-2 appeartoformafeedback controlloop:while
p53
Thepublicationcostsofthis articleweredefrayedin partbypagecharge
payment. Thisarticlemusttherefore beherebymarked"advertisement"
in accordance with 18U.S.C.§1734solelytoindicate this fact.
caninduce mdm-2transcription (12, 13),highlevelsofmdm-2 protein inhibit p53 transcriptional transactivation activity (13, 14). Theresults reportedheredemonstrate thattreatment ofmurinecellswith UVlight resultsinasuppression ofDNA synthesisthatis independent ofthe presenceofp53. Incells expressing functional wild-type p53, the mdm-2 gene is induced following a lag time that increases with UV dose. Thisenhanced expression of mdm-2 precedes the appearance ofcellsentering S phase at normal rates of DNA synthesis. These data are consistent with the idea that mdm-2 may reversethe possible negative regulatory effects of p53 in the G1phaseof thecell cycleand/orpromotethe entryof cells into S phase after DNA damage is presumably repaired.
MATERIALS
ANDMETHODS
Cell Culture. Cells were grown in Dulbecco's modified Eagle's medium supplemented with 10% heat-inactivated fetal bovine serum in an atmosphere of 10%Co2.
UV Light Treatment. Cells were treated with UV light essentially asdescribed (5).The medium was removed and thecells were irradiated with an 8-Wgermicidal lamp deliv-ering 1.9
J'm-2 sec-1
at a distance of 40 cm. The dose was quantitated with a J-225 short-wave UV meter (Ultraviolet Products, SanGabriel, CA).FlowCytometry.Cells were labeled for 30 min with 10 ,uM BrdUrdatvarious timesfollowing UV irradiation. The cells were trypsinized,
fixed
in 70% ethanol (-20°C), and later stainedwith anti-BrdUrd antibodies conjugated to fluores-ceinisothiocyanate (Becton Dickinson) essentiallyas recom-mendedbythemanufacturer. Sampleswereanalyzedwithan EPICS 753 cell sorter (Coulter) with an MDADS II data system. An argon laser(488 nm, 500 mW) was used for themeasurementofthelogarithmoffluorescein fluorescenceat 515-530nmand linearpropidium iodide fluorescenceat>610
nm.
Western Analysis. Following incubation of 3 mg of each lysatewith the monoclonalantibodiesPAb419 (specific for simian virus 40largeTantigen)orPAb421(specific forp53)
or with anti-mdm-2 serum, the immunoprecipitates were
separated by
SDS/polyacrylamide
gelelectrophoresis along with high molecular weight prestained markers (Bethesda ResearchLaboratories) and blotted onto Hybond ECL ni-trocellulose(Amersham). The membrane was sliced atthe 68-kDa prestained marker and the half of the membrane containingthe higher molecular weight proteins was incu-bated with the anti-mdm-2 serumwhile the other halfwasincubated withPAb421. Antibody bindingwasdetected with Abbreviations: CAT, chloramphenicol acetyltransferase; MCK,
muscle-specificcreatine kinase.
*Present address: Centre National de la Recherche Scientifique, France.
tPresentaddress:RockefellerUniversity, 1230YorkAvenue,New York,NY10021.
*To
whomreprintrequests should beaddressed. 11623125I-protein
A (NEN), andquantitation was on aMolecular Dynamics Phosphorlmager.Chloramphenicol
Acetyltransferase
(CAT) Assays. The transfection andUV treatmentprotocol is essentially that of Devary et al. (2). A total of 10 ug of DNA (5t.g
ofCAT constructand 5pg of salmonspermcarrierDNA)wasused, and CAT assays were as described(15). Datawere quanti-tated on aPhosphorImagerandnumbersquoted in the textare the average oftwo duplicates. The total conversion of
[14C]chloramphenicol
to acetylatedproductwas<40%oin all reactions.RESULTS
Wild-Typep53 Protein Increases inaDose-Dependent
Man-ner inResponse to UV Irradiation.Maltzman andCzyzyk (5) showed thatp53 levelsin
BALB/C3T3
cellsrose withinthe first2 hrafterUVtreatment.Twodifferentimmortalmurine cell lines (C127 and 12.1) expressing wild-type p53protein
(16, 17)were treatedwithincreasing doses ofUVlightand analyzed for changes in p53 levels. Both cell lines showed increasing p53protein with increasing doses
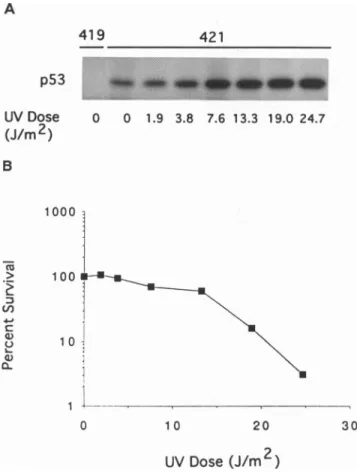
(Fig.
1anddata notshown).At thehighest dose used (24.7J/m2,
3% colony-forming ability, Fig. 1) C127 cells exhibiteda4-fold increase inthe incorporation of[35S]methionine
into the p53protein
immediatelyfollowing UVtreatment. The greatestincrease inp53expressionin thefirst 2 hr after UVtreatmentisincells sustainingthemostdamage.Fritscheetal.(6)haveindicated
apossible conformation change inthep53
protein
stabilized A 419p53
UV Dose (J/m 2) 421 *:~~~~~~~~~~~~~~~~~~~~~~~~~~~... .. .. .. ... ... -O O 1.9 3.8 7.6 13.3 19.0 24.7 B 1000 'a) V1) 40~ 1 00 1 0 1 0 10 20 UV Dose(J/m2
)FIG. 1. Increaseinp53as afunction of UV dose andinhibition
ofcolonyformationability. (A)C127 cellswereirradiatedwithaUV
lampatthe indicated dosesand thenimmediatelylabeled for2 hr with
[35S]methionine. Normalizedaliquots of labeledcellextractswere
incubated with eitherPAb421, ap53-specificmonoclonalantibody,
orPAb419, anegativecontrol. (B) C127cellswereplatedatalow
density, irradiated with various doses of UV light, incubated in
growthmedium for 1week,andscored forcolonyformation.
in response to some types of DNA
damage.
Monoclonalantibody PAb246,
whichrecognizes
thewild-type
murinep53
protein
(18), reactedwithvirtually
all thep53
presentinthese cells before and after UV treatment.Antibody PAb240,
whichrecognizesamutantor
denaturation-specific epitope,
didnot reactafter UV treatment, andso noevidence forsuch a conformational
change
ofp53
protein
could be demon-strated in this system.mdm-2 Protein Increases in Response to UV Irradiation. Sincethe mdm-2 geneis
regulated
by
thep53 protein (12, 13)
and themdm-2
protein
is knowntobindtothep53protein
and inhibitp53transcriptional
transactivation(13, 14),
itwasof interest to determine whether mdm-2 levels increased in responseto UVtreatment. Thesteady-state
levels of bothp53
andmdm-2,
aswellastheproportion
of each boundincomplex
withoneanother,
weremeasuredby
Western blotanalysis.
C127 cellsweretreated witheitheralow(3.8
J/m2,
94%
colony-forming ability)
or ahigh (19 J/m2,
16%colony-forming ability)
UVdose.The cellswereharvestedatvarious times afterUV treatment,lysed,
andincubated with eitherap53-specific antibody
(PAb421)
or an antiserumagainst
mdm-2. The
immunoprecipitates
wereelectrophoresed
and blottedontoamembrane,
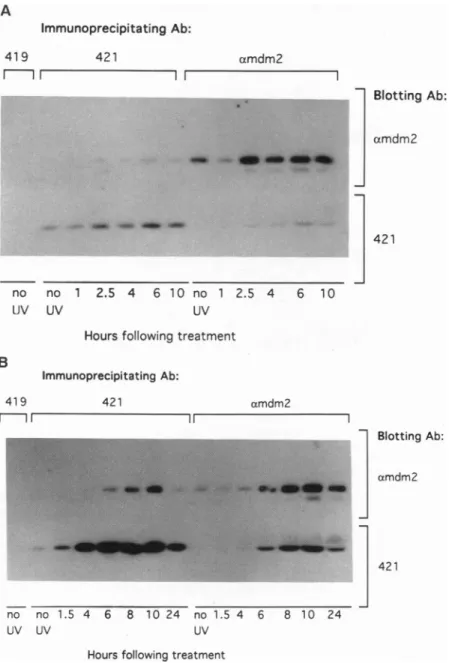
thetophalfof whichwasincubated withthe anti-mdm-2serumandthebottom half withPAb421. WhenC127 cellsweretreated withthe lowdoseofUV,
the levelofp53 protein
rose within 1.5hr,
and mdm-2protein
levels increased and
peaked by
2.5 hrfollowing
treatment(Fig. 2A).
Bothp53
and mdm-2proteins
increased about3-to4-foldin responsetothe lowdose ofUV
(Fig. 2A). However,
the
proportion
ofp53
bound to mdm-2(20-30%)
did notchange
significantly
withtime,
andat notimewas amajority
ofthe
p53
boundtomdm-2.When C127cells weretreated with the
high
doseofUV,
p53
levelsrose moredramatically:
atthe lowdose ofUV(3.8
J/m2),
the maximalincreaseinp53
levelswasabout3-fold,
whereasatthe
high
dose(19
J/m2),
p53
levels increasedby
asmuchas60-foldat8hr. The
steady-state
levels(Fig. 2B)
reflectalarger
increaseinp53
than theresults withlabeling
with
[35S]methionine
(Fig. 1),
consistent withthe demonstra-tionby
Maltzman andCzyzyk
(5)
that thep53
protein
is stabilizedby
UVtreatment. Theincreasein mdm-2protein
wasalsogreateratthe
high
doseandpeaked
at13-fold10 hr after UVtreatment(Fig. 2B). Thus,
the doseofUVaffects both thetiming
andthe levelof induction of mdm-2.Com-parison
ofthe levelsofp53
and mdm-2at4 hraftertreatmentclearly
showsthat theinduction ofmdm-2expression
atthehigh
doselags
behind theincreaseinp53
levels.Although p53
levelsare
higher
1.5hr aftertreatmentwithahigh
dose than after treatment with a lowdose,
the induction ofmdm-2expression
isdelayed
afterthehigh
dose.The Western blots
(Fig. 2)
quantitate
thesteady-state
levelsof
p53
and mdm-2 in cells treatedwithUVlight.
Similarexperiments
following
theincorporation
of[35S]methionine
intop53
andmdm-2proteins
in these cells demonstratedalag
inincreasedexpression
ofmdm-2protein following
stabili-zation ofp53
athigh
UVdoses. Athigh
UVdoses,
p53
levels roserapidly (Fig. 1A),
but mdm-2protein
levelsincreasedonly
afterasignificant delay
(data
notshown).
mdm-2mRNA Is Induced
by
UVLight
inap53-Dependent
Manner. Northern blotanalysis
ofRNA from C127 cells treated with low andhigh
doses of UV and harvested at various times after treatment showed agood
correlation between increased mdm-2 message andprotein
levels(data
notshown).
At a low doseofUVlight (3.8
J/m2),
mdm-2 mRNAincreasedandpeaked
2hraftertreatment, whichwasthetime atwhich the amountof
protein
also wasmaximal(Fig. 2A).
Athigher
UVdoses,
thepeak
increase in mdm-2protein
wasdelayed (Fig.
2B)
and thesubsequent
increase inmdm-2
protein
levelswasaccompanied by
increased mdm-2mRNA
(data
notshown).
This UV-stimulated increase in 421Proc. Natl.Acad. Sci. USA 90(1993) 11625 A ImmunoprecipitatingAb: 419 421 amdm2 7 Blotting Ab: |amdm2 ..~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~.... 421 no no 1 2.5 4 6 10 no 1 2.5 4 6 10 UV UV B UV Hours following treatment Immunoprecipitating Ab: 419 421 amdm2
ml1
-i A Blotting Ab: amdm2 , . ... ... ...:.i:S.:.... ...."''...S'1'' ... i BSt ,~~~~~~~~~~~~~~~~~~~~~~~~~~~~...i *,...:.-;... T= IMI~ ~ ~~~421 no no 1. 5 4 6 8 10 24 no 1.5 4 6 8 10 24 UV UV UVHours following treatment
FIG.2. Levels ofp53and mdm-2changeas afunction of dose. (A)C127cellsweretreated with a UV doseof 3.8J/m2,harvestedatthe indicatedtimesfollowing treatment,andincubated witheitherPAb419(negative control),PAb421(specific forp53),or mdm-2antiserum (right sideofblot).Theimmunoprecipitates wereelectrophoresedin apolyacrylamidegelandblotted onto amembrane,thetophalfofwhich was incubated with themdm-2 antiserum while the bottomhalfwasincubated withPAW21.(B)C127 cells wereexposedto 19J/m2andanalyzed asinA.
both mdm-2 message andproteinwas notobserved in cells lacking functionalp53. Neither (10)3 cells (16), whichlack p53 protein, nor (12)1 cells containing T antigen, which expresswild-typep53 proteinin the presence of the simian virus40largeTantigen, induceexpression ofmdm-2 follow-ing UV treatment. Therefore, the induction ofmdm-2
ex-pression appears tobe dependentupon the transcriptional transactivation activity of
p53
protein.p53
Transcriptional Activity
inCeils
Treatedwith UVLight. Next, thetranscriptional transactivationactivity
of endoge-nousmurinep53
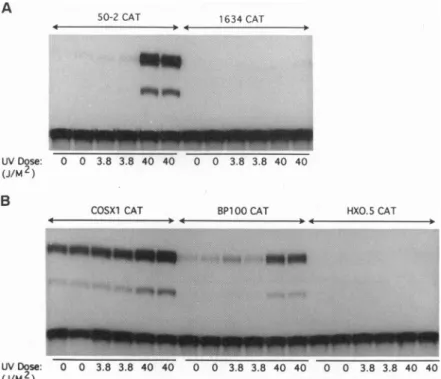
wasmeasured aftertreatmentofC127cells with UV light. A construct containing two copies of theconsensus
p53
DNA binding site from the murine muscle-specificcreatine kinase(MCK)
promoterlinkedto areporterconstructcontainingtheCAT gene(15)wasinduced 39-fold aftertreatmentofC127cells with 40
J/m2
(Fig.
3A)whereasasimilarconstructlackingtheMCK sequences
(1634CAT)
was notinduced more than2-fold. Sinceexpression of the mdm-2 gene is regulated by
p53
through ap53-binding
element in thefirst intron of the gene(13), three fragments fromthefirst intron of the mdm-2gene were tested for UV inducibility. Constructs containing the entire intron (COSX1CAT)or an85-bp fragmentincludingthetwo imper-fect p53 consensus binding sites (BP100CAT) (13) were
induced3.3- and8.4-foldbyaUVdoseof40J/m2 (Fig.3B). Athird construct, containing400bp from theintron exclud-ing thep53bindingsites(HXO.SCAT),was not UVinducible (<10% increase in CAT activity) (Fig. 3B). Although the 85-bpfragmentconferred UVinducibilityonthe CATgene in C127 cells, it did not do so in (10)3 cells, which lackp53 protein (<2-fold induction at 40 J/m2; data not shown). Because this UV response element is also ap53response element(13)and it isnotinduced in cellslacking p53 protein, theseexperimentsindicate that mdm-2 isap53-inducibleUV response gene.
mdm-2 Expression Precedes Normal EntryofCells into S Phase.Undersomecircumstances,overexpression ofthep53 proteincausescellsto arrestin theG1phaseof the cellcycle Cell
Biology: Perry
etal.50-2 CAT 1634 CAT
10 0 .0
_.*_n ...
M.
M-0 0 3.8 3.8 40 40 0 0 3.8 3.8 40 40
COSX1 CAT BPI00CAT HXO.5 CAT
.. .>4.It . 0 4 0
0 0 3.8 3.8 40 40 0 0 3.8 3.8 40
FIG.3. p53transcriptional transactivation activity is increased inresponsetoUVlight.(A) C127cellsweretransfectedwithaDNAconstruct
containing the CATreportergenefusedtoaminimalpromoter(1634CAT)ortothep53responseelementfrom the murineMCKpromoter
(50-2CAT).Twenty-four hours after transfection,cellsweretreated withahighor alowdoseof UVlight and after another 24hr,thecells were
harvested andlysed. Equalamountsofcellextractswereassayed for CAT activity. (B) Three fragments from the first intron of the mdm-2gene weretested for UVinducibility in C127 cells by the protocol described in A; COSX1CAT contains the entire intron, BP100CATcontains 85
bp that includetwoimperfectp53consensusbinding sites, and HXO.5CATcontains 400 bp from the first intronexcludingthep53bindingsites. (9, 19). While the function of mdm-2 is not understood,
overexpression oftheproteinhasbeen showntoinhibitp53 transcriptional transactivation activityintransient
transfec-tionexperiments (13, 14). Ithasbeenproposedthat
mdm-2-mediated inhibition of p53 transcriptional transactivation activitymayleadtoprogression ofcellsfromtheG1phaseto theS phase ofthecycle (20). Kastanetal.(10)have shown
that, after yirradiation,p53blocks cells in G1. It ispossible
that mdm-2 functions to overcome this G1 block. To see
whetherasimilar blockoccursinUV-treated cells, cellswere
labeled with thethymidine analogue BrdUrd and analyzedfor
No uv 4hr 0 0 c 0 0 0 - .-0 ie E z
BrdUrdcontentbyfluorescence-activatedcytometryusinga
fluorescein-conjugated monoclonal antibody specific for Brd-Urd. Cellswerealso stained withpropidium iodide, which
reflects the total amount of DNA in each cell, whereas
BrdUrd incorporation reflects nucleotide incorporation or
DNAsynthesisineachcell.Flowcytometryprofiles of C127
cells treated withamoderatelyhighUV dose of 14J/m2 (Fig.
4) showed aclear suppression in BrdUrd incorporation in
early,mid,andlate Sphase by4hr afterUVtreatment.The percentage ofUV-treated cells in G1, S, and G2 remained fairly constant (propidium iodide). Six to 8 hr after UV
Shr 12hr
q6 rA
24hr
I
PI fluorescence
FIG.4. Expressionofmdm-2precedestheresumptionof normal rates ofDNAsynthesisfollowingUVtreatment.C127 cellstreatedwith
aUVdoseof14J/m2werelabeled with 10,uMBrdUrd for 30minattheindicated timesfollowingtreatment.Cellswereharvested,processed
forfluorescence-activatedcytometry,and stainedwithanti-BrdUrdconjugatedtofluoresceinisothiocyanate (FITC) and counterstainedwith
propidiumiodide(PI).For each timepoint,the BrdUrdprofileis shownabovethepropidiumiodideprofile. Fifty thousandcellswereanalyzed
for each timepoint.
A UV Dose: (J/M2) B UVDose: (J/M2) .A
Proc. Natl. Acad. Sci. USA 90
(1993)
11627 irradiation, mdm-2 expression was maximal (data notshown), and at 8 hr there was a pronounced increase in the incorporation of BrdUrd into cells with a
Gl-phase
contentof DNA(cells in early S phase) as seen by a recovery ofhigh BrdUrd fluorescence (Fig. 4). In fact, there was a greater percentage of cells in early S phase 8 hr after UV treatment than in untreated asynchronous cells. There was also a shoulder on theG,
peak of cells stained for DNA content (propidium iodide), indicating an increase in the number of cells in early to mid S phase at 8 hr. This trend continuedasthiswaveof cells proceeded through S phase (Fig. 4). Thus, during the 6- to 8-hr lag before the resumption of normal rates of DNA synthesis, either there must be an accumulation of cells in G1 which synchronously enter S phase 8 hr following UV treatment or, alternatively, the progressionofcells from G1 to S phase must be accelerated by some signals provided prior to 8 hr following treatment. The waveof cells moving through S phase with normal rates ofDNA synthesis might well have resulted from p53 and mdm-2interactions, since (10)3 cells, which lack p53 protein, also exhibited a decrease in DNA synthesis following UV irradiation but failed to recover from this suppression with a wave ofcells incorporating normal levels of BrdUrd (Fig. 4B).The(10)3 cells also failed to induce comparable levels of mdm-2 message and protein after UV irradiation (data not shown).
To substantiate the correlation between recovery of nor-mal S-phase DNA synthesis with expression of mdm-2 pro-tein, C127 cellsweretreated with a UV dose of 3.8 J/m2. At this low dose, mdm-2 levels peak at 2-2.5 hr following treatment(Fig. 2A). Asignificant increase in cells with a
G,
DNAcontent incorporating a large amount of BrdUrd ap-peared2hrafterUV treatment (data not shown), so mdm-2 expressionprecedes the resumption of a normal rate of DNA synthesisfollowing UV treatment with either a high or low UVdose. Athighdoses, enhanced expression of mdm-2 and a return to normal S-phase levels of DNA synthesis were delayed when compared with the response at lower UV doses.
DISCUSSION
Irradiation of mammalian cells with UV light results in a transient inhibition of DNA and RNA synthesis with a coordinate induction of expression of several immediate early genes such as c-jun and c-fos that in turn appear to regulate expression of other UV response genes (1). It has beenproposedthat this transient decrease in DNA and RNA synthesis following UV treatment reflects the time during which the UV-induced damage is repaired before the cells continue through the cycle (1, 21). This decrease could be brought about byacheckpoint control mechanism(21), and thep53tumor-suppressor geneproduct, which accumulates inresponse to DNAdamage (5-7, 10), acts as acell cycle checkpointin the response to yirradiation (9).
Thewild-typep53 proteinplays a role inblocking progres-sion through the G1 phaseof thecellcycle afteryirradiation, resulting in a complete inhibition of DNA synthesis in S-phase cells following treatment(9, 10). Incontrast, after UVirradiation,there is a decreased rate of DNA synthesis both in cells containing wild-type p53 protein and in cells lacking
p53
protein. p53 levels increase within 1-2 hrafter UV irradiation and continue toaccumulate when cells are treatedwithahigh doseof UV. Thehighlevels ofp53 protein, upto60-foldnormallevels,mightbeexpectedtoblockcells fromprogressingoutof G1 into S phase (19, 22), but there is no directevidence foraG1 blockintheseexperiments. Thetranscriptional transactivation activity of p53 isdramatically increased following UV treatment, and one target of p53 function in the UV response is the mdm-2 gene, which contains ap53 response element in the first intron (13). At low doses of UV irradiation, theresponsiveness ofthis element is notapparent in a transient transfection assay (Fig. 3B), but theinduction of mdm-2 occurs rapidly (see Fig. 2A) and is dependent on the presence ofp53, since it does not occur in cells expressing simian virus 40 large T antigen (data not shown) or in cells lackingp53 protein. At higher doses, the synthesis of mdm-2 mRNA and protein are delayed several hours after the stimulation of high p53 protein levels in response to irradiation. Thus, at high UV doses other factors, inaddition top53 protein, are required for mdm-2 induction and these factors are rate-limiting for mdm-2 expression. Presumably, mdm-2 synthesis is delayed to permit repair synthesis to occur before normal rates of scheduled DNA synthesis can resume, and this delay is lengthened with increased amounts of DNA damage.
We aregrateful to Dr. T.Silhavyforthe useof hisUV light source and to Dr. J. Momand for stimulating discussions duringthe early stages of this work. We thank Dr. J. Chenforadvice on Western blotting and Drs. C. Finlay and X. Wuforhelpfuland supportive discussions. We acknowledge Dr. X. Wu forcritically readingthe manuscript.
1. Holbrook, N. J. & Fornace, A. J., Jr. (1991) New Biol. 3, 825-833.
2. Devary, Y., Gottlieb, R. A., Lau, L. F. & Karin, M. (1991) Mol. Cell. Biol. 11, 2804-2811.
3. Fields, S. & Jang, S. K. (1990)Science 249, 1046-1049. 4. Raycroft, L., Wu, H. & Lozano, G. (1990) Science 249,
1049-1051.
5. Maltzman, W. &Czyzyk, L. (1984) Mol. Cell. Biol. 4, 1689-1694.
6. Fritsche, M., Haessler, C. & Brander, G.(1993) Oncogene 8, 307-318.
7. Hall, P. A., McKee, P. H., Menage, H. duP., Dover, R. & Lane, D. P. (1993) Oncogene 8, 203-207.
8. Zhan, Q., Carrier, F. & Fornace,A. J., Jr. (1993)Mol. Cell. Biol. 13, 4242-4250.
9. Kuerbitz, S. J., Plunkett, B. S., Walsh, V. W. & Kastan, M. B.(1992) Proc.Natl. Acad. Sci. USA 89, 7491-7495. 10. Kastan, M. B., Zhan, Q.,El-Deiry, W. S.,Carrier,F.,Jacks,
T.,Walsh, W. V., Plunkett, B. S., Vogelstein, B. &Fornace, A. J., Jr. (1992) Cell 71,587-597.
11. Fornace,A. J., Jr.,Alamo,I., Jr., &Hollander,M. C.(1988) Proc.Natl.Acad. Sci. USA 85,8800-8804.
12. Barak, Y., Juven, T.,Haffner,R. &Oren, M. (1993)EMBOJ. 12,461-468.
13. Wu, X., Bayle, J. H., Olson, D. & Levine, A. J. (1993) Genes Dev. 7,1126-1132.
14. Momand, J., Zambetti, G. P., Olson, D. C., George, D. & Levine, A. J. (1992)Cell69, 1237-1245.
15. Zambetti, G. P., Bargonetti, J., Walker, K., Prives, C. & Levine, A. J. (1992) Genes Dev. 6, 1143-1152.
16. Harvey, D. & Levine, A. J. (1991) Genes Dev. 5,2375-2385. 17. Sherley, J. L. (1991) J. Biol. Chem. 266,24815-24828. 18. Gannon, J. V., Greaves, R., Iggo, R. & Lane, D. P. (1990)
EMBO J.9, 1595-1602.
19. Martinez, J.,Georgoff, I., Martinez, J. & Levine, A. J. (1991) Genes Dev. 5, 151-159.
20. Olson, D., Marechal, V.,Momand,J., Chen, J.,Romocki, C. &Levine, A. J. (1993) Oncogene8,2353-2360.
21. Brown, M., Garvik, B., Hartwell, L., Kadyk, L., Seeley, T. & Weinert,T.(1992) in The Cell Cycle, eds. Beach, D.,Stiliman, B. &Watson, J. D. (ColdSpringHarbor Lab. Press,Plainview, NY), Vol. 56, pp. 359-365.
22. Michalovitz, D., Halevy, 0. & Oren, M. (1990) Cell 62, 671-680.