Procedia Computer Science 23 ( 2013 ) 137 – 145
1877-0509 © 2013 The Authors. Published by Elsevier B.V.
Selection and peer-review under responsibility of the Program Committee of CSBio2013 doi: 10.1016/j.procs.2013.10.018
ScienceDirect
4th International Conference on Computational Systems-Biology and Bioinformatics, CSBio2013
Apriori gene set-based microarray analysis for disease classification
using unlabeled data
Worrawat Engchuan and Jonathan Hoyin Chan*
Data and Knowledge Engineering Laboratory (D-Lab)Bangkok, Thailand 10140
Abstract
Gene set-based microarray analysis allows researchers to better analyze the gene expression data for studying complex diseases like cancer. By transforming gene expression data into another form using gene set information, the biomarkers will have higher discriminative power and should result in more accurate disease classification. This work compares two techniques for applying our previously developed NCFS-i-based method to deal with unlabeled data, i.e. to make predictive diagnosis. Seven cancer datasets that include 4 breast cancer and 3 lung cancer datasets were used in this study. The results show that inferring gene set activity using curated phenotype-correlated genes (PCOGs) sets of training data is a more robust method for applying NCFS-i-based method to work with unlabeled data, providing biologically relevant gene sets.
© 2013 The Authors. Published by Elsevier B.V.
Selection and peer-review under responsibility of the Program Committee of CSBio2013. Keywords: gene set activity; gene expression; microarray analysis; cancer classification; feature selection
1. Introduction
Presently, there are many gene set-based microarray analysis approaches proposed for achieving improved understanding of complex diseases, driven by multiple mechanisms1. The benefit of studying those complex diseases is to provide the opportunity for scientists to be able to find the treatments for those patients with complex diseases like cancers. Gene set-based microarray analysis is categorized into two main approaches in this work. The first is the traditional approach, which will directly use microarray data along with application of gene set
* Corresponding author. Tel.: +662-470-9819. E-mail address: [email protected]
© 2013 The Authors. Published by Elsevier B.V.
Selection and peer-review under responsibility of the Program Committee of CSBio2013
Open access under CC BY-NC-ND license. Open access under CC BY-NC-ND license.
knowledge. The traditional approach is to use gene set knowledge to select an appropriate subset of genes that remove the undesirable redundant genes for further analysis, as in the work of2. The second approach of utilizing
gene set knowledge is called transform-approach. This approach will use gene set information to transform the microarray data into another form of data. The previous transform-approaches are found to work well in identifying biomarkers for cancer classification tasks. In 2008, Lee et al.3 proposed method for selecting the relevant genes
called Conditional-Responsive Genes (CORGs) for transforming gene expression data to pathway activity, which represent the level of association of that pathway to disease development or a condition of interest. The pathway activity data inferred by CORGs provided higher discriminative power than using gene expression data or pathway activity that is inferred by all gene members in the pathway. In a follow-up work in 2011, Sootanan et al. 4 proposed
a modified approach called Negatively Correlated Feature Sets using ideal markers (NCFS-i) to search for a set of genes called Phenotype-Correlated Genes (PCOGs) to be used in pathway activity inference. The NCFS-i-based method would include all relevant genes even those genes have a small effect on disease development. This method was shown to be more robust than the CORG-based method.
The current pathway activity inferring methods require that prior information (classes) of samples be known, or estimated, before it can transform gene expression to pathway activity. So, the classifier built using this pathway activity data may not be applied easily for disease diagnosis of unlabeled microarray data (from subject with unknown condition). This work compares different apriori methods to apply NCFS-i pathway activity inferring method for disease diagnosis of unlabeled microarray data. Also, instead of focusing only on pathway information alone as in previous works, multiple gene sets were examined in order to find out the most informative gene set for inferring gene set activity.
2. Methodology 2.1 Dataset
Seven gene expression datasets were collected from Gene Expression Omnibus (GEO) (Table 1)5. First four
datasets are breast cancer datasets, which compose of two well-known metastasis datasets (GSE1456 with 159 samples, GSE2034 with 286 samples) and two tumor/normal datasets (GSE5764 with 30 samples, GSE7904 with 62 samples). The metastasis dataset is the most appropriate for researching as it has large sample size, which may be the representative of the population. So, it was used in first part of this study to find an appropriate way to apply NCFS-i-based method for classification of unlabeled cancer data. Then three datasets (GSE2109 with 71 samples, GSE10245 with 58 samples, and GSE18842 with 48samples) of lung cancer were applied for validation of the technique. The standard z-score normalization process was applied before transformation.
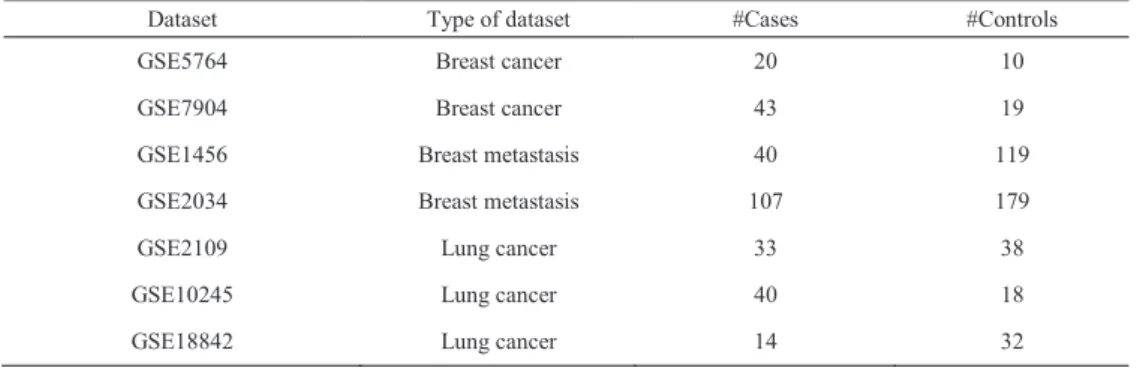
Table 1. Gene expression datasets used in this study
Dataset Type of dataset #Cases #Controls
GSE5764 Breast cancer 20 10
GSE7904 Breast cancer 43 19
GSE1456 Breast metastasis 40 119
GSE2034 Breast metastasis 107 179
GSE2109 Lung cancer 33 38
GSE10245 Lung cancer 40 18
2.2 NCFS-i-based gene set activity inferring method
In this work, we further evaluate the NCFS-i-based method of inferred gene set activity by using a larger number of gene sets from the preprocessed Molecular Signature DataBase (MSigDB)6. The MSigDB consists of multiple
gene sets, which are categorized by different criteria (see Table 2). In this work, all five gene sets were used for comparison, in order to choose the most informative gene sets for activity inference. As before, to calculate gene set activity of each gene set, the gene members were ranked by their discriminative power to differentiate case and control, evaluated by the Student t-test. A greedy search algorithm was then used, started by picking the most positive (top) and the most negative (bottom) rank genes from the t-test. The subtracted value of these two genes was assigned to be the interim gene set activity. In the next iteration, another top and bottom gene pair was selected and the subtracted value was added to the previous gene set activity to calculate a new gene set activity. Next, discriminative power of the two gene set activity values was compared. If the new gene set activity has higher discriminative power, then repeat the previous steps and iterate. The greedy search would stop when the new gene set activity has lower discriminative power than previous iteration4.
Table 2. Information of gene sets from MSigDB
Code Gene sets Description
C1 Positional gene sets For each human chromosome and cytogenetic band. C2 Curated gene sets From online pathway databases, publications in PubMed, and
knowledge of domain experts.
C3 Motif gene sets Based on conserved cis-regulatory motifs from a comparative analysis of the human, mouse, rat, and dog genomes.
C4 Computational gene sets Defined by mining large collections of cancer-oriented microarray data. C5 GO gene sets Consist of genes annotated by the same GO terms. 2.3 Applying NCFS-i-based method to deal with unlabeled dataset
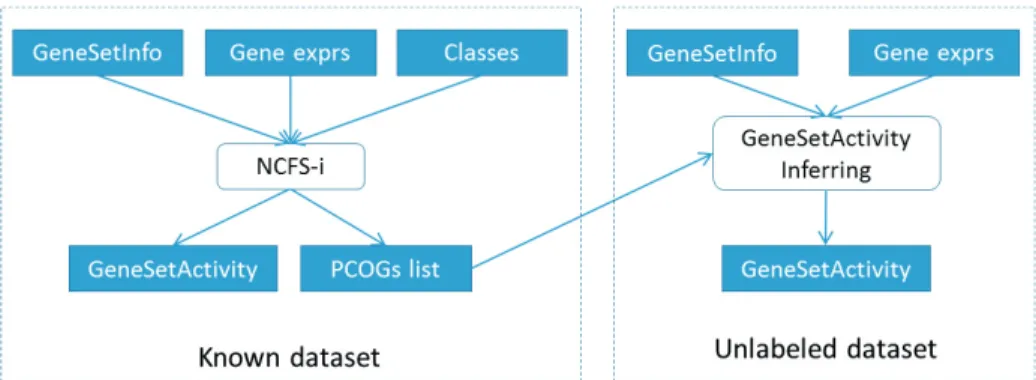
Here, we compared two methods of applying NCFS-i-based method to work with unlabeled datasets. The first method does not require the use of prior information of samples in unlabeled datasets. Basically, NCFS-i-based method will do greedy search for each gene set in order to find the gene subset that response to disease or condition of interest (PCOGs set) as before. Then, instead of finding PCOGs set of the unlabeled set, we simply use the same PCOGs set with known or training dataset for inferring gene set activity in the unlabeled dataset (see Fig. 1). This was the method used by both CORG and PCOG previously. The PCOGs of each gene sets are composed of positive and negative sets. The gene set activity will be calculated as following formula.
np ip nn in in ip
np
Z
nn
Z
ivity
geneSetAct
1 1))
(
(
/
))
(
(
(1) where, np is number of genes in positive set of PCOGs, Zipis gene expression of geneip, nn is number of genes inFig. 1. Gene set activity inference in unlabeled dataset using PCOGs set of known dataset
An alternative way of applying NCFS-i-based method is to estimate the apriora i information first. The method considered is by doing pre-classification (PC) using gene expression data, then using that estimated prior information for inferring gene set activity. A comparison of classification algorithms for gene expression data was constructed in order to find a suitable algorithm and the appropriate number of top genes to use in classification to estimate apriora i information. Three classic classification algorithms were used for the comparison using the breast metastasis datasets: K-Nearest Neighbor (KNN), Random Forest (RF), and Support Vector Machine (SVM). The top genes were evaluated and ranked by SVMAttributeEval of WEKA7. The number of genes used was varied as
done previously8. (See Fig. 2.)
Fig. 2. Workflow to find a suitable classifier and appropriate number of genes for classification 3. Results
3.1 Comparison of gene sets
The classification performance by using different gene sets to infer activity was evaluated with two breast cancer datasets (GSE1456 and GSE2034). The comparison was done using SVM with 10-fold cross-validation. The Single Pathway Classification (SPC) ranker method proposed by Chan et al. was used in featff tture selection8. The results
showed that classification of gene set activity data inferred using C2, C4 and C5 were more robust (Fig.3). In particular, C2, which is curated gene sets, provided the best overall performance. Consequently, the curated gene sets from C2 were used in subsequent analysis of this work.
Fig. 3. 10-fold cross-validation result using different gene sets. 3.2 Comparison of classification algorithm
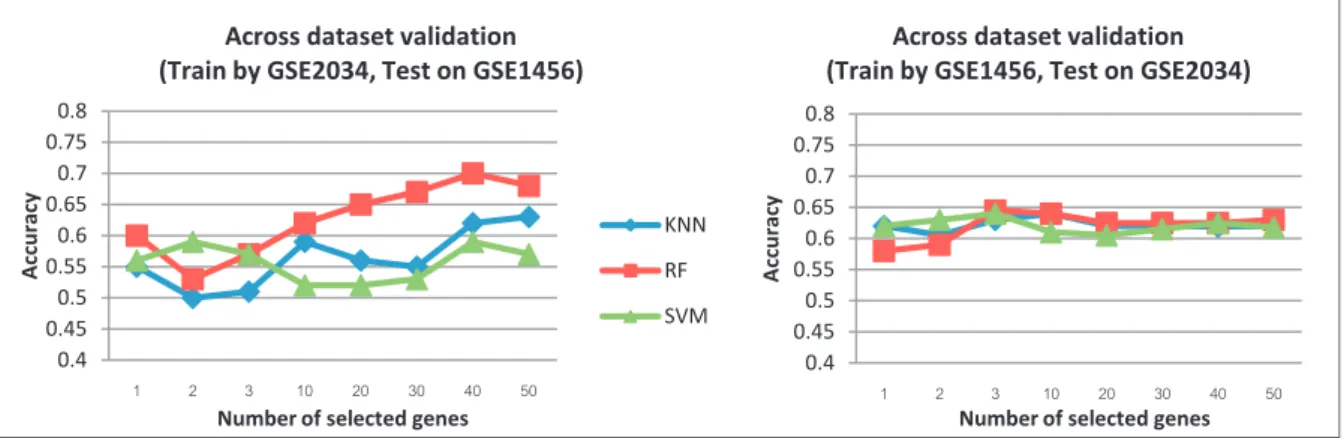
Comparison of the classification algorithms was done to choose a suitable classifier and appropriate number of genes for pre-classification. The breast metastasis datasets (GSE1456, GSE2034) were used in this comparison by alternately choosing one dataset as training data and the other dataset as testing data. Fig. 4 shows the comparative results of different classifiers and number of top genes used in the feature selection step.
Referring to Fig. 4, the Random Forest (RF) classifier, the red line with square blocks, performed better than the other two classifiers when the number of top genes used in classification was more than 10 genes. Then to find the suitable number of top genes used, for the GSE2034 dataset, RF was used. The results were quite similar when using number of top genes from 10-50 genes. However, when tested by GSE1456, RF performed the best when using 40 top genes. Overall, we decided to choose RF algorithm with 40 top genes for apriora i estimation.
Fig. 4. Classification result of different classifiers using gene expression data. 3.3 Comparison of classification using gene set activity inferred from different methods
In order to be able to apply NCFS-i-based method for inferring gene set activity in an unlabeled dataset (test dataset), we propose two possible ways here. First is the use of PCOGs set from training data and the second is doing pre-classification to estimate the prior information. Here, we compared classification performance using gene set activity inferred by the two different methods. Furthermore, we also compared our results to the biased method of gene set activity inferring when the validating datasets used in the study were all known. So, PCOGs of validating datasets can be identified by using its information.
That is, we compared the classification performance using gene set activity inferred in three different ways: using PCOGs of training data (labeled as PCOGs); doing pre-classification (labeled as PC); and using known or labeled data as test dataset (labeled as Bias). The best classifier, random forest was used as classifier in this comparison.
0.4 0.45 0.5 0.55 0.6 0.65 0.7 0.75 0.8 A ccurac y
Number of selected genes
Across dataset validation (Train by GSE2034, Test on GSE1456)
KNN RF SVM 0.4 0.45 0.5 0.55 0.6 0.65 0.7 0.75 0.8 A ccurac y
Number of selected genes
Across dataset validation (Train by GSE1456, Test on GSE2034)
Also, the number of top gene sets (1, 2, 3, 10, 20, 30, 40, 50) used in classification was evaluated and ranked by Single Pathway Classification (SPC) ranker8using a single gene set. Breast cancer and breast metastasis datasets
were used for evaluation. Similar classification results were found using gene set activity of both breast cancer datasets (GSE5764, GSE7904).
The classification results (Fig. 5) show that inferring gene set activity using PCOGs set from training data and select only top 2-3 gene sets for classification would give the best classification performance ((Accuracy=0.81, when using top 2 gene sets for testing in GSE7904; Accuracy=0.72 when use top 3 gene sets in testing with GSE5764). This result is consistent with the prior work8.
Fig. 5. Across dataset validation of Breast cancer dataset (GSE5764 and GSE7904)
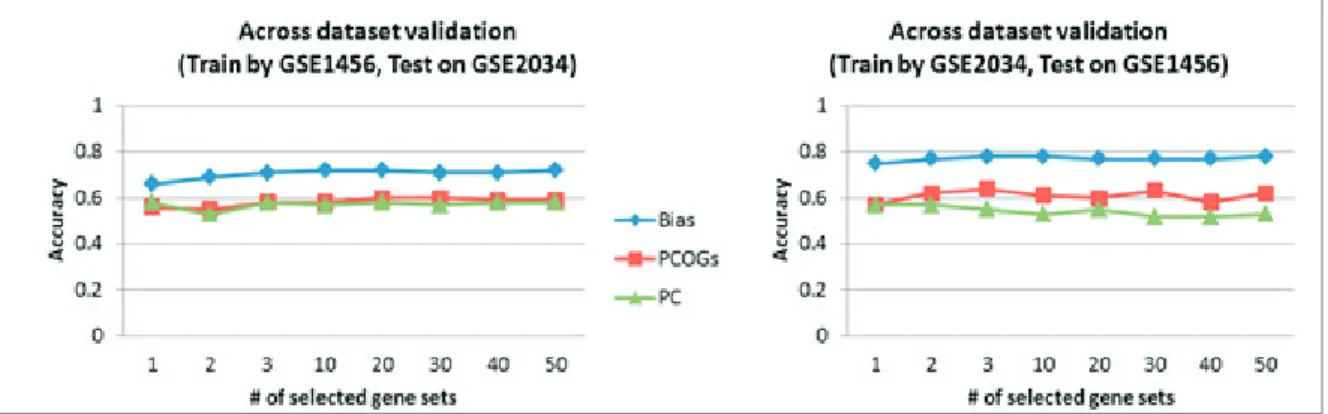
Fig.6 shows another classification result of gene set activity data from breast metastasis datasets (GSE1456, GSE2034). The result shows that even though the use of PCOGs set of training data for inferring gene set in testing data did not give the best performance in classification, it is still better than doing pre-classification for estimate prior information. Taking into account all results, inferring gene set activity in unlabeled set using PCOGs set from training data was more appropriate when applying the NCFS-i-based method.
3.4 Applying NCFS-i-based method in classification of lung cancer unlabeled data
The use of PCOGs set of training data for inferring gene set activity in unlabeled data did quite well in both breast cancer and breast metastasis datasets. Then, we applied NCFS-i-based method to another 3 lung cancer datasets (GSE2109, GSE10245, and GSE18842) to validate our result. In the validation step, GSE2109 was assumed to be known data and was used for training classifier. Then, the classifier was used to classify an assumed-unlabeled datasets (GSE10245, GSE18842). Fig. 7 shows the validation result on gene set activity data of lung cancer datasets. The validation results confirm that using the same PCOGs set with training data produced the most robust results.
Fig. 7. Validation result of applying NCFS-i-based method to lung cancer unlabeled datasets 3.5 Biological interpretation of result
From the total of 169 gene sets, the SPC ranker was used to select the top 10 gene sets from lung cancer dataset (GSE2109) for biological analysis. A list of top 10 gene sets is shown in Table 3. There is evidence from publications in well-known open-access journals support that eight out of top 10 gene sets are related to the lung cancer development and/or treatment. The Gene sets in cancer (Table 3; rank number 10), is the collection of genes that are related to any cancer, including lung cancer. This analysis shows that using SPC ranker on gene set activity data inferred by NCFS-i is powerful enough to significantly select the related gene sets for the cancer classification. Therefore, this supports that the PCOGs information from training data can be used for classification of unlabeled data.
Table 3. List of top gene sets in GSE2109 dataset, selected by SPC ranker
Ranking Gene set References
1 Cytokine, Cytokine receptor interaction Van Dyke et al.9
2 Melanogenesis Bellei et al.10
3 WNT signaling pathway Mazieres et al.11
4 ECM receptor interaction Devaraj et al.12
5 Phosphotidylinositol signaling system Hanai et al.13, Tsurutani et al.14
6 Calcium signaling pathway Yang et al.15
7 Long term potentiation -
8 Leukocyte transendothelial migration Lu et al.16
9 NOTCH signaling pathway Westhoff et al.17
10 Pathway in cancer -
4. Conclusions
This work confirms that NCFS-i-based method is a robust method to perform gene set-based microarray analysis, using PCOGs set of training data for inferring gene set activity in unlabeled data. Our results also showed that the use of C2 gene sets from MSigDB, with SPC ranker feature selection on gene set activity inferred by NCFS-i-based
0 0.2 0.4 0.6 0.8 1 Accuracy
Number of selected gene sets
Across dataset validation (Train by GSE2109, Test on GSE10245)
Bias PCOGs PC 0 0.2 0.4 0.6 0.8 1 A ccuracy
Number of selected gene sets
Across dataset validation (Train by GSE2109, Test on GSE18842)
method, is able select biologically significant gene sets. Future work includes further validation of the developed methodology, as well as identification of the best classification algorithm for gene set activity data. The methodology will also be adapted to our current multi-class classification work. The transformed datasets will also be made available publicly.
Acknowledgments
Worrawat Engchuan would like to thank National Center for Genetic Engineering and Biotechnology, Thailand logy Thonburi for scholarship support.
References
1. Pang H and Zhao H. Building gene set cluster from Random Forest classification using class votes. BMC Bioinformatics 2008; 9: 87. 2. Bandyopadhyay N, Kahveci T, Goodison S, Sun Y, and Ranka S. Gene set-based feature selection algorithm for cancer microarray data Advance in Bioinformatics 2009.
3. Lee E, Chuang H, Kim J, Ideker T, and Lee D. Inferring gene set activity toward precise disease classification, PLOS computational biology 2008; 4
4. Sootanan P, Prom-on S, Meechai A, and Chan JH. Gene set-based microarray analysis for robust disease classification, Neural Computing and Applications 2013; 21: 649-660.
5. Edgar R, Domrachev M, and Lash AE. Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acid Res 2002.
6. Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Elbert BL, Gillete MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, and Mesirov JP. Geneset enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. PNAs 2005; 102: 15545-15550.
7. Hall M, Frank E, Holmes G, Pfahringer B, Reutemann P, and Witten IH. The WEKA Data mining software: An update. SIGKDD Exploration 2009; 11.
8. Chan JH, Sootanan P, and Larpeampaisarl P. Feature selection of gene set markers for microarray-based disease classification using negatively correlated feature set. IJCNN 2011; 3293-3299.
9. Van Dyke AL, Cote ML, Wenzlaff AS, Chen W, Abrams J, Land S, Giroux CN, and Schwatz AG. Cytokine and cytokine receptor single-nucleotide polymorphisms predict risk for non-small cell lung cancer among women. Cancer Epidemol Biomarker Prev 2009; 18: 1829-40.
10. Bellei B, Pitisci A, Izzo E, and Picardo M. Inhibition of Melanogenesis by the Pyridinyl Imidazole Class of Compounds: Possible Involvement of the Wnt/ß-Catenin Signaling Gene set. PLoS ONE 2012; 7: e33021.
11. Mazieres J, He B, You L, Xu Z, and Jablons DM. Wnt signaling in lung cancer. Cancer Lett. 2005; 222: 1-10.
12. Devaraj S, Natarajan J. miRNA-mRNA network detects hub mRNAs and cancer specific miRNAs in lung cancer. In Silico Biol. 2011; 11: 281-95.
13. Hanai J, Doro N, Sasaki AT, Kobayashi S, Cantley LC, Seth P, and Sukhatme VP. Inhibition of lung cancer growth: ATP citrate lyase knockdown and statin treatment leads to dual blockade of mitogen-activated protein kinase (MAPK) and phosphatidylinositol-3-kinase (PI3K)/AKT gene sets. J Cell Physiol. 2012; 227: 1709-20.
14. Tsurutani J, West KA, Sayvah J, Gills JJ, and Dennis PA. Inhibition of the phosphatidylinositol 3-kinase/Akt/mammalian target of rapamycin gene set but not the MEK/ERK gene set attenuates laminin-mediated small cell lung cancer cellular survival and resistance to imatinib mesylate or chemotherapy. Cancer Res. 2005; 65: 8423-32.
15. Yang H, Zhang Q, He J, and Lu W. Regulation of calcium signaling in lung cancer. J Thorac Dis. 2010; 2: 52-56.
16. Lu Y, Wang L, Liu P, Yang P, and You M. Gene-expression signature predicts postoperative recurrence in stage I non-small cell lung cancer patients. PLoS ONE 2012; 7: e30880.
17. Pelosi G, Spaggiari L, Mazzarol G, Viale G, Pece S, and Di Fiore PP. Alterations of the Notch gene set in lung cancer. Proc Natl Acad Sci USA. 2009; 106: 22293-9.