0095-1137/08/$08.00⫹0 doi:10.1128/JCM.00464-08
Copyright © 2008, American Society for Microbiology. All Rights Reserved.
Evaluation of a Multilocus Variable-Number Tandem-Repeat Analysis
Scheme for Typing Human
Brucella
Isolates in a Region of
Brucellosis Endemicity
䌤
†
Mireille M. Kattar,
1,2* Rola F. Jaafar,
3George F. Araj,
1,2Philippe Le Fle
`che,
4,5Ghassan M. Matar,
3Roland Abi Rached,
1,2Simon Khalife,
1,2and Gilles Vergnaud
4,6*
Department of Pathology and Laboratory Medicine1and Department of Microbiology and Immunology,3American University of
Beirut, Beirut, Lebanon; World Health Organization Regional Collaborating Center on Human Brucellosis, Beirut, Lebanon2;
Universite´ Paris-Sud 11, CNRS, UMR8621, Institut de Ge´ne´tique et Microbiologie, Orsay 91405, France4; Department of
Analytical Microbiology, Centre d’Etudes du Bouchet, Vert le Petit 91710, France5; and DGA/D4S-Mission pour la
Recherche et l’Innovation Scientifique, Bagneux 92220, France6
Received 9 March 2008/Returned for modification 6 July 2008/Accepted 30 September 2008
Brucellosis remains an important anthropozoonosis worldwide. Brucellaspecies are genetically
homoge-neous, and thus, the typing ofBrucellaspecies for epidemiological purposes by conventional molecular typing
methods has remained elusive. Although many methods could segregate isolates into the phylogenetically recognized taxa, limited within-species genetic diversity has been identified. Recently, multilocus variable-number tandem-repeat analysis (MLVA) was found to have a high degree of resolution when it was applied to
collections ofBrucellaisolates from geographically widespread locations, and an assay comprising 16 such loci
(MLVA-16) was proposed. This scheme includes eight minisatellite loci (panel 1) and eight microsatellites (panel 2, which is subdivided into panels 2A and 2B). The utility of MLVA-16 for the subtyping of human
Brucellaisolates from geographically restricted regions needs to be further evaluated, and genotyping data-bases with worldwide coverage must be progressively established. In the present study, MLVA-16 was applied
to the typing of 42 human Brucella isolates obtained from 41 patients recovered from 2002 to 2006 at a
tertiary-care center in Lebanon. All isolates were identified asBrucella melitensisby MLVA-16 and were found
to be closely related to B. melitensis isolates from neighboring countries in the Middle East when their
genotypes were queried against those in the web-based Brucella2007 MLVA database (http://mlva.u-psud.fr/). Panel 2B, which comprised the most variable loci, displayed a very high discriminatory power, while panels 1 and 2A showed limited diversity. The most frequent genotype comprised seven isolates obtained over 7 weeks in 2002, demonstrating an outbreak from a common source. Two isolates obtained from one patient 5 months apart comprised another genotype, indicating relapsing disease. These findings confirm that MLVA-16 has a
good discriminatory power for species determination, typing of B. melitensis isolates, and inferring their
geographical origin. Abbreviated panel 2B could be used as a short-term epidemiological tool in a small region of endemicity.
Brucellae are facultative intracellular pathogens that infect a wide variety of animal species and humans. Brucellosis is the most common anthropozoonosis, with more than 500,000 cases reported annually worldwide (28). The genus Brucella cur-rently encompasses nine recognized species (seven terrestrial and two marine mammal species) that display animal host specificity, among which three present veterinary and public health concerns (11, 27, 31).Brucella melitensispredominantly infects sheep and goats,B. abortusinfects cattle, andB. suis
infects swine and a range of wild animals; but cross-infection of
other mammalian species, including humans, may occur (10). Animal brucellosis causes abortion and infertility in livestock (cattle, goats, and sheep), resulting in serious economic losses. Human brucellosis is a subacute or chronic febrile illness that can involve multiple organs and that can result in a wide variety of manifestations and significant morbidity if the diagnosis is overlooked and treatment is not promptly initiated (5). Animal brucellosis has been successfully eradicated in most developed countries, resulting in the virtual disappearance of the human disease in North America, Northern Europe, and Northwest Asia, where most cases are now due to either travel to areas of endemicity, accidental laboratory exposure, or occasionally, exposure to wild animals (10). However, human brucellosis remains endemic and a major public health problem in many developing countries and some developed countries in Latin America, Southern Europe, Africa, Southeast Asia, and the Middle East (28). The most common cause of human brucel-losis isB. melitensis, withB. abortusandB. suisaccounting for smaller proportions of cases (19, 28).Brucellaspp. are consid-ered potential military, agricultural, and civilian category B biological threat agents (30) due to their relative ease of
dis-* Corresponding author. Mailing address for Mireille M. Kattar: Department of Pathology and Laboratory Medicine, American Uni-versity of Beirut Medical Center, Cairo Street, Beirut, Lebanon. Phone: 961-1-374374, ext. 5175. Fax: 961-1-370845. E-mail: [email protected]. Mailing address for Gilles Vergnaud: Universite´ Paris-Sud 11, CNRS, UMR8621, Institut de Ge´ne´tique et Microbiologie, Orsay 91405, France. Phone: 33-1-69156208. Fax: 33-1 -69156678. E-mail: [email protected].
† Supplemental material for this article may be found at http://jcm .asm.org/.
䌤Published ahead of print on 15 October 2008.
3935
on May 16, 2020 by guest
http://jcm.asm.org/
semination and costly eradication if they were spread in a possible bioterrorism event.
Species identification and subtyping ofBrucellaculture iso-lates is important for epidemiologic surveillance; investigation of outbreaks in regions of both endemicity and nonendemicity; and distinguishing cases of human reinfection from relapse, thereby influencing clinical therapeutic decisions (3).
Terrestrial Brucella spp. are homogeneous and harbor ⬎80% interspecies homology by DNA-DNA hybridization studies (31, 38), identical 16S rRNA sequences (15), and ⬎98% sequence similarity by comparative genomics (18, 29). Molecular methods commonly used for the subtyping of iso-lates of other bacterial species, such as multilocus enzyme electrophoresis (12), pulsed-field gel electrophoresis (4), ran-dom amplified polymorphic DNA analysis (34), enterobacte-rial repetitive intergenic consensus sequence PCR (26, 35), repetitive intergenic palindromic sequence PCR (25), ampli-fied fragment length polymorphism analysis (42), and mono-locus (such as omp2a and omp2b) sequence analysis (9) or multilocus sequence typing (40), are able to segregateBrucella
isolates into the recognized species and certain species biovars at best. However, these methods are not sufficiently discrimi-natory for the routine subtyping of isolates for epidemiological trace-back purposes.
Tandemly repeated sequences used in forensic investiga-tions have also been found in bacteria (for a review, see reference 39). Minisatellites have repeat unit sizes of 9 bp or greater, and microsatellites have repeat unit sizes of up to 8 bp (39). Combinations of minisatellite and microsatellite repeats in multilocus variable-number tandem-repeat anal-ysis (MLVA) have proven highly discriminatory in the epide-miological subtyping of isolates belonging to monomorphic bacterial species, such asBacillus anthracis,Yersinia pestis(8, 23), and more recently, Brucella spp. In Brucella, MLVA schemes with 21 loci (MLVA-21) and MLVA-16 (1, 22, 41) that use a combination of repeat markers distributed across the
Brucellagenome were able to distinguish isolates of Brucella
spp. of widespread temporal and geographical origins or of very close origins (13). Furthermore, with some rare excep-tions, which might be due to a lack of resolution and incorrect clustering produced by the biotyping methods, the isolates formed MLVA groups that corresponded to the known Bru-cellaspecies. A study of 128 human isolates ofB. melitensisby MLVA-16 identified the Americas, West Mediterranean, and East Mediterranean groups and detected remarkable degrees of diversity within each group (1). The relative stability of MLVA loci has been estimated directly by repetitive subcul-turing of a few strains in the laboratory (41) and by the typing ofB. melitensis isolates of the Rev.1 vaccine strain obtained from different sources (13), so that the most recent MLVA data analyses have started to assign different weights to differ-ent markers, taking into account the mutation rate and the associated levels of homoplasy (1). The MLVA schemes that have been devised are very promising, but the strength of a typing method depends as much on the technique itself as on the associated genotyping database. These typing assays still need to be applied to larger numbers of isolates from as many countries as possible to validate their reliability in investigating strain relatedness both in countries of endemicity, especially where efforts to eradicate human disease may be attempted,
and in brucellosis-free countries where a cluster of infections may be suspected to be due to a bioterrorism event. The primary purpose of high-resolution typing in a format compat-ible with the production of large-scale international databases is the long-term surveillance of infectious diseases and the early detection of abnormal events, including temporal or geo-graphical shifts in the population structure, e.g., the emergence and spread of new clones or lineages. The current Brucel-la2007 MLVA database, hosted at http://mlva.u-psud.fr, con-tains data derived from more than 500 animal and human
Brucellaisolates. It is expected that in the future, similar and compatible databases with data for isolates from wider geo-graphical origins will be developed and made jointly and freely accessible to querying via the use of web-based analysis tools (16).
In this study, we applied the MLVA-16 scheme (1, 22) to a series of clinical isolates of Brucella accrued over a 5-year period at a single tertiary-care center in Lebanon, a small country where human brucellosis is endemic. The genotypes of these isolates were compared to the genotypes previously de-termined by MLVA-16 typing and are included in the Bru-cella2007 MLVA database.
(This study was presented in part at the 107th General Meeting of the American Society for Microbiology, Toronto, Ontario, Canada, 2007.)
MATERIALS AND METHODS
Bacterial strains.Forty-two clinicalBrucellaisolates (isolates AUB-BRUP-S1 to AUB-BRUP-S42 [isolates S1 to S42, respectively]) obtained from clinical specimens from 41 different patients submitted to the clinical microbiology laboratory at the American University of Beirut Medical Center from February
2002 to December 2006 were investigated. These represented allBrucellaisolates
collected during that time period. Thirty-nine of the isolates were isolated from blood, and three were from tissue specimens. Two of the 39 blood isolates were obtained from cultures of blood from one patient 5 months apart. All isolates
were identified asBrucellaspecies on the basis of colonial morphology, positive
oxidase and catalase tests, positive agglutination with brucella-specific antiserum, and positive amplification by real-time PCR, as described previously (21). In addition, all patients from whom these isolates were obtained had positive
Brucellaserology titers by Wright’s standard tube agglutination and the anti-human globulin Coombs test (5).
BrucellaMLVA-16 genotyping scheme.MLVA was performed with all 42
isolates according to the scheme initially proposed by Le Fle`che et al. (22), which
includes 15 tandem-repeat loci (MLVA-15), and modified by Al-Dahouk et al. to include 1 additional locus, bruce19 (MLVA-16) (1). PCR amplification of eight minisatellite loci in panel 1 (the bruce06, bruce08, bruce11, bruce12, bruce42, bruce43, bruce45, and bruce55 loci), three microsatellite loci in panel 2A (the bruce18, bruce19, and bruce21 loci), and five microsatellite loci in panel 2B (the bruce04, bruce07, bruce09, bruce16, and bruce30 loci) was carried out by a protocol similar to the one described previously (1).
DNA preparation.DNA was extracted from one loopful of bacterial cells grown for 48 h on chocolate agar, and single colonies were isolated by using the tissue protocol of the QIAamp DNA minikit (Qiagen, Hilden, Germany). DNA concentrations were measured by UV spectrophotometry (Shimadzu, Japan).
PCR amplification.PCR amplification was performed in a total volume of 15
l containing 5 ng of DNA, 1⫻PCR buffer, 1 U ofTaqDNA polymerase, 200
M of each deoxynucleoside triphosphate, 0.3 mM each flanking primer, and 1
M of betaine (catalog no. B2629; Sigma-Aldrich). The PCR cycling parameters were as follows: initial denaturation at 96°C for 5 min, followed by 30 cycles of denaturation at 96°C for 30 s, primer annealing at 60°C for 30 s, and extension at 70°C for 1 min, with a final extension step at 70°C for 5 min. For detection of
the PCR products, 5l of each of the PCR amplification products was subjected
to gel electrophoresis with a 2% 3:1 HRB agarose gel (catalog no. E776;
Am-resco) for panel 1 and a 3% HRB agarose gel for panel 2 in 0.5⫻
Tris-borate-EDTA buffer. Electrophoresis was carried until the bromophenol blue had run at least 20 cm. The size markers used were a 100-bp ladder (EZ Load 100-bp
on May 16, 2020 by guest
http://jcm.asm.org/
PCR molecular ruler; Bio-Rad) for panel 1 and a 20-bp ladder (EZ Load 20-bp molecular ruler; Bio-Rad) for panel 2. Gels that had been stained with ethidium
bromide (0.5g/ml) were digitally acquired with a gel documentation system
(DigiDoc-It; UVP, Upland, CA).
Analysis of MLVA data.Minisatellite repeat numbers (panel 1 [loci bruce06, bruce08, bruce11, bruce12, bruce42, bruce43, bruce45, and bruce55]) and mic-rosatellite repeat numbers (panel 2A [loci bruce18, bruce19, and bruce21] and panel 2B [loci bruce04, bruce07, bruce09, bruce16, and bruce30) were calculated on the basis of the band sizes after the gel images were normalized by use of the Bionumerics (version 5.1) software package (Applied Maths, Belgium) and the previously published allele numbering convention (22). The reproducibilities of the MLVA profiles were assessed by comparing the typing results for experi-ments performed independently at two laboratory testing sites in Lebanon and France. The similarity matrix for each panel was calculated with Bionumerics software by using the repeat number at each locus as a character type with the categorical similarity coefficient. This categorical approach considers all alleles at each locus and within each panel to be equally distant (37). For MLVA-16 clustering analysis, an aggregated similarity matrix was produced by using a composite data set and giving weights of 20, 5, and 1 to panels 1, 2A, and 2B, respectively, according to the principles reported previously (1). Weights were assigned to the individual loci in each panel because all loci do not evolve at the same speed but have different mutation rates, as determined empirically. Clus-tering was done by use of the unweighted paired group method for arithmetic averages algorithm. Genetic diversity was determined by calculating the Hunter-Gaston (or Simpson’s) diversity index (HGDI) of each MLVA marker and for the combination of markers for each of panel 1, panel 2A, panel 2B, panel 2 (panel 2A plus panel 2B), and panels 1 and 2 (20). The 95% confidence intervals
of the HGDIs for indices of⬎0.85 were calculated by the method of Grundmann
et al. (17). The MLVA-16 genotypes, defined by the combination of alleles, of the 42 isolates were compared to the corresponding data obtained for the reference strains and field isolates investigated previously (1, 13, 14, 22, 31, 32, 33). The present and previous data have been incorporated into a single database designated Brucella2007, which can be queried at http://mlva.u-psud.fr by the use of web-based analysis tools.
RESULTS AND DISCUSSION
The purpose of this study was to evaluate the polymorphisms of the MLVA-16 loci in a series ofBrucella isolates and the relatedness of the strains from a geographically restricted re-gion where Brucella is endemic. Furthermore, we sought to validate the established Brucella2007 genotyping database as-sociated with MLVA-16 as a reliable investigation tool in this context. In the present study, the MLVA-16 scheme was ap-plied to type 42 human isolates ofBrucella obtained over a 5-year period at the largest tertiary-care center in Lebanon. Lebanon is a small country in the eastern Mediterranean re-gion with a geographical surface area of 10,452 km2, an
esti-mated population of 4,000,000, and a reported incidence of brucellosis of 50 per 1 million inhabitants (28).
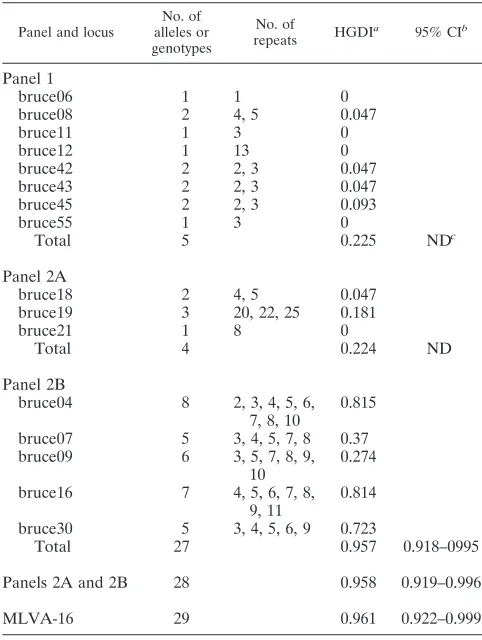
The typeability of the various MLVA markers in this series approached 1 (37). Only the bruce04 locus in strain S29 and the bruce09 locus in strain S19 could not be amplified, despite multiple attempts. All other amplification reactions were suc-cessful. The reproducibility of MLVA-16 was excellent, as ex-periments performed in two different laboratories in Lebanon and France by use of the same protocol and with the results interpreted separately in a blinded fashion yielded 100% con-cordant results. This is not surprising for a sequence-based approach and was recently confirmed by a ring trial for the panel 1 loci among 15Brucella reference laboratories (http: //mlva.u-psud.fr/BRUCELLA/). In that trial, the few discrep-ancies in typing results observed were due to errors in allele calling (24a). The 42 isolates in the series used in this study formed a homogeneous group for which the differences did not exceed 15% (Fig. 1). Limited diversity was observed with the
panel 1 and panel 2A loci (Table 1), as would be expected for strains originating from a very restricted geographical area and by the use of markers that are the most phylogenetically infor-mative. MLVA-16 yielded a total of 29 genotypes with seven clusters and 22 singleton genotypes. Previous work identified a total of 68, 52, and more than 300 genotypes for panel 1, panel 2A, and panel 2B, respectively (1, 13, 14, 22, 31, 32, 33) (see Fig. S1 in the supplemental material). Twenty-six panel 1 ge-notypes, numbered 41 to 66 (1, 22), corresponded to B. melitensis. No new panel 1 genotypes were identified in the present study. Thirty-seven isolates shared panel 1 genotype 43. Panel 1 genotypes 42, 44, and 57 were observed in a single isolate each; and genotype 60 was observed in isolates S18 and S24, which were derived from the same patient. Therefore, only 5 different panel 1 genotypes were observed among the 26 panel 1B. melitensisgenotypes observed so far. Accordingly, the bruce06, bruce11, bruce12, and bruce55 loci from panel 1 and the bruce21 locus from panel 2 showed no diversity among the 42 isolates (HGDI⫽0). All five panel 1 genotypes repre-sented in the present study correspond to the East Mediterra-nean group proposed previously (1) (see Fig. S1 in the supple-mental material). The panel 2B markers displayed the most diversity in the present collection. This was as expected, given the high mutation rate of some panel 2B markers (13), with bruce04 and bruce16 exhibiting the highest HGDIs, followed by bruce30 (HGDIs⬎0.7). Bruce07 and bruce09 in panel 2B had HGDIs of⬍0.5. Panel 2B and panels 2A and 2B yielded 27 and 28 genotypes, respectively, and had similar HGDIs. The combined panel 1 and 2 markers yielded 29 genotypes and an HGDI that was only slightly higher than that for panel 2B alone. The HGDIs of panel 2B, panels 2A and 2B, and panels 1 and 2 were all⬎0.95, which is considered optimal for a typing system (37). These data might indicate that in the setting of a local outbreak investigation, panel 1 and panel 2A could be omitted and the highly polymorphic panel 2B would be suffi-cient. However, typing with panel 1 and 2A is necessary before typing with panel 2B is used in order to check that the isolate is indeed of the expected local type, as it is important to keep in mind that the HGDI value is not a sufficient indicator for the selection of tandem repeat loci and validation of an MLVA assay. Three of the five panel 2B loci (bruce04, bruce09, bruce30) are part of the hypervariable octameric oligonucleo-tide fingerprint variable-number tandem repeats (HOOF-Prints) MLVA panel of loci that have been described by Bricker et al. (6, 7) and that have proven to be highly mutable in terrestrialBrucella spp. (6, 7, 36, 41). The most frequent genotype (largest cluster) comprised seven isolates (isolates S3 to S9) obtained over 7 weeks in the late spring and early summer of 2002, demonstrating an outbreak from a common source. Another genotype was shared by isolates S18 and S24, obtained 5 months apart from one patient who had no at-risk occupational history and who experienced a recurrence of his febrile illness. The two isolates from this patient differed from the rest of the isolates at the bruce45 minisatellite locus from panel 1 (panel 1 genotype 60, two versus three repeats), indi-cating relapsing disease in this patient. Two isolates obtained 2 months apart (isolates S37 and S41) and four pairs obtained over 1 year apart (isolates S25 and S30, S12 and S39, S2 and S23, and S28 and S42) had identical genotypes. These five seemingly unrelated pairs may represent either
on May 16, 2020 by guest
http://jcm.asm.org/
cally unrelated isolates with homoplasy at MLVA-16 loci (most likely panel 2B) or persistent circulating strains causing spo-radic infections. All other isolates had distinct genotypes re-flecting sporadic cases. In a previous study (1), two pairs of epidemiologically related humanB. melitensisisolates in Ger-many displayed identical MLVA-16 profiles. One pair of iso-lates was obtained from one patient: one isolate was obtained before treatment for brucellosis and one isolate was obtained 3 months later when he experienced a relapse related to inad-equate therapy. Another pair of isolates came from a married couple who visited Turkey, where they contracted their infec-tion from the consumpinfec-tion of unpasteurized cheese. Three isolates from a small outbreak ofB. suisinfections in pigs in Spain also shared the same MLVA-16 genotype (14). The clustering of epidemiologically related isolates identified in the current and previous studies support the use of MLVA-16 as a valuable tool for investigations of outbreaks of both human and animal brucellosis.
In comparison to previous data and by the use of all 16 loci,
23 of the 29 genotypes obtained were unique (see Fig. S1 in the supplemental material). Eight isolates had genotypes identical to those of strains from Syria (n⫽2), Turkey (n⫽2), Israel (n⫽
1), Lebanon (n ⫽ 1), and Germany (n ⫽ 2). The last two isolates probably originated from Turkey (2). Thirty-one (31) isolates were single-locus or double-locus variants of closely relatedB. melitensis isolates from neighboring countries, in-cluding Syria, Israel, and Turkey (see Fig. S1 in the supple-mental material). The unique fingerprinting profiles for most of theB. melitensisisolates in this series and their close relat-edness by MLVA compared to the relatrelat-edness of other human
B. melitensisisolates (1) probably reflect microevolution due to few mutational events among indigenous strains derived from a common ancestor rather than the introduction of foreign strains from other areas, since most of these variations were in panel 2B loci.
[image:4.585.46.535.68.428.2]Although phenotypic biovar typing was not performed in this study, previous studies have shown that the results of the MLVA-16 or the MLVA-21 typing schemes that have been
FIG. 1. Dendrogram based on the MLVA-16 genotyping assay showing the relationships of the 42 human isolates ofBrucella.Under panels 1, 2A, and 2B are shown the individual MLVA-16 loci and the number of tandem repeats for each isolate. Key, the serial number for the isolate in the Brucella2007 MLVA database (http://mlva-u-psud.fr/) (see Fig. S1 in the supplemental material); strain, the local strain designation; panels 1, 2A, and 2B, genotypes corresponding to each isolate in the database for each set of loci; date isolation, the dates (day/month/year) that samples were received in the laboratory for culturing. The information under comments provides the specimen source-patient gender (M, male; F, female)/patient age (in years).
on May 16, 2020 by guest
http://jcm.asm.org/
devised (1, 41) show no correlation with those of biovar typing forB. melitensis. Our isolates were closely related toB. meliten-sisisolates from various countries that belonged to all three biovars (biovars 1, 2, and 3). Neither MLVA nor multilocus sequence typing distinguished theB. melitensisbiovars, which are mere serotypes (1, 40, 41). This indicates that variable-number tandem-repeat loci and the single-nucleotide polymor-phisms which provide congruent data evolve independently of the putative genetic determinants for these biovars. This is also the case forB. abortus, but to a lesser extent. In contrast, both MLVA-16 and MLVA-21 separateB. suis biovar 2 from the otherB. suisbiovars, biovars 1, 3, and 4 (see Fig. S1 in the supplemental material) (14, 41).
Both MLVA-16 and MLVA-21 segregated Brucellastrains into groups corresponding to the recognized phylogenetic lineages in the genusBrucella(22, 41). When it was applied to more than 500 strains of terrestrialBrucella, panel 1 in MLVA-16 subdivided these isolates into 65 genotypes that were, with one exception, exclusive to each of the Brucella
species (see Fig. S1 in the supplemental material). Similarly, by MLVA-21, six minisatellites were found to be sufficient for species designation (41). In both schemes,B. canisclustered with B. suis biovar 4. These data suggest that MLVA is a generally useful method for primary species assignment of
terrestrial Brucella spp., especially in regions of endemicity whereB. melitensis,B. abortus, andB. suisare the predominant causes of both human and animal brucellosis.
The diversity of the panel 1 loci and most panel 2 loci in our series parallels that found in a study of 24 humanB. melitensis
isolates from Sicily in Italy (24). The two studies are compa-rable in that they focused on a restricted geographic area. Two panel 1 genotypes (genotypes 49 and 51) were observed in the Sicilian study, and both were from the West Mediterranean group (see Fig. S1 in the supplemental material). We found higher levels of diversity for bruce30 (panel 2B) and bruce19 (panel 2A) and a lower level of diversity for bruce08 (panel 1). The Sicilian isolates formed a homogeneous group that could be distinguished from the East Mediterranean group at three of the panel 1 minisatellite loci. These data illustrate the use-fulness of panel 1 not only for the species identification of
Brucella isolates but also for inference of their geographical origins. In contrast, panel 2 markers afford a higher discrimi-natory power for investigation of strain relatedness in regions of endemicity. An abbreviated panel consisting of five micro-satellite loci, panel 2B, can be used as a practical short-term epidemiological tool in this setting, but the species cannot be deduced from the data produced by this panel only. Con-versely, in regions of nonendemicity or whenever the introduc-tion of an exogenous strain might be suspected, all loci should be used to pinpoint the region where the isolates may have originated. For that reason, there is a need to build expanded searchable and shared international databases of MLVA fin-gerprints to enable such comparisons. The currently existing Brucella2007 MLVA database (http://mlva.u-psud.fr) provides one constituent within the framework of such a concerted effort (16).
In summary, MLVA-16 displayed a good discriminatory power for the typing of B. melitensis isolates from a small region of endemicity, and either panel 2 or panels 1 and 2 may be used as epidemiological tools for the resolution of strains and for distinguishing relapses from reinfections in patients with brucellosis.
ACKNOWLEDGMENTS
This study was funded by the University Research Board and Med-ical Practice Plan, American University of Beirut, and by the French De´le´gation Ge´ne´rale pour l’Armement.
REFERENCES
1.Al-Dahouk, S., P. Le Fle`che, K. No¨ckler, I. Jacques, M. Grayon, H. C. Scholz, H. Tomaso, G. Vergnaud, and H. Neubauer.2007. Evaluation ofBrucella
MLVA typing for human brucellosis. J. Microbiol. Methods69:137–145.
2.Al-Dahouk, S., H. Neubauer, A. Hensel, I. Sho¨neberg, K. No¨ckler, K. Alpers, H. Merzenich, K. Stark, and A. Jansen.2007b. Changing epidemiology of
brucellosis, Germany, 1962–2005. Emerg. Infect. Dis.13:1895–1900.
3.Al-Dahouk, S., R. M. Hagen, K. No¨ckler, H. Tomaso, M. Wittig, H. C. Scholz, G. Vergnaud, and H. Neubauer.2005. Failure of a short-term antibiotic therapy for human brucellosis using ciprofloxacin. A study on in vitro
sus-ceptibility ofBrucellastrains. Chemotherapy511:352–356.
4.Allardet-Servent, A., G. Bourg, M. Ramuz, M. Pages, M. Bellis, and G. Roizes.1988. DNA polymorphism in strains of the genusBrucella. J.
Bacte-riol.170:4603–4607.
5.Araj, G. F. 1999. Human brucellosis: a classical infectious disease with
persistent diagnostic challenges. Clin. Lab. Sci.12:207–212.
6.Bricker, B. J., and D. R. Ewalt.2005. Evaluation of HOOF-Print assay for
typingBrucella abortusstrains isolated from cattle in the United States:
results with four performance criteria. BMC Microbiol.5:37.
7.Bricker, B. J., D. R. Ewalt, and S. M. Halling.2003.Brucella‘HOOF-Prints’: strain typing by multi-locus analysis of variable number of tandem repeats
[image:5.585.42.283.89.409.2](VNTRs). BMC Microbiol.3:15.
TABLE 1. Summary of MLVA-16 results for 42 human isolates ofB.melitensis
Panel and locus
No. of alleles or genotypes
No. of
repeats HGDI
a 95% CIb
Panel 1
bruce06 1 1 0
bruce08 2 4, 5 0.047
bruce11 1 3 0
bruce12 1 13 0
bruce42 2 2, 3 0.047
bruce43 2 2, 3 0.047
bruce45 2 2, 3 0.093
bruce55 1 3 0
Total 5 0.225 NDc
Panel 2A
bruce18 2 4, 5 0.047
bruce19 3 20, 22, 25 0.181
bruce21 1 8 0
Total 4 0.224 ND
Panel 2B
bruce04 8 2, 3, 4, 5, 6,
7, 8, 10
0.815
bruce07 5 3, 4, 5, 7, 8 0.37
bruce09 6 3, 5, 7, 8, 9,
10
0.274
bruce16 7 4, 5, 6, 7, 8,
9, 11
0.814
bruce30 5 3, 4, 5, 6, 9 0.723
Total 27 0.957 0.918–0995
Panels 2A and 2B 28 0.958 0.919–0.996
MLVA-16 29 0.961 0.922–0.999
aHGDI, Hunter-Gaston diversity index.
bCI, confidence interval.
cND, not done.
on May 16, 2020 by guest
http://jcm.asm.org/
8.Ciammaruconi, A., S. Grassi, R. De Santis, G. Faggioni, V. Pittiglio, R. D’Amelio, A. Carattoli, A. Cassone, G. Vergnaud, and F. Lista.2008.
Field-able genotyping ofBacillus anthracisandYersinia pestisbased on 25-loci
multi locus VNTR analysis. BMC Microbiol.8:21.
9.Cloeckert, A., N. Vizcaino, J.-Y. Paquet, R. A. Bowden, and P. H. Elzer.2002.
Major outer membrane proteins ofBrucellaspp.: past, present and future.
Vet. Microbiol.90:229–247.
10.Corbel, M. J.1997. Brucellosis: an overview. Emerg. Infect. Dis.3:213–221. 11.Foster, G., B. S. Osterman, J. Godfroid, I. Jacques, and A. Cloeckaert.2007.
Brucella cetisp. nov., andBrucella pinnipedialissp. nov. forBrucellastrains with cetaceans and seals as their preferred hosts. Int. J. Syst. Evol Microbiol.
57:2688–2693.
12.Gandara, B. A., L. Merino, and M. A. Rogel, and E. Martinez-Romero.2001.
Limited genetic diversity ofBrucellaspp. J. Clin. Microbiol.39:235–240.
13.García-Yoldi, D., P. Le Fle`che, C. M. Marín, M. J. de Miguel, P. M. Mun˜oz, G. Vergnaud, and I. Lo´pez-Gon˜i.2007. Assessment of genetic stability of
Brucella melitensisRev. 1 vaccine strain by multiple-locus variable number of
tandem repeat analysis. Vaccine25:2858–2862.
14.García-Yoldi, D., P. Le Fle`che, M. De Miguel, P. M. Mun˜oz, J. M. Blasco, Z. Cvetnik, C. M. Marín, G. Vergnaud, and I. Lo´pez-Gon˜i.2007. Comparison of multiple-locus variable-number tandem-repeat analysis with other
PCR-based methods for typingBrucella suisisolates. J. Clin. Microbiol.45:4070–
4072.
15.Gee, E. J., B. K. De, P. N. Levett, A. M. Whitney, R. T. Novak, and T. Popovic.
2004. Use of 16S rRNA gene sequencing for rapid confirmatory
identifica-tion ofBrucellaisolates. J. Clin. Microbiol.42:3649–3654.
16.Grissa, I., P. Bouchon, C. Pourcel, and G. Vergnaud.2007. On-line resources for bacterial micro-evolution studies using MLVA or CRISPR typing.
Bio-chimie90:660–668.
17.Grundmann, H., H. Satoshi, and G. Tanner.2001. Determining confidence intervals when measuring genetic diversity and the discriminatory abilities of
typing methods for microorganisms. J. Clin. Microbiol.39:4190–4192.
18.Halling, S. M., B. D. Peterson-Burch, B. J. Bricker, R. L. Zuerner, Z. Qing, L.-L. Li, V. Kapur, D. P. Alt, and S. C. Olsen.2005. Completion of the
genome sequence ofBrucella abortusand comparison to the highly similar
genomes ofBrucella melitensisandBrucella suis. J. Bacteriol.187:2715–2726.
19.Hubbard, D. J., R. K. Porschen, and K. Van Horn.2007. Evaluation of dye
tolerance media for the identification ofBrucellaspecies, abstr. C-170. Abstr.
107th Gen. Meet. Am. Soc. Microbiol. American Society for Microbiology, Washington, DC.
20.Hunter, P. R., and M. A. Gaston.1988. Numerical index of the discrimina-tory ability of typing systems: an application of Simpson’s index of diversity
J. Clin. Microbiol.26:2465–2466.
21.Kattar, M. M., P. Zalloua, G. F. Araj, J. Samaha-Kfoury, H. Shabklo, S. Khalife, S. S. Kanj, and M. Deeb.2007. Development and evaluation of real-time PCR assays for rapid diagnosis of human brucellosis. Diagn.
Microbiol. Infect. Dis.59:23–32.
22.Le Fle`che, P., I. Jacques, M. Grayon, A. Al-Dahouk, P. Bouchon, F. Denoeud, K. No¨ckler, H. Neubauer, L. A. Guilloteau, and G. Vergnaud.2006.
Evalu-ation and selection of tandem repeat loci for aBrucellaMLVA typing assay.
BMC Microbiol.143:2913–2921.
23.Lista, F., G. Faggioni, S. Valjevac, A. Ciammaruconi, J. Vaissaire, C. le Doujet, O. Gorge´, R. De Santis, A. Carattoli, A. Ciervo, A. Fasanella, F. Orsini, R. D’Amelio, C. Pourcel, A. Cassone, and G. Vergnaud.2006.
Geno-typing of Bacillus anthracisstrains based on automated capillary 25-loci
multiple locus variable-number tandem repeats analysis. BMC Microbiol.
6:33.
24.Marianelli, C., C. Graziani, C. Santangelo, M. T. Xibilia, A. Imbriani, R. Amato, D. Neri, M. Cuccia, S. Rinnone, V. Di Marco, and F. Ciuchini.2007. Molecular epidemiologic and antimicrobial susceptibility characterization of
Brucellaisolates from humans in Sicily, Italy. J. Clin. Microbiol.45:2923– 2928.
24a.Melzer, F., G. Vergnaud, and A. Whatmore.2008. The results of the first
international ring trial of MLVABrucellatyping. Abstr. Int. Brucellosis Res.
Conf., London, United Kingdom, abstr. 035.
25.Mercier, E. E. Jumas-Bilak, A. Allardet-Servent, D. O’Callaghan, and M.
Ramuz.1996. Polymorphism inBrucellastrains detected by studying
distri-bution of two short repetitive DNA elements. J. Clin. Microbiol.34:1299–
1302.
26.Michaux-Charachon, S., G. Bourg, E. Jumas-Bilak, A. Guigue-Talet, P. Guigue-Talet, A. Allardet-Servent, D. O’Callaghan, and M. Ramuz.1997.
Genome structure and phylogeny in the genusBrucella. J. Bacteriol.179:
3244–3249.
27.Moreno, E., A. Cloeckaert, and I. Moriyo´n.2002.Brucellaevolution and
taxonomy. Vet. Microbiol.90:209–227.
28.Pappas, G., P. Papadimitriou, N. Akritidis, L. Christou, and E. V. Tsianos.
2006. The new global map of human brucellosis. Lancet Infect. Dis.6:91–99.
29.Ratushna, V. G., D. M. Sturgill, S. Ramamoorthy, S. A. Reichow, Y. He, R. Lathigra, N. Sriranganathan, S. M. Halling, S. M. Boyle, and C. J. Gibas.
2006. Molecular targets for rapid identification ofBrucellaspp. BMC
Mi-crobiol.6:13.
30.Roth, L. D., A. S. Khan, S. R. Lillibridge, S. M. Ostroff, and J. M. Hughes.
2002. Public health assessment of potential biological terrorism agents.
Emerg. Infect. Dis.8:225–230.
31.Scholz, H. C., Z. Hubalek, I. Sedlacek, G. Vergnaud, H. Tomaso, S. Al-Dahouk, F. Melzer, P. Ka¨mpfer, H. Neubauer, A. Cloeckert, M. Maquart, M. S. Zygmunt, A. M. Whatmore, E. Falsen, P. Bahn, C. Go¨llner, M. Pfeffer, B. Huber, H.-J. Basse, and K. No¨ckler.2008.Brucella microtisp. nov. isolated
from the common voleMicrotus arvalis. Int. J. Syst. Evol. Microbiol.58:375–
382.
32.Scholz, H. C., Z. Hubalek, J. Nesvadbova, H. Tomaso, G. Vergnaud, P. Le Fle`che, A. M. Whatmore, S. Al Dahouk, M. Kru¨ger, C. Lodri, and M. Pfeffer.
2008. Isolation ofBrucella microtifrom soil. Emerg. Infect. Dis.14:1316–
1317.
33.Scholz, H. C., E. Hofer, G. Vergnaud, P. Le Fleche, A. Whatmore, S. Al Dahouk, M. Pfeffer, M. Kru¨ger, A. Cloeckaert, and H. Tomaso.30 October
2008, posting date. Isolation ofBrucella microtifrom mandibular lymph
nodes of red foxes,Vulpes vulpes, in Lower Austria. Vector Borne Zoonotic
Dis. [Epub ahead of print.]
34.Tcherneva, E., N. Rijpens, B. Jersek, and L. M. F. Herman.2000.
Differen-tiation ofBrucellaspecies by random amplified polymorphic DNA analysis.
J. Appl. Microbiol.88:69–80.
35.Tcherneva, E., N. Rijpens, C. Naydensky, and L. M. F. Herman.1996. Repetitive element sequence based polymerase chain reaction for typing of
Brucellastrains. Vet. Microbiol.51:169–178.
36.Valdezate, S., I. Cervera, P. Hernandez, A. Navarro, and J. A. Sae´z Nieto.
2007. Characterization of human outbreaks of brucellosis and sporadic cases by the use of hyper-variable octameric oligonucleotide fingerprinting
(HOOF) variable number repeats. Clin. Microbiol. Infect.13:887–892.
37.Van Belkum, A., P. T. Tassios, L. Dijkshoorn, S. Haeggman, B. Cookson, N. K. Fry, V. Fussing, J. Green, E. Feil, P. Gerner-Smidt, S. Brisse, and M. Struelens.2007. Guidelines for validation and application of typing methods
for use in bacterial epidemiology. Clin. Microbiol. Infect.13(Suppl. 3):1–46.
38.Verger, J. M., F. Grimont, P. A. D. Grimont, and M. Grayon.1985.Brucella, a monospecific genus as shown by deoxyribonucleic acid hybridization. Int. J.
Syst. Bacteriol.35:292–295.
39.Vergnaud, G., and C. Pourcel.2006. Multiple locus VNTR (variable number
of tandem repeat) analysis (MLVA), p. 83–104.InE. Stackebrandt (ed.),
Molecular identification, systematics and population structure of pro-karyotes. Springer-Verlag, Berlin, Germany.
40.Whatmore, A. M., L. L. Perrett, and A. P. MacMillan.2007. Characterization
of the genetic diversity ofBrucellaby multilocus sequencing. BMC
Micro-biol.7:34.
41.Whatmore, A. M., S. J. Shankster, L. L. Perrett, T. J. Murphy, S. D. Brew, R. E. Thirlwall, S. J. Cutler, and A. P. MacMillan.2006. Identification and
characterization of variable-number tandem-repeat markers for typing
Bru-cellaspp. J. Clin. Microbiol.44:1982–1993.
42.Whatmore, A. M., T. J. Murphy, S. J. Shankster, E. Young, S. Cutler, and A. P. Macmillan.2005. Use of amplified fragment length polymorphism to
identify and typeBrucellaisolates of medical and veterinary interest. J. Clin.
Microbiol.43:761–769.