www.impactjournals.com/oncotarget/ Oncotarget, Vol. 6, No. 17
Ccdc6 knock-in mice develop thyroid hyperplasia associated to
an enhanced CREB1 activity
Vincenza Leone1, Concetta Langella1, Francesco Esposito1, Claudio Arra2, Giuseppe Palma2, Domenica Rea2, Orlando Paciello3, Francesco Merolla1, Davide De Biase3, Serenella Papparella3, Angela Celetti1, Alfredo Fusco1,4
1 Istituto per l’ Endocrinologia ed Oncologia Sperimentale del CNR e/o Dipartimento di Medicina Molecolare e Biotecnologie
Mediche, Università degli Studi di Napoli “Federico II”, Naples, Italy
2Istituto Nazionale per lo Studio e la Cura dei Tumori “Fondazione Giovanni Pascale”, IRCCS, Naples, Italy
3Department of Pathology and Animal Health, University of Naples “Federico II”, Naples, Italy
4Instituto Nacional de Câncer - INCA, Rua André Cavalcanti, Rio de Janeiro, CEP RJ, Brazil
Correspondence to:
Alfredo Fusco, e-mail: [email protected]
Keywords: Ccdc6, thyroid, CREB1, knock-in mice
Received: February 03, 2015 Accepted: April 14, 2015 Published: April 27, 2015
ABSTRACT
CCDC6 was originally identified upon rearrangement with RET in human thyroid papillary carcinomas generating the RET/PTC1 oncogene. We have previously reported
that CCDC6 interacts with CREB1 and represses its transcriptional activity.
Since the function of at least one allele of CCDC6 is lost following RET/PTC1
rearrangements, we aimed at the generation of mice, carrying a CCDC6 mutant gene. Previous studies suggested that the coiled-coil domain of CCDC6, mainly encoded by
human exon 2, is required for the protein function. Therefore, we engineered a murine
Ccdc6 construct, carrying a deletion of the exon 2, that was able to exert only a mild repression on CREB1 transcriptional activity, with respect to the wild type Ccdc6. Subsequently, we generated Ccdc6-ex2 knock-in mice. These mice developed thyroid hyperplasia associated with an enhanced CREB1 activity and an increased expression of the CREB-1 regulated genes.
These results strongly support a CCDC6 promoting role, ascribed to its
functional impairment, in the development of thyroid papillary carcinomas harboring
the RET/PTC1 oncogene.
INTRODUCTION
CCDC6 was identified upon rearrangement with RET in papillary thyroid carcinomas (PTC) generating the RET/PTC1 oncogene detectable in about 20% of PTCs [1]. This rearrangement has been recently identified also in lung adenocarcinomas even though at a much lower frequency (less than 5%) [2]. Moreover, CCDC6 gene rearrangements with genes other than RET have been reported in solid and not solid tumours [3–5] suggesting that CCDC6 gene has a high susceptibility for recombination. CCDC6 gene product is an ubiquitously expressed 65 kDa nuclear and cytosolic protein, phosphorylated by extracellular signal-regulated protein kinase following serum stimulation with no signifi-cant homology to known genes [6, 7]. Recent studies
These results support the hypothesis that even the impairment of one CCDC6 allele, following RET/PTC1 rearrangement, could contribute to papillary thyroid carcinogenesis enhancing thyroid cell proliferation through the activation of the CREB1 transcriptional activity.
In order to confirm our hypothesis we planned to generate mice carrying a mutant Ccdc6 gene. It has been demonstrated that the fragment of 60 amino acid of the CCDC6 coiled-coil domain included in the RET/PTC1 product is necessary for homo-dimerization, constitutive activation and transforming ability of the oncoprotein. Then, we hypothesized that the CCDC6 coiled-coil domain, mainly encoded by exon 2, is required for the function of the protein. Therefore, we first investigated whether the deletion of the murine exon 2 in Ccdc6 (Ccdc6-ex2) could affect the Ccdc6 ability to act as a negative modulator of the CREB1 transcriptional activity, with respect to the Ccdc6 wild type [9]. Subsequently, we proceeded to the generation of mice carrying a Ccdc6 gene deleted of exon 2 (Ccdc6-ex2 mice).
These mice developed thyroid hyperplasia likely due to an increased CREB1 activity and consequent expression of the CREB1-regulated genes, as expected from the described effect of wt CCDC6 to repress CREB1 transcriptional activity.
RESULTS
Ccdc6-ex2 protein increases CREB binding to CRE element but exerts a milder repression on CREB1 transcriptional activity in comparison to Ccdc6 wt
We have previously demonstrated that the CCDC6 protein was able to interact with CREB1 and modulate its activity by reducing the binding of CREB to CRE element in a dose-dependent manner [9].
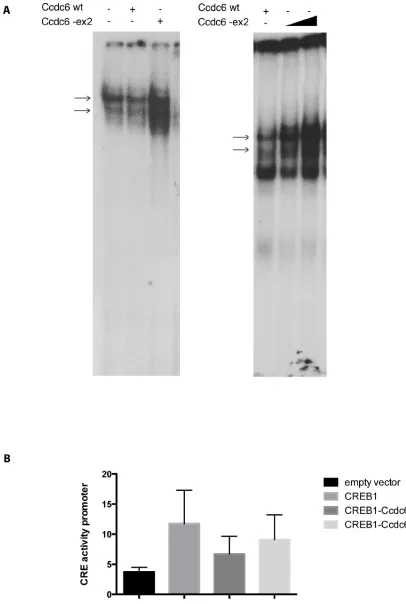
Therefore, we investigated the ability of a Ccdc6 deprived of the exon 2 to repress the CREB1 transcriptional activity. We analyzed the effect of Ccdc6-ex2 on the binding of CREB to the CRE element by Electrophoretic Mobility Shift Assay (EMSA). Nuclear extracts from control cells or HeK-293 cells, overexpressing Ccdc6 wt or Ccdc6-ex2, were incubated with a CRE radiolabelled oligonucleotide. As shown in Figure 1A, nuclear proteins from Ccdc6-ex2 extracts were able to bind the CRE oligonucleotide in EMSA (left panel, lane 1). The Ccdc6-ex2 expression increased the binding of CREB to CRE element (left panel, lane 3) in a dose-dependent manner (lanes 2 and 3, right panel), compared to the expression of the Ccdc6 wt that showed a reduced binding of CREB to CRE element (left panel, lane 2). As Ccdc6-ex2 was able to increase the binding of CREB to CRE element, we analyzed the effect of Ccdc6-ex2 on CREB1 transcriptional activity. HeK-293 cells were transfected with a CRE-luc
reporter gene [10] in which tandem three CRE universal sites were fused upstream to a luciferase cDNA together with Ccdc6 wt or Ccdc6-ex2 in the presence of CREB1. As shown in Figure 1B, the CREB1 overexpression enhanced the reporter CRE-luc activity more than 3-fold, whereas co-transfection of Ccdc6 resulted in a decrease of CREB1-mediated transcriptional activity. However, the inhibition of CREB1 transcriptional activity after co-transfection of the Ccdc6 with its ex2-deleted mutant construct is significantly less evident in comparison to that obtained co-transfecting the wt Ccdc6 construct. This result suggests that the deletion of the coiled-coil domain, mainly coded for by the murine exon 2, could impair the Ccdc6 protein function at least for its ability to downregulate CREB1 activity.
Generation of Ccdc6-ex2 mice
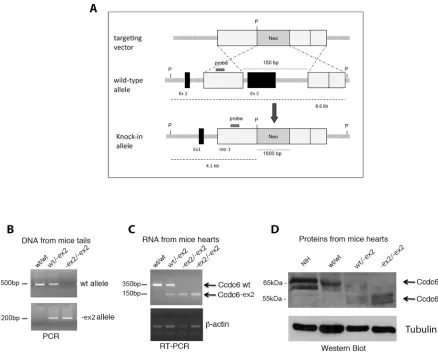
These findings led us to proceed to the generation of the Ccdc6-ex2 knock-in mice. We used gene targeting techniques in embryonic stem (ES) cells to generate a mutation at the murine Ccdc6 genomic locus. We deleted exon 2 which contains most of the coiled-coil domain and replaced it with a neomycin-resistance cassette (Figure 2A) (see Materials and Methods). Cell clones resistant to G418 have been isolated and screened for Ccdc6 homologous recombination. The positive clones were expanded and injected into blastocysts from the C57BL/6 mice strains. The resulting chimaeric blastocysts were transferred to uteri of foster mothers of the same strain. The chimaeric offspring was crossed with wt mice to obtain germ line transmission with the generation of mice heterozygous for Ccdc6 gene deletion. The heterozygous mice were then crossed each other to generate homozygous mice.
Ccdc6-ex2/-ex2 mouse embryonic fibroblasts
(MEFs) showed a reduced rate of growth and an increased susceptibility to apoptosis compared to the Ccdc6 wt counterparts
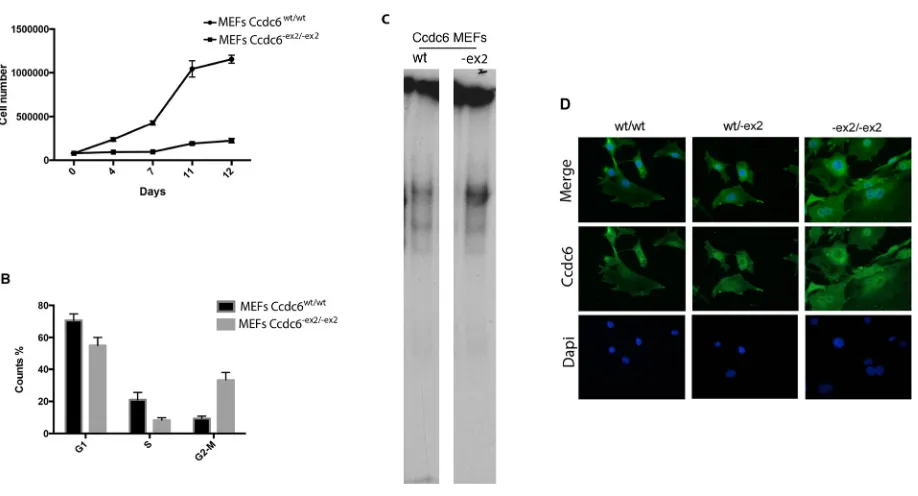
To investigate the role of CCDC6 in cellular proliferation in vivo, MEFs from Ccdc6wt/wt and Ccdc6-ex2/-ex2 embryos at 12.5 days post coitum (dpc) were obtained and the growth rate and cell-cycle distribution were analyzed.
[image:4.612.80.518.46.404.2]As shown in Figure 3A, the Ccdc6-ex2/-ex2 MEFs showed a proliferation rate significantly slower than the Ccdc6wt/wt controls. To assess whether the slow rate of growth of Ccdc6-ex2/-ex2 MEFs was due to deranged progression through the phases of the cell cycle, we examined asynchronously growing MEFs by flow cytometry. Ccdc6-ex2/-ex2 MEFs accumulated in the G2 phase of the cell cycle, compared to Ccdc6wt/wt MEFs cell cycle distribution (Figure 3B). Nevertheless, the percentage of the Ccdc6-ex2/-ex2 fibroblasts Figure 2: Generation of Ccdc6-ex2/-ex2 knock-in mice. A. Schematic representation of the endogenous wt allele, the targeting vector and
[image:4.612.58.557.521.601.2]the resulting Ccdc6-ex2 allele. P, Pst1; Neo, neomycin; Ex, exon, Intr, intron. B. The genotyping of Ccdc6 mice was performed using PCR analysis of DNA extracted from mice tails. C. RT-PCR analysis of total RNA extracted from hearts of Ccdc6 mice. D. Western blot analysis of Ccdc6 proteins in hearts of wt and knock-in mice. Gamma-tubulin was analyzed to evaluate equal protein loading. NIH lysate extracts have been utilized as a control of the murine Ccdc6 weight, recognized by a specific anti-CCDC6 antibody, that we generated (anti-C16-R).
Table 1: The table indicated the frequency of wt, heterozygous, and homozygous offspring after
heterozygote matings in Ccdc6 knock-in mice
heterozygote matings offspring = 198 mice
N° mice %
Ccdc6 wt/wt 81 41
Ccdc6 wt/-ex2 95 48
in the G1 and S phases was reduced with respect to MEFs Ccdc6 wild type.
Recent studies reported the involvement of the CCDC6 gene in apoptosis and the ability of its truncated mutant CCDC6 (1–101), which corresponds to the portion of CCDC6 included in RET/PTC1, to act as dominant negative on the wt CCDC6-induced apoptosis [7].
Therefore, in order to evaluate the susceptibility of the Ccdc6-ex2/-ex2 MEFs to apoptosis we performed FACS analysis of MEFs derived from Ccdc6wt/wt and Ccdc6-ex2/-ex2 embryos grown in complete medium or starved for 24 h or 48 h after annexin staining. As shown in Figure 4A (left panel) a consistent fraction of Ccdc6-ex2/-ex2 MEFs were annexin positive, 8-fold more than Ccdc6wt/wt MEFs. Then, to confirm that Ccdc6-ex2/-ex2 MEFs had an increased susceptibility to apoptosis, we detected in unstressed Ccdc6-ex2/-ex2 MEFs, a significant increase in 14–3-3σ transcription, a regulator of apoptosis and cell cycle checkpoints, whose activity has been already linked to CCDC6, compared to wild type MEF [12]. Moreover, in Ccdc6-ex2/-ex2 MEFs, we also observed increased transcription of Killer/DR5, an effector of TRAIL, whose pathway is known to be active in thyroid, as already reported [13]. In Ccdc6wt/-ex2 MEFs the fold inductions of both the apoptotic proteins looked very similar to wild type MEFs (Figure 4B).
Next, we analyzed the susceptibility to the apoptosis of Ccdc6wt/wt and Ccdc6-ex2/-ex2 MEFs by cell viability assay, evaluated by water-soluble tetrazolium salt (WST-1) analysis, after treatment with high doses of H2O2. As shown in Figure 4C the Ccdc6-ex2/-ex2 MEFs resulted sensitive to high doses of H2O2 (800 mM) showing a lower percentage (60%) of viable cells with respect to Ccdc6wt/wt MEFs (78%) (Figure 4C). Nevertheless, it has been reported that MEFs show a high susceptibility to high doses of oxidative damage [14]. Finally, nuclear extracts from MEF Ccdc6-ex2/-ex2 showed an increased binding to CRE elements with respect to the extracts from the wt fibroblasts (Figure 3C). Interestingly, at immunofluorescence in MEFs Ccdc6-ex2/-ex2 the Ccdc6 protein showed a nuclear localization and a reinforced cytosolic staining, with respect to the wt and to the heterozygotes MEFs (Figure 3D).
Ccdc6-ex2 knock-in mice develop thyroid
hyperplasia associated with an increased CREB1 transcriptional activity
[image:5.612.79.538.56.304.2]Either Ccdc6-ex2/-ex2 or Ccdc6wt/-ex2 mice did not show any evidence of illness up to 12 months of age. However, histopathological analysis of aged (17–22 month-old) mice revealed the presence of thyroid hyperplasia in 15 out of 30 Ccdc6-ex2/-ex2 mice. Histologically, atypical follicles Figure 3: Analysis of Ccdc6-ex2/-ex2 and wt MEF growth. A. MEFs were prepared from Ccdc6wt/wt and Ccdc6-ex2/-ex2 embryos at 12.5
dpc. At culture passage 4, they were plated and counted daily for 12 days to extrapolate growth curves. The mean values + SE of three different cell clones (each originating from a different embryo) for each genotype are reported. B. Propidium iodide flow cytometry of asynchronous growing Ccdc6wt/wt and Ccdc6-ex2/-ex2 MEFs. The percentage (expressed as mean value + SE) of cells in each phase of the cell
cycle is indicated. *P < 0.05. C. EMSA assay was performed with a radiolabelled oligonucleotide containing the CRE element incubated with nuclear extracts from Ccdc6wt/wt and Ccdc6-ex2/-ex2 MEFs. D. Immunofluorescence of Ccdc6 in MEF wt, MEFwt/-ex2 and MEF-ex2/-ex2 was
contained eosinophilic secretory and inactive material (colloid). Follicular cells were cuboidal to columnar with indistinct cell borders, moderate amount of eosinophilic cytoplasm, one basally located oval nucleus and indistinct nucleoli (Figure 5A). In Ccdc6wt/wt mice the thyroids did not show any alteration, as well as in Ccdc6 wt/-ex2 mice (data not shown). Consistently, we found low levels of TSH and high levels of T3 and T4 hormone levels in Ccdc6-ex2/-ex2 mice with respect to Ccdc6wt/wt analyzed at the 13 month of age (Figure 5B).
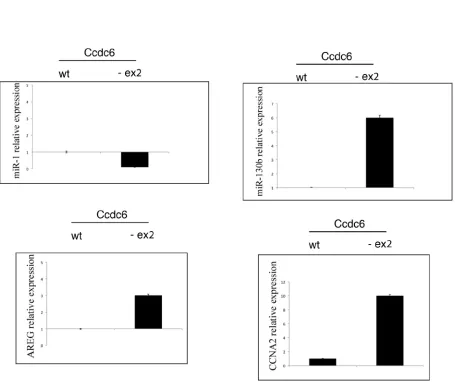
We have previously demonstrated that CCDC6 was able to act as negative regulator of the transcriptional activity of CREB1 and of its phosphorylation [9]. As shown in Figure 6A, western blot showed increased levels of phosphorylation of CREB1 protein (at Ser 133) in extracts from hyperplastic thyroids obtained by Ccdc6-ex2/-ex2 mice with respect to the extracts from normal thyroid from wt mice. In order to investigate whether the increase in the phosphorylation status may reflect the transcriptional ability of the thyroid cells, we have analysed the levels of some CREB1 target genes, such as AREG, Cyclin A, miR-130b (positively regulated by CREB1) and miR-1 (negatively regulated by CREB1) in hyperplastic thyroids of Ccdc6-ex2/-ex2 mice and controls. As shown in Figure 6B, the CREB
transcriptional targets AREG, Cyclin A and miR-130b showed an upregulated profile, whereas miR-1 appeared downregulated in Ccdc6-ex2/-ex2 mice with respect to the Ccdc6wt/wt mice. These results confirm that the impairment of the Ccdc6 coiled-coil domains influences the CREB1 transcriptional activity in a positive fashion.
DISCUSSION
[image:6.612.85.539.46.351.2]Previous studies have demonstrated that the expression of CCDC6 is able to reduce CREB1 activity by increasing the PP1 activity on CRE element, with a consequent decrease of binding of CREB1 to the responsive regions and by activating HDAC1, affecting the acetylation status of histone H3 at the CREB1 responsive promoters [9]. Therefore, the negative modulation of the CREB1 activity, exerted by CCDC6, may account for the inhibition of cell growth as CREB1 activation represents the final step of the TSH activation pathway that exerts a critical role in the thyroid cell growth and differentiation [15, 16]. Thus, CCDC6 would work as tumor suppressor in inhibiting the CREB1-dependent gene expression. Therefore, the loss of one allele in the PTCs carrying the RET/PTC1 oncogene might amplify the RET/ PTC1 oncogenic activity in leading thyroid cells to the Figure 4: Ccdc6-ex2/-ex2 MEFs are more susceptible to apoptosis than wt MEFs. A. Apoptosis analysis in Ccdc6wt/wt and Ccdc6 -ex2/-ex2 MEFs grown in complete medium or starved for 24 h or 48 h. Apoptosis was calculated as the percentage of cells positive at annexin
V-FITC. B. 14–3-3σ and DR5 relative expression in MEFwt, MEFwt/-ex2 and in MEFex2/-ex2 were analysed by qRT-PCR. Data are the mean
± SD of three independent experiments. C. Percentage of cell viability evaluated by WST-1 analysis on Ccdc6wt/wt and Ccdc6-ex2/-ex2 MEFs
malignant state, acting in synergism with the oncogene. The tumor suppressive role of CCDC6 in thyroid carcinogenesis is also supported by recent investigations showing that CCDC6 is involved in the protection of genome integrity. Indeed, silencing of CCDC6 or the mutation in the recognition site for the ATM kinase in primary GC1 germ cells increases the rate of survival, impairs the phosphorylation of histone H2AX on S139, affects ROS production and makes the cells more resistant to oxidative damage and genotoxic stress [17, 18].
In order to demonstrate a tumour suppressive role of the CCDC6 gene in vivo we first planned to generate a Ccdc6 null mice. Unfortunately, we could not generate mice null for Ccdc6 since we did not get germ-line transmission starting from the chimeric founder mice derived from embryonic stem cells harboring a targeted mutation in a single Ccdc6 allele (data not shown).
Next, we decided to generate Ccdc6 mice carrying a mutant Ccdc6 gene, altered in its function. To impair CCDC6 activity we deleted the exon 2 of the Ccdc6 gene that codes for almost the entire coiled-coil domain that
[image:7.612.68.532.45.392.2]Figure 6: CREB1 transcriptional activity is increased in thyroids of Ccdc6-ex2/-ex2 mice. A. Western blot analysis of
pCREB protein in total cell extracts from Ccdc6wt/wt and Ccdc6-ex2/-ex2 thyroids. Tubulin expression was used to normalize the amount
of loaded proteins. B. AREG, CCNA2, miR-1 and miR-130b genes expression by qRT-PCR from Ccdc6wt/wt and Ccdc6-ex2/-ex2 thyroids.
of Ccdc6-ex2/-ex2 mice showed higher levels of CREB1 phosphorylation in Ccdc6-ex2/-ex2 mice in comparison with the wt counterpart at immunoblot with the anti-pS133 CREB1 antibody. Accordingly, in hyperplastic thyroids of Ccdc6-ex2/-ex2 mice, the expression analysis of CREB1 target genes revealed the upregulation of CREB1 associated to increased levels of AREG and Cyclin A, or decreased levels of miR-1, confirming the modulation of these gene by an enhanced CREB1 activity.
We have also analyzed other organs of the Ccdc6 -ex2/-ex2 mice. In Ccdc6-ex2/-ex2 mice, out of the head and neck region, left ventricular hypertrophy has been rarely observed, even though without a statistical significance (data not shown). Moreover, a testicular phenotype has been observed occasionally in few animals deleted of exon 2 and a CCDC6 involvement in testicular germ cell population at normal and pathologic level has been reported [18]. Conversely, the Ccdc6wt/-ex2 mice analyzed did never show any pathology.
Thus, the animal studies confirmed that the loss of CCDC6 activity is able to increase the thyroid cell proliferation by amplifying the CREB1 activation and overall synergizing with the activity of the RET/ PTC1 oncogene in the induction of the PTCs. The same mechanisms could be, of course, ascribed to the different oncoproteins that carries a truncated CCDC6 at their aminoterminus.
By the analysis of the MEF Ccdc6-ex2/-ex2 we detected a replication rate slower than the MEFs wild type. However, it has been well documented that to an enhanced CREB1 activity corresponded an increase of the growth rate in thyroid cells and a decrease in that of fibroblasts, due to apoptosis. Indeed, we observed an increase of the CREB activity that enhances the growth rate of thyroid cells, while decreases fibroblast proliferation [19–22]. Therefore, Ccdc6-ex2 leads to an increased CREB activity, but has a negative effect on MEFs proliferation. In conclusion, the results we report here are in support of a likely role of the haploinsufficiency of CCDC6 expression in the development of PTCs carrying the RET/PTC1 rearrangements.
MATERIALS AND METHODS
Expression vectors
The expression Ccdc6 wt (nt 1–1410) and Ccdc6-ex2 (Δnt 220–372) plasmids were obtained by amplifying the Ccdc6 full lenght or the ccdc6 deleted, in the nucleotides 220–372, by PCR and then inserting them into the mcs of the pcDNA4ToB (Invitrogen).
Electrophoretic mobility shift assay
EMSA was performed as previously described [9]. CREB consensus oligonucleotide probe was from
Santa Cruz Biotechnology Inc (TransCruz™ Gel Shift Oligonucleotides).
Transactivation assay
B-CPAP cells were transiently transfected with the reporter construct [9]. Co-transfections were carried out in the presence of 200 ng of reporter construct and 500 ng of Renilla construct and 2 μg of pCMVSport6-CREB1 with or without the indicated amounts of wt Ccdc6 or mutant Ccdc6-ex2 constructs. Luciferase and Renilla activities were measured with the dual-luciferase reporter assay kit (Promega).
Generation and genotyping of mutant mice
The mouse Ccdc6 genomic locus (ENSMUSG00 000048701) was cloned screening a phage library of mouse 129/Sv genomic DNA (Stratagene, La Jolla, California) using a mouse Ccdc6 cDNA as a probe, which was obtained by cross-hybridization experiments with human CCDC6 cDNA. A targeting vector was constructed from subcloned genomic fragments. For the selection of targeted clones, a neomycin-resistance (neo) cassette was placed in the opposite transcriptional orientation to the endogenous Ccdc6 gene. The final product consists of a 5′ genomic fragment (1.66 kb) containing exon 1, the neo cassette (1.7 kb) and a downstream CCDC6 genomic fragment (5.73 kb). Thus, a ~12 kb Ccdc6 genomic region containing exon 2 was replaced with the neo cassette vector.
Linearized targeting vector DNA was introduced by electroporation into 129/Sv embryonic stem (ES) cells. After G418 selection, genomic DNA of resistant clones was analyzed by Southern blot analysis using a probe lying within the 5′ homology region (generated with primers 5′-GCCCTTGTTGGTGGAGTGTT-3′ and 5′-ACCCTCTCTCCCTACCTACA-3′) hybridized to PstI digested genomic DNA. To generate chimeras, cells of targeted clones were injected into host blastocysts of C57BL/6J mice then transferred into uteri of pseudo-pregnant females. Male chimeras were mated with C57BL/6J females and F1 heterozygous progeny were intercrossed. Mice were genotyped for the Ccdc6 deletion by PCR. Primers used for genotyping are as follows: CCDC6 ln3R1 5′-GGAGGCAGATGAGTTCCTAAGG-3′ NEO5′R 5′-CTAAAGCGCATGCTCCAGACTGCC-3′ CCDC6U2 5′-CAGTAACACTTTATTCAAGAAAATCC AG-3′
Growth and cell cycle analysis of MEFs
penicillin/streptomycin and 1% gentamicin (GIBCO). The cells (4 x 105/dish) were plated in a series of 6-cm culture dishes and counted daily with a hemocytometer for 12 consecutive days to extrapolate growth curves. Cell cycle analysis was performed using a FACS Calibur cytofluorimeter (Becton Dickinson, San Jose, CA, USA). For cell cycle analysis, MEFs in logarithmic growth were trypsinized, fixed in 70% ethanol and stored at 4°C for a few days. Cells were then washed with PBS without Ca2+ and Mg2+, stained with 50 μg/ml propidium iodide containing RNAse (20 μg/ml) and analyzed by FACS. Cell debris and fixation artefacts were gated out, and G1, S and G2/M populations were quantified using CellQuest software (Becton Dickinson). A similar number of events was analyzed in each experiment.
Protein extraction, western blotting
Protein extraction and Western blotting procedures were carried out as reported elsewhere [23]. The indicated antibodies have been used for Western blotting: anti-CCDC6 antibody (C-16-R) has been generated upon rabbit immunization with the following polypeptide (Cys-Gly- Leu-His-Val-Gln-His-Met-Gly-Thr-Ser-His-Gly-Ile-Thr-Arg), synthetized by the NeoSystem Group, (Strasbourg, France) and affinity purified from immunized serum; anti-CREB1 has been obtained from Upstate Biotechnology; anti-pCREB Ser133 was from Cell Signaling. Anti-γ-tubulin (sc-17787) and anti-Actin (sc-1615) were from Santa Cruz Biotechnology. Blots were visualized using the ECL chemiluminescence system (Amersham/Pharmacia).
RNA extraction, cDNA preparation, semiquantitative and quantitative reverse transcription-PCR
Total RNA isolation, RT-PCR and qRT-PCR from tissues or from cells were performed as previously described [24]. Primers used are as follows:
AREG(f) 5′-gcgaatgcagatacatcgag-3′; AREG(r) 5′-ccacaccgttcaccaaagta-3′; CCNA2(f) 5′-cttggctgcaccaacagtaa-3′; CCNA2(r) 5′-caaactcagttctcccaaaaaca-3′; 14–3-3(f) 5′-tgtggacagccgacagtg-3′; 14–3-3(r) 5′-cttcagatgtgggggtcatc-3′; Killer/DR5(f) 5′-ccctgagatctgccagtcat-3′; Killer/DR5(r) 5′-tgggggtacaggaagtcagt-3′. Indirect immunofluorescence
The indirect immunofluorescence was performed as previously described [7].
WST-1 colorimetric assays
To determine cell viability, a tetrazolium salt WST-1 assay was performed as directed by the manufacturer (Roche).
Annexin assay
For Annexin V apoptosis assay (BD, Pharmingen, EremBodegem, Belgium), we stained cells simultaneously with fluorescein isothiocyanate (FITC)-Annexin V and non-vital dye PI according to the manufacturer’s instructions. The percentage of the M-phase cells was determined by staining with PI and antibody to phospho-histone H3 (P-H3) (Cell Signaling, Beverly, MA, USA), followed by FITC-conjugated secondary antibody (Jackson Immunoresearch Laboratories, West Grove, PA, USA). Samples were analyzed with a CyAnTM ADP flow cytometer (Dako Cytomation, Glostrup, Denmark) using an argon-ion laser tuned to 488 nm measuring forward and orthogonal light scatter. Data were acquired by Summits software and analyzed with Modfits software.
ACKNOWLEDGMENTS
This work was supported by grants from: Associazione Italiana per la Ricerca sul Cancro-AIRC (IG 11477), Ministero dell’Università e della Ricerca Scientifica e Tecnologica–MIUR (PRIN 2011), Progetto CREME supported by P.O.R. Campania FSE 2007–2013, CUP B25B09000050007, CNR Epigenomics Flagship Project “EPIGEN”.
REFERENCES
1. Grieco M, Santoro M, Berlingieri MT, Melillo RM, Donghi R, Bongarzone I, Pierotti MA, Della Porta G, Fusco A, Vecchio G. PTC is a novel rearranged form of the RET proto-oncogene and is frequently detected
in vivo in human thyroid papillary carcinomas. Cell. 1990; 60:557–563.
2. Takeuchi K, Soda M, Togashi Y, Suzuki R, Sakata S, Hatano S, Asaka R, Hamanaka W, Ninomiya H, Uehara H, Lim Choi Y, Satoh Y, Okumura S, et al. RET, ROS1 and ALK fusions in lung cancer. Nat Med. 2012; 18:378–381. 3. Kulkarni S, Heath C, Parker S, Chase A, Iqbal S,
Pocock CF, Kaeda J, Cwynarski K, Goldman JM, Cross NC. Fusion of H4/D10S170 to the platelet-derived growth factor receptor beta in BCR-ABL-negative myelo-proliferative disorders with a t(5;10)(q33;q21). Cancer Res. 2000; 60:3592–3598.
4. Schwaller J, Anastasiadou E, Cain D, Kutok J, Wojiski S, Williams IR, LaStarza R, Crescenzi B, Sternberg DW, Andreasson P, Schiavo R, Siena S, Mecucci C, et al. H4(D10S170), a gene frequently rearranged in papillary thyroid carcinoma, is fused to the platelet-derived growth factor receptor beta gene in atypical chronic myeloid leu-kemia with t(5, 10)(q33;q22). Blood. 2001; 97:3910–3918. 5. Puxeddu E, Knauf JA, Sartor MA, Medvedovic M,
involved in regulation of the immune response. Endocr Relat Cancer. 2005; 12:319–313.
6. Grieco M, Cerrato A, Santoro M, Fusco A, Melillo RM, Vecchio G. Cloning and characterization of H4 (D10S170), a gene involved in RET rearrangements in vivo. Oncogene. 1994; 9:2531–2535.
7. Celetti A, Cerrato A, Merolla F, Vitagliano D, Vecchio G, Grieco M. H4(D10S170), a gene frequently rearranged with RET in papillary thyroid carcinomas: functional character-ization. Oncogene. 2004; 23:109–121.
8. Merolla F, Luise C, Muller MT, Pacelli R, Fusco A, Celetti A. Loss of CCDC6, the first identified RET partner gene, affects pH2AX S139 levels and accelerates mitotic entry upon DNA damage. PLoS One. 2012; 7:e36177. 9. Leone V, Mansueto G, Pierantoni GM, Tornincasa M,
Merolla F, Cerrato A, Santoro M, Grieco M, Scaloni A, Celetti A, Fusco A. CCDC6 represses CREB1 activity by recruiting histone deacetylase 1 and protein phosphatase 1. Oncogene. 2010; 29:4341–4351.
10. De Angelis R, Iezzi S, Bruno T, Corbi N, Di Padova M, Floridi A, Fanciulli M, Passananti C. Functional interac-tion of the subunit 3 of RNA polymerase II (RPB3) with transcription factor-4 (ATF4). FEBS Lett. 2003; 547:15–19. 11. Morra F, Luise C, Merolla F, Poser I, Visconti R, Inuzuka H,
Ilardi G, Guggino G, Monaco R, Colecchia D, Monaco G, Cerrato A, Chiariello M, Denning K, Claudio P, Staibano S, Celetti A. “FBXW7 and USP7 regulate CCDC6 turnover during the cell cycle and affect cancer drugs susceptibility in NSCLC”. Oncotarget. 2015; Mar 30. [Epub ahead of print]. 12. Thanasopoulou A, Stravopodis DJ, Dimas KS, Schwaller J,
Anastasiadou E. Loss of CCDC6 affects cell cycle through impaired intra-S-phase checkpoint control. PLoS One. 2012; 7:e31007.
13. Ahmad M, Shi Y. TRAIL-induced apoptosis of thyroid can-cer cells: potential for therapeutic intervention. Oncogene. 2000; 19:3363–3371.
14. Huo YY, Li G, Duan RF, Gou Q, Fu CL, Hu YC, Song BQ, Yang ZH, Wu DC, Zhou PK. PTEN deletion leads to dereg-ulation of antioxidants and increased oxidative damage in mouse embryonic fibroblasts. Free Radic Biol Med. 2008; 44:1578–1591.
15. Nguyen LQ, Kopp P, Martinson F, Stanfield K, Roth SI, Jameson JL. A dominant negative CREB (cAMP response element-binding protein) isoform inhibits thyrocyte growth,
thyroid-specific gene expression, differentiation, and func-tion. Mol Endocrinol. 2000; 14:1448–1461.
16. Hashimoto K, Zanger K, Hollenberg AN, Cohen LE, Radovick S, Wondisford FE. cAMP response element- binding protein-binding protein mediates thyrotropin-releasing hormone signaling on thyrotropin subunit genes. J Biol Chem. 2000; 275:33365–33372.
17. Staibano S1, Ilardi G, Leone V, Luise C, Merolla F, Esposito F, Morra F, Siano M, Franco R, Fusco A, Chieffi P, Celetti A. Critical role of CCDC6 in the neo-plastic growth of testicular germ cell tumors. BMC Cancer. 2013; 13:433.
18. Merolla F, Pentimalli F, Pacelli R, Vecchio G, Fusco A, Grieco M, Celetti A. Involvement of H4(D10S170) protein in ATM-dependent response to DNA damage. Oncogene. 2007; 26:6167–6175.
19. Rozengurt E. Early signals in the mitogenic response. Science. 1986; 234:161–166.
20. Dumont JE, Jauniaux JC, Roger PP. The cyclic AMP-mediated stimulation of cell proliferation. Trends Biochem Sci. 1989; 14:67–71.
21. Heldin NE, Paulsson Y, Forsberg K, Heldin CH, Westermark B. Induction of cyclic AMP synthesis by forskolin is followed by a reduction in the expression of c-myc messenger RNA and inhibition of 3H-thymidine incorporation in human fibroblasts. J Cell Physiol. 1989; 138:17–23.
22. Magnaldo I, Pouyssegur J, Paris S. Cyclic AMP inhibits mitogen-induced DNA synthesis in hamster fibroblasts, regardless of the signalling pathway involved. FEBS Lett. 1989; 245:65–69.
23. Fedele M, Pentimalli F, Baldassarre G, Battista S, Klein-Szanto A JP, Kenyon L, Visone R, De Martino I, Ciarmiello A, Arra C, Viglietto G, Croce CM, Fusco A. Transgenic mice overexpressing the wild-type form of the HMGA1 gene develop mixed growth hormone/prolactin cell pituitary adenomas and natural killer cell lymphomas. Oncogene. 2005; 24:3427–3435.