Tumor cell adhesion and migration supported by interaction of a receptor protease complex with its inhibitor
Full text
Figure
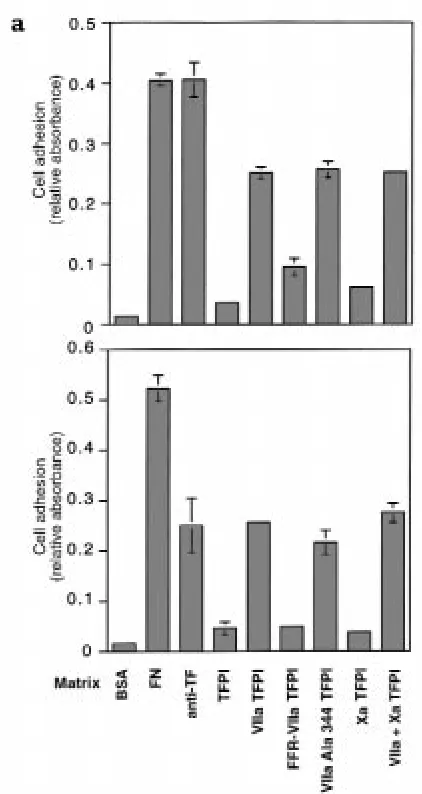
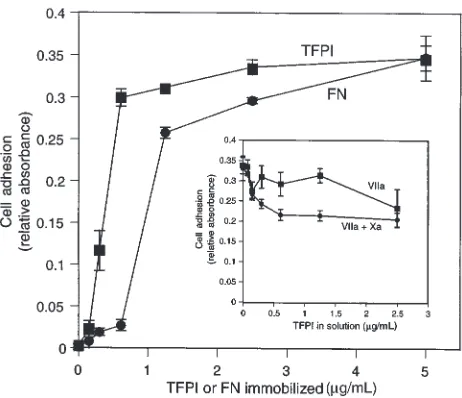
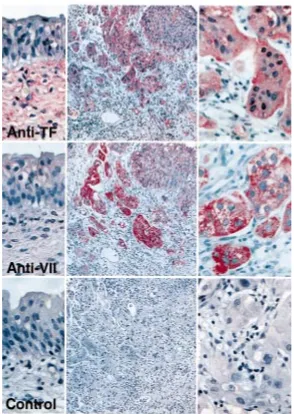


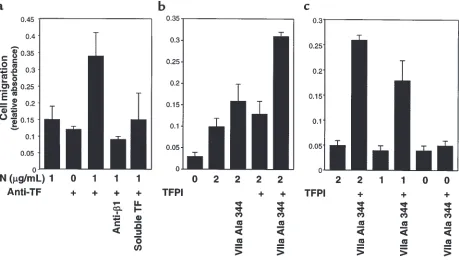
Related documents
The use of nonambiguous functional and phylogenetic criteria derived from the TCDB classification system has allowed the identification within the Hemiascomycete phylum of 97
Load Frequency Control of Two Area Multi Unit Power System Using Three Parameter Tunable Tilt-Integral-Derivative
In present study, isolated compounds from Nigella sativa seeds were screened for their antibacterial activity by disc diffusion method against four
The Chair should be familiar with the Board’s policies or charter, and the Board’s written delegations to the chief executive.. They should ensure the Board acts
4,5 Because of the unique crystal quality and photonic properties, in particular, one dimensional (1D) ZnO nanomaterials have been used as functional units in the
Endophytic fungi possess a storehouse of various bioactive metabolites with anti microbial, anti viral, anti cancerous or anti oxidant actions.. Some of the
Due to human demand and greedy towards the environment, various alternatives applied in human, namely science in law, science and policy, science and
The intent of this review is to shed light on teaching and learning patterns during general surgery residency training that not only guarantee trainees’ success on standardized
