ABSTRACT
MA, CHUNXIAO. Redesigning Procaspase-8 Dimer Interface Improves its Dimerization. (Under the direction of Clay Clark.)
Caspase-8 is a member of the cysteine-dependent aspartate-directed proteases
fam-ily and activated at the cytosolic face of the cell membrane. Its activation depends on
adaptor-induced dimerization of caspase-8 monomer and the following limited
autopro-teolysis at the inter-subunit linker which separates the large subunit and small subunit
of the catalytic domain. In contrast, the executioner caspase, caspase-3 is a stable dimer
in physiological conditions. However, it is not known why procaspase-8 is a monomer
while procaspase-3 is a dimer. In order to define the role of the dimer interface in the
dimerization and activation of procaspase-8, we have generated several mutants that
mimic the dimer interface of executioner caspase. We show that redesigning the interface
improves the dimerization of procaspase-8. Procaspase-8 is activated by sodium citrate,
and we show that procaspase-8 with the mutated interface requires lower salt
concen-tration to acquire full activity, which indicates that the mutated procaspase-8 is more
readily dimerized. Kinetic simulation also suggests that these mutations accelerate the
first step of dimerization. However, in cellulo studies in HEK293 cells show that cells
transfected with mutated procaspase-8 do not exhibit a significant increase in apoptosis,
which means that more modifications are needed. In conclusion, the reconstruction of
executioner caspase interface in procaspase-8 interface removes the negative design
ele-ment from the interface, which help the dimerization of procaspase-8. However, there are
©Copyright 2013 by Chunxiao Ma
Redesigning Procaspase-8 Dimer Interface Improves its Dimerization
by Chunxiao Ma
A dissertation submitted to the Graduate Faculty of North Carolina State University
in partial fulfillment of the requirements for the Degree of
Doctor of Philosophy
Biochemistry
Raleigh, North Carolina
2013
APPROVED BY:
Michael Goshe Bob Rose
Stephen Franzen Clay Clark
DEDICATION
BIOGRAPHY
Chunxiao Ma was born in Hebei, China. She graduated from Nankai University in Jun
2007 with a Bachelor of Science degree in Biotechnology. In Aug 2007, She moved to the
United states and started graduate school in pursuit of a Ph.D. in Biochemistry at North
Carolina State University under the direction of Dr. A. Clay Clark. During the same
time, she also learned a Biomedicals statistics masters degree in the Statistic department
ACKNOWLEDGEMENTS
I would like to thank Dr. A. Clay Clark for his passion and commitment to making his
students the best they can be. You are truly an outstanding mentor and teacher. I would
also like to thank my committee members Dr. Michael Goshe, Dr. Robert Rose and Dr.
Stefan Franzen for your support and encouragement for the past six years. I would also
like to thank Dr. Maxwell and Dr. Hemenway for giving their time and sitting in on my
prelim defense.
I would like to thank my family far away in China. Although during these six years,
the time we been together is less than 3 months, the internet and phone calls provide
intimate contact between us. Your unwavering support has enabled me to become the
person that I am. I am blessed to have such an amazing family.
I would like to thank my friends for making the last six years some of the best times
in my life. Sarah MacKenzie, thanks for all your help during my graduate life. You are
a excellent colleague in lab work as well as a very good friend in my life. And Dongli
Wang, it is very good that we came from the same university in China and came to the
same one here. Thanks for your help in my prelim and also in my research. We find a
lot of interesting topics to discuss, both about research and about China. To my fellow
labmates, Joe, Bob, Brian, Matt and Christie, I have learned so much from all of you.
I would also like to thank Rose’s lab, Xun and Greg, you help me a lot as we are good
TABLE OF CONTENTS
LIST OF TABLES . . . vii
LIST OF FIGURES . . . viii
Chapter 1 Introduction . . . 1
1.1 Abstract . . . 1
1.2 Caspase-8 is an initiator in caspase family . . . 2
1.2.1 Structure of Caspase-8 . . . 2
1.2.2 Activation mechanism of caspase-8 . . . 4
1.3 Caspase-8 has multiple functions in the cell lifecycle . . . 7
1.3.1 Caspase-8 participates in both extrinsic and intrinsic apoptosis pathways . . . 7
1.3.2 Necroptosis . . . 10
1.3.3 Non-apoptotic caspase-8 functions . . . 12
1.4 Caspase-8 is under regulation at several levels . . . 16
1.4.1 Regulation by FLIP . . . 16
1.4.2 Regulation by post-translational modifications . . . 17
1.5 Project aims . . . 19
1.6 Figures . . . 20
Chapter 2 Materials and Methods . . . 26
2.1 Materials . . . 26
2.2 Stock Solutions . . . 27
2.3 Methods . . . 27
2.3.1 Plasmid construction and mutagenesis . . . 27
2.3.2 Protein Purification . . . 28
2.3.3 Equilibrium folding studies . . . 30
2.3.4 Size Exclusion Chromatography . . . 33
2.3.5 Procaspase-8 Activity Assay . . . 34
2.3.6 Kinetics of procaspase-8 activation . . . 35
2.3.7 Chemical cross-linking . . . 36
2.3.8 Analytical ultracentrifuge(AUC) . . . 36
2.3.9 Caspase-8 crystalization . . . 39
2.3.10 Cell culture and transfection . . . 39
2.3.11 Western blot . . . 41
2.4 Figure and Tables . . . 42
3.1 Abstract . . . 45
3.2 Introduction . . . 46
3.3 Results . . . 49
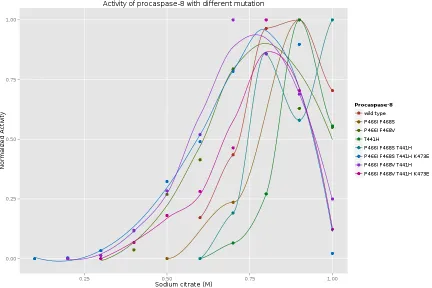
3.3.1 The procaspase-8 with mutated dimer interface have less depen-dence on sodium citrate for activation . . . 50
3.4 The redesigned dimer interface accelerates the dimerization of procaspase-8 55 3.5 Caspase-8(P466I, F468V, T441H, K473E) is not enough to trigger more apoptosis in cell culture . . . 59
3.6 Figures and Table . . . 62
Chapter 4 Discussion . . . 90
4.1 Figures . . . 95
References . . . 97
Appendices . . . 109
Appendix A Equilibrium folding studies of procaspase-8 . . . 110
A.1 Abstract . . . 110
A.2 Introduction . . . 111
A.3 Results . . . 112
A.3.1 Denaturation of procaspase-8 . . . 113
A.3.2 Equilibrium unfolding of procaspase-8 . . . 114
A.3.3 Refolding of procaspase-8 . . . 114
A.3.4 Using a titrator for study the folding of procaspase-8 . . . . 115
A.4 Discussion . . . 116
A.5 Figures . . . 118
Appendix B SAS code for drug screening . . . 129
B.1 Introduction . . . 129
B.2 SAS code . . . 129
Appendix C Cross-linking experiment for detecting procaspase-8 dimerization 132 C.1 Abstract . . . 132
C.2 Results . . . 133
C.3 Figures . . . 135
Appendix D Crystallography condition screening of caspase-8 . . . 140
D.1 History of caspase-8 crystallography . . . 140
D.2 Purification of mutated caspase-8 . . . 142
D.3 Crystallography of mutated caspase-8 . . . 143
LIST OF TABLES
Table 2.1 Primer used in cloning and mutagenesis . . . 43
Table 2.2 Partial specific volume of each amino acid . . . 44
Table 3.1 Activation rate simulation result . . . 57
LIST OF FIGURES
Figure 1.1 Structure of caspase-8 . . . 20
Figure 1.2 Structure of the caspase-8 active site . . . 21
Figure 1.3 Constructs of caspase-8 . . . 22
Figure 1.4 Notation of caspase-8 active site . . . 22
Figure 1.5 Structure changes during maturation . . . 23
Figure 1.6 Dimer-interface of caspase-3 and caspase-8 between β6-strand . . 24
Figure 1.7 Dimer-interface of caspase-3 and caspase-8 between α5-helix and loop5 . . . 25
Figure 2.1 A diagram of constructing pcDNA3.1(-)+FLAG vector . . . 42
Figure 3.1 Overlay structures of caspase-8 and caspase-3 . . . 62
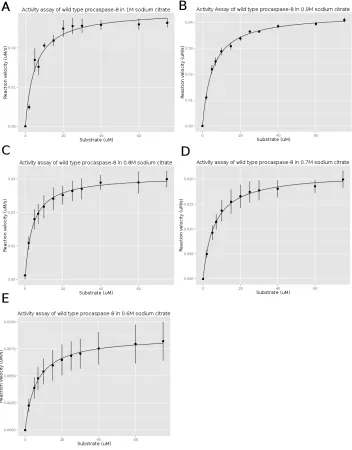
Figure 3.2 The activity assay of wild type procaspase-8. . . 63
Figure 3.3 The activity assay of procaspase-8 (P466I F468S) mutation . . . . 64
Figure 3.4 Structure of caspase-8 dimer interface with mutations . . . 65
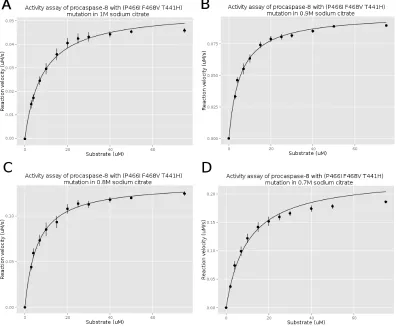
Figure 3.5 The activity assay of procaspase-8 (P466I F468V) mutation . . . 66
Figure 3.6 Structure of caspase-8 α5-helix with mutations . . . 68
Figure 3.7 The activity assay of procaspase-8 (T441H) mutation . . . 69
Figure 3.8 The activity assay of procaspase-8 (P466I F468S T441H) mutation 70 Figure 3.9 Structure of caspase-8 with mutation (P466I, F468V, T441H) . . 71
Figure 3.10 The activity assay of procaspase-8 (P466I F468V T441H) mutation 72 Figure 3.11 The activity assay of procaspase-8 with (P466I F468S T441H K473E) mutation . . . 74
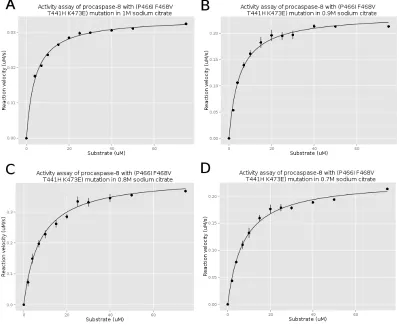
Figure 3.12 The activity assay of procaspase-8 (P466I F468V T441H K473E) mutation . . . 76
Figure 3.13 Thekcat/Kmof procaspase-8 at different sodium citrate concentration 78 Figure 3.14 The normalizedkcat/Km of procaspase-8 at different sodium citrate concentration . . . 79
Figure 3.15 The highest kcat/Km of the procaspase-8 mutants at the most suit-able citrate concentration . . . 80
Figure 3.16 Activation simulation of wild-type procaspase-8 . . . 81
Figure 3.17 Activation simulation of procaspase-8(P466I F468S) mutation . . 82
Figure 3.18 Activation simulation of procaspase-8(P466I F468V) mutation . . 83
Figure 3.19 Activation simulation of procaspase-8(P466I F468V T441H K473E) mutation . . . 84
Figure 3.20 The mutation in procaspase-8 dimer interface are not enough to trigger apoptosis . . . 85
Figure 3.22 Western blot of transfected cell lysate . . . 87
Figure 4.1 The dimer interface of caspase-8 with Thr441His mutation . . . . 95
Figure 4.2 The dimer interface of mutated caspase-8 . . . 96
Figure A.1 The excitation scan of procaspase-8. . . 118
Figure A.2 Fluorescence emission spectra of procaspase-8 at pH 7.0 with dif-ferent incubation time. . . 119
Figure A.3 Fluorescence emission spectra of procaspase-8 at pH 7.0 with dif-ferent incubation time. . . 120
Figure A.4 Unfolding fluorescence emission spectra of procaspase-8 at pH 7.0 with different urea concentration. . . 121
Figure A.5 Refolding fluorescence emission spectra of procaspase-8 at pH 7.0 with different urea concentration. . . 122
Figure A.6 The average emission wavelength(AEW) of equilibrium unfolding and refolding data. . . 123
Figure A.7 Fluorescence emission scan of procaspase-8 with different urea con-centration and different incubation time. . . 124
Figure A.8 Average emission wavelength plot of 1 µM procaspase-8. . . 125
Figure A.9 The normalized AEW of procaspase-8 excited by 280 nm with dif-ferent protein concentrations. . . 126
Figure A.10 The normalized AEW of procaspase-8 excited by 295 nm with dif-ferent protein concentrations. . . 127
Figure A.11 Fitting of the two state folding energy curve . . . 128
Figure B.1 Drug screen result . . . 131
Figure C.1 DMP reaction . . . 135
Figure C.2 Cross-link of wild-type procaspase-8 with 5 µM DMP . . . 135
Figure C.3 Cross-link of wild-type procaspase-8 with 10 µM DMP . . . 136
Figure C.4 Cross-link of wild-type procaspase-8 with 20 µM DMP . . . 136
Figure C.5 Cross-link of wild-type procaspase-8 with 50 µM DMP . . . 137
Figure C.6 Cross-link of wild-type procaspase-8 with 100 µM DMP . . . 137
Figure C.7 Cross-link of procaspase-8 (P466I F468V) with 5 µM DMP . . . . 138
Figure C.8 Cross-link of procaspase-8 (P466I F468V T441H) with 5 µM DMP 138 Figure C.9 Cross-link of procaspase-8 (P466I F468V T441H K473E) with 5 µM DMP . . . 139
Figure D.1 The SDS-PAGE of fractions from Q-sepharose . . . 145
Chapter 1
Introduction
1.1
Abstract
Caspase-8 is an aspartate specific protease which belongs to the caspase family. It is
involved in the regulation of apoptosis pathway during normal development as well as in
removing unhealthy cells in adult life. As observed in other caspases, caspase-8 exists as an
inactive prozymogen and needs to be activated by removing the long prodomain and the
intersubunit linker. The difference is that besides the well known functions of caspase-8
in initiating apoptosis pathways, both the extrinsic and intrinsic, caspase-8 is also shown
to function in proliferation and necroptosis. Since caspase-8 is involved in various signal
pathways, the expression and function of caspase-8 is under restricted regulation. The
dysregulation of caspase-8 expression or dysfunction of caspase-8 leads to a variety of
human diseases. For example, caspase-8 has been found to be inactivated mutated or
inhibited in several human cancers. Therefore, the activation of procaspase-8 presents
a promising method for cancer therapy through restoring the defective apoptosis.
and biophysical approaches, could provide information in understanding the activation
mechanism of procaspase-8, so as to provide a strategy for cancer therapy.
1.2
Caspase-8 is an initiator in caspase family
Caspases are a family of conserved cysteine-dependent aspartate-directed proteases [1].
Since the first caspase was identified [2], fifteen other caspases had been found [3], eleven
of which are found in humans. According to their function in the life and death of a cell,
caspases are classified into two groups, apoptotic caspases and inflammatory caspases
[4]. Caspase-1, -4, -5 are cytokine activators involved in the inflammatory response [5, 6].
The apoptotic caspases are further divided into two groups, the initiator and effectors
caspases, depending on when they enter the apoptotic pathway as well as the length of
their N-terminal prodomain [7]. For example, caspase-8, 9, 10 are initiator caspases, whose
prodomain contains 2 tandom death effector domains (DED) (Caspase8,10) [8, 9, 10] or a
caspase recruitment domain (CARD) (Caspase-9) [11] The initiator caspases exist in the
cell as monomers that need to dimerize to be fully activated. In contrast, caspase-3, 6, 7
are executioner caspases [12] that contain a short N-terminal domain. Because they exist
as a dimer, the activation mechanism requires the cleavage of the inter-subunit linker.
1.2.1
Structure of Caspase-8
There is no structure for the full length caspase-8, because it is not soluble when expressed
in E.coli [13]. A truncated version of caspase-8, without the prodomain, contains 263
amino acids and is soluble inE.coli[14]. The truncated variant of caspase-8 is subdivided
to the formation of substrate-binding groove. The catalytic domain of caspase-8 follows
the classical α/β motif which is composed by a central six parallel β-strands, five are parallel and one is anti-parallel [15] (Figure 1.1). The anti-parallel β strand lies on the edge of the β-sheet and contributes to the dimer interface. Five α-helices surround the
β-sheet core with two on one side (H2, H3) and three (H1, H4, H5) on the other side. Thisα/βstructure is called a caspase fold. Overlayed with the structure of caspase-3 [16], the overall topology of the two structures are quiet similar. This phenomenon provides
evidence for the high structural conservation throughout the caspase family. Despite the
similarity in the main structure, slight differences are also found in the loop region, near
the active site. In loop 1, there is a short α helix insertion between strand β1 and helix
α1. The length of loop 5 is different between caspase-3 and caspase-8. These differences make the conformation different in this region and influence the specificity in substrate
selection [17].
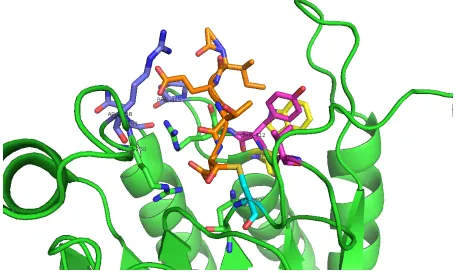
The active site of caspase-8 possesses the conserved catalytic histidine-cysteine dyad
and a large pocket which is capable to accommodate four residues from its substrates. We
name them as P4-P1 residues [18](Figure 1.4), and their corresponding binding pocket is
named as S4-S1. Taken the advantage from high-throughput technologies, about 50-100
caspase-8 substrates have been identified [19, 20]. A common recognizable sequence was
extracted by comparing the sequence of all substrates. The specific sequence for
caspase-8 is (Leu/Val)-Glu-X-Asp [21]. Caspase-caspase-8 cleaves after the Asp. In S1 pocket, the Asp
from inhibitor forms hydrogen bond with Gln358 and salt bridge with Arg413 and Arg260
(Figure 1.2). This interaction pattern is conserved and also observed in caspase-1 and
caspase-3. The ”X” position prefers hydrophobic amino acids, because the S2 pocket is
comprised by two hydrophobic amino acid Tyr and Val [22]. The S3 and S4 pockets are
Arg413, Arg258 and Asn261. Arg 413 forms salt bridges with Glu in the substrate. In S4
pocket, the surface composed of aromatic groups of Trp420 and Tyr412 sandwich with
Ile through nonpolar interactions. These hydrophobic amino acids make a cleft which
prefer aliphatic amino acids. Leu, Val and Ile can all appear at this position. However,
there are no absolute rules in defining whether an Asp-containing peptide or a full length
protein is a substrate [24]. For example, the movement of the backbone of loop 3 makes
Asp an unfavorable residues in S4 pocket of caspase-8. However, the crystal structure of
caspase-8-Z-DEVD-CHO suggests that the Asp can still fit into the S4 pocket [25].
1.2.2
Activation mechanism of caspase-8
The activation mechanism of initiator caspases is different from that of effector caspases.
Effector caspases, 3,6,7 are cleaved by initiator caspases to become activated [26, 27].
However, initiator caspase-8 does not have an upstream protease, so activation occurs
by a distinct mechanism involving dimerization and proteolysis. The proteolysis of the
inter-subunit linker is also important for the full activation as discussed below.
Because the KD of procaspase-8 (micromolar range) [28], is much higher than its
cytosolic concentration(≤ 50nM), procaspase-8 cannot dimerize by itseft in the cell [29].
Therefore, upon a cell death signal, the recruitment of procaspase-8 by its N-terminal
tandom DED domains into the death inducing signaling complex (DISC) increase the
local concentration, which promotes dimerizaton [30]. The DISC provides a platform for
enriching the local concentration of procaspase-8, leading to the autoproteolytic cleavage
of the inter-subunit linker [29].
This model seems to intimate two things. One, procaspase-8 should have some activity
Asp, Asp374 and Asp384, is necessary for activation of caspase-8. The first theory has
been proved in the experiments by using an inducible proximity system [31], which has
been confirmed by several researchers using different techniques [29, 28, 32]. The second
theory was proved several years later both in vitro and in cell culture by mutating the
two cleaved Asp residues at the linker region [33, 34]. The uncleavable procaspase-8 is
not able to cause apoptosis in caspase-8 deficient cells [35]. Activity assays of caspase-8,
which only contains one cleavable Asp at the inter-subunit linker, shows that cleavage at
the Asp384 can restore more activity than cleavage at Asp374 [36].
The biochemical data explained above provided evidence as to the importance of
dimerization and proteolysis at the linker region, but the detailed mechanism of activation
is still unknown. Recently, an NMR (nuclear magnetic resonance) structure of
procaspase-8 was solved [36]. From the structure, it was observed that procaspase-procaspase-8 formed the
core structure of caspases, the caspase fold. Most of the immature parts come from
the loops, which rearrange to form the conserved substrate binding site. This discovery
explains why procaspase-8 obtains some basal activity before processing at the
inter-subunit linker. However, whether the linker can be processed by the other protomer
is not clear. Previous data shows that procaspase-8 is more likely to be processed by
another dimeric caspase-8 [33, 32] or another enzyme, such as granzyme B [37]. Using
an inducible-dimeric system, it has been shown that dimerized procaspase-8 molecules
serve as a substrate for other proximate dimeric procaspase-8 or caspase-8 molecule.
This model is also supported by biological research which shows that a highly oligomeric
Fas-FADD complex is needed for the formation of DISC [38, 39] and the DISC which is
visible under high-resolution confocal microscopy [40]. Solution NMR was used to detect
the change from procaspase-8 to caspase-8[41]. Kelley and his colleagues used active
that the intra-dimeric model (cleavage between the two protomer in one dimer) is also
a potential proteolysis mechanism of procaspase-8. Whether procaspase-8 is cleaved by
intramolecular proteolysis is still unknown. However the structure of procaspase-8 shows
that the linker region is very flexible and might form a conformer which puts the cleavage
site near the active site. Therefore, procaspase-8 has multiple modes of activation.
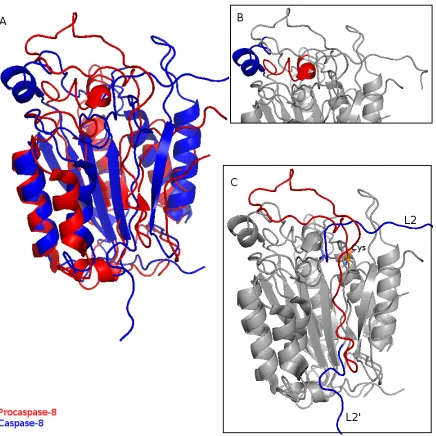
After the cleavage and removal of 10 amino acids from the inter-subunit linker, the
5 loops (L1-L5) undergo conformational rearrangement and forms a loop bundle, which
provides a functional active site. The loop bundle is conserved by all known crystalized
caspases [42, 43, 44]. Comparing the structures of procaspase-8 and caspase-8, loop 1(L1)
is rotated by about 90◦ and moved out from the active site (Figure 1.5(B)). The shortα -helix in L1 occupies the substrate binding site. This movement provides enough space for
substrate binding. L3 shifts toward the binding groove. The cleavage of the inter-subunit
linker makes loop 2 into two separate fragments, one is still called loop 2 (L2), which
represent the C-terminal of the p18 subunit, loop 2’ (L2’) indicates the N-terminal of
the small subunit. Loop 2, which contains the catalytic cysteine extends along the top of
the molecule toward the other monomer and shifts the Cys towards the binding site by
about 6 A◦ (Figure 1.5(C)). Spectrum shifts from procaspase-8 to caspase-8 show that
all these movements might happen in a induced-fit model during binding of substrate or
inhibitor [41]. The movements around the active site even change the substrate preference
of caspase-8 [35].
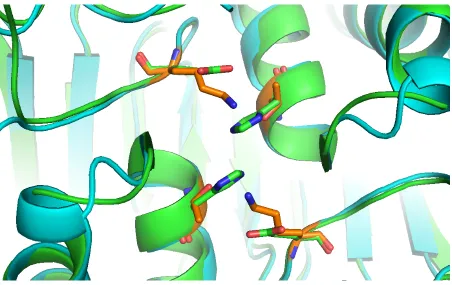
The dimer interface of caspase-8 obtains some different properties with other caspases.
In caspase-1, the inflammatory initiate caspase, Arg286 on loop2 forms a salt-bridge with
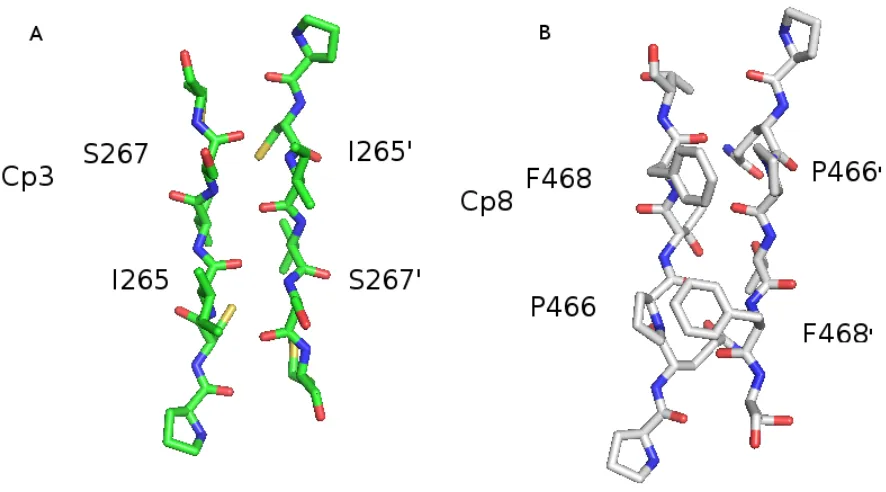
Glu390 across the dimer interface [45]. The effector caspase-3 does not have an equivalent
flat anti-parallel β-sheet (Figure 1.6(A)). In caspase-8, its dimer interface provides some obstacle for the dimerization of caspase-8. In α-helix 5, the corresponding position are replaced by Thr441 and Lys473, which cannot form any strong interaction. In the two
β-strand provided by each monomer, two bulky hydrophobic residues Pro466 and Phe468 make the dimer interface unflat (Figure 1.6(B)). The proline from one monomer and the
phenylalanine in the other monomer form a new hydrophobic dimer interface and require
more precise matching pattern in dimerization. The dimerization caused by recruiting to
the DISC or other complex make caspase-8 have better alignment in the dimer interface.
Completion the loop rearrangement allows caspase-8 to be fully activated. Subsequent
cleavage at Asp210 and Asp216 remove the two DEDs through an inter-dimer cleavage
mechanism, which completes the maturation of procaspase-8.
1.3
Caspase-8 has multiple functions in the cell
life-cycle
1.3.1
Caspase-8 participates in both extrinsic and intrinsic
apop-tosis pathways
Apoptosis, or programmed cell death, is a well known evolutionarily conserved, tightly
regulated process of cell suicide in all higher organisms [46, 47]. It is involved in several
tissues and different life stages. For example, the shaping of nervous system, organs and
extremities during cell development and differentiation needs the correct regulation of
apoptosis [48, 49]. Later, apoptosis participates in cell turnover to maintain homeostasis
of healthy tissues and also eliminates unhealthy cells which are under various signals,
Two classic apoptosis signaling pathways have been reported, the extrinsic pathway
and the intrinsic pathway [52]. The role of caspase-8 in the extrinsic pathway is most
clear. The extrinsic pathway starts by the recognition and binding of extracellular
apop-totic ligands to their corresponding receptors in the outer membrane. Ligands belong to
the tumor necrosis factor (TNF) superfamily cytokines, such as FasL (CD95) [53], Apo2
(TRAIL) [54], TNF [55] and TL1A [56]. Their receptors are all death domain (DD)
con-taining proteins that form high aggregation complexes after ligand binding [57, 58, 59].
Five death receptors were found and divided into two groups depending on whether they
can recruit procaspase-8. Receptors such as Fas, TRAIL-R1 and R2 recruit the adaptor
protein FADD into the complex through the homologous interaction of DD in FADD
with the cytoplasmic DD domain of receptors [60, 61]. With this same principle, caspase
8 and 10, which contain DED domains, are recruited to the complex through association
with FADD [60, 62]. The huge complex is called DISC (death induced signaling
com-plex). Upon DISC association, caspase-8 is activated by dimerization and autocleavage
through the mechanism discribed above. One unclear point is whether the fully activated
caspase-8 is released into the cytosol. This is unclear because it is possible that activated
caspase-8 becomes monomeric after release from the DISC [33]. There are evidence for
both observations. Using new approaches to characterize the spatial-temporal activity of
caspase-8, it was found that caspase-8 can cleave its substrates until associated with the
DISC. Furthermore, caspase-8 has higher activity before removing the prodomain [63].
On the other hand, polyubiquitination of caspase-8, instead of leading to the degradation
of caspase-8, has been reported to have another function, which is to stabilize the dimeric
form of caspase-8 after release from the DISC [64]. Therefore caspase-8 has two locations
suggests that caspase-8 participates in the intrinsic apoptosis pathway as well as the
previously characterized extrinsic pathway.
The intrinsic apoptotic pathway was originally thought to be an alternative
apop-totic pathway, which evolved from the extrinsic apopapop-totic pathway. Alternatively, the
two pathways form a dual fail-safe mechanism to ensure that the unwanted cells will
be removed for sure. However, using the Fas ligand to treat various cell lines revealed
that different cell lines prefered one of the two apoptotic signaling pathways [67]. The
discriminator between the two pathways is XIAP [68], the potent inhibitor of caspases
[69]. In type I cell, such as MCF-7, caspase-8 and caspase-3 were activated within 30 min
after the treatment of Fas. In type II cell lines, such as Jurkat, the cleavage of caspase-3
was delayed to about 60 min. The choice in the cell signaling pathway is that in type
I cell, the apoptosis pathway follows the extrinsic pathway discribed above. In type II
cells, very little DISC and active caspase-8 is observed, so these cells favor the intrinsic
pathways for apoptosis [70]. The role of caspase-8 in the intrinsic apoptotic pathways is
discribed below.
One role of caspase-8 in the intrinsic pathway is that it can mediate the cleavage of
Bid, a pro-apoptotic Bcl-2 family protein [66]. Bid is a BH3-only protein in Bcl-2 family
[71]. Full length Bid is generally thought to be inactive in the cytosol [72]. The N-terminus
of Bid is an inhibitor of the interaction of Bid with other BH domain-containing proteins
[73]. After cleavage by caspase-8, the C-terminal tBid can translocate and insert into the
outer membrane of mitochondria and induce the permeabilization of the mitochondria
outer membrane (MOMP) [74, 75]. Cytochrome c is released and initiates the formation of
the apoptosome thereby initiating the intrinsic apoptotic pathway. However, this model
is not so strictly executed. In caspase-independent apoptosis, fluorophore labeled Bid
membrane potential [76]. This phenomenon is consistent with the discovery that
caspase-8 can also be recuited to the mitochondria where it can directly cleave Bid. Several
mechanisms have been published about the recruitment or translocation of capsase-8 into
mitochondria. One caspase-8 binding protein, FLICE-associated huge protein (FLASH)
was reported as a subsidiary in facilitating caspase-8 activation on mitochondria [77]. A
mitochondria-specific lipid, cardiolipin, provides an anchor for caspase-8 to embed in and
oligomerize, [78]. The cardiolipin-rich domain performs as both a lipid-raft and the DISC,
which are activating platform for caspase-8. No matter which mechanism caspase-8 uses,
the activated caspase-8 will cleave Bid to initiate the intrinsic pathway.
In summation, caspase-8 is an initiator in both the extrinsic and intrinsic apoptotic
pathways. In the extrinsic pathway, after activation by the DISC, caspase-8 directly
activates the effector caspase-3. In the intrinsic pathway, procaspase-8 is activated by
lipid rafting and causes the release of cytochrome c and then activates another initiator
caspase-9. These two mechanisms guarantee that both type I and type II cells will trigger
programmed cell death when triggered by a death signal.
1.3.2
Necroptosis
Necroptosis, formerly called necrosis, is an alternative cell death pathway that has been
viewed as an accidental and unregulated cellular event. It was first discovered in murine
L929 fibrosarcoma cells where it was shown that if caspases were inhibited by the broad
caspase inhibitor zVAD-FMK, the cell would die via the necrosis pathway [79, 80].
Var-ious studies showed that necrosis is under strict control and regulation, and so necrosis
was renamed necroptosis. A genome wide siRNA screen was performed on necrotic cells
kinase RIPK1 was first identified as the initiator of necroptosis [82], although the latest
research shows that RIPK1 is not necessary in TNF induced necrosis[83]. Later,
multi-ple techniques were used to find that RIPK3 is downstream of RIPK1 and able to form
a necrosis-induced complex [84]. Further studies showed that RIPK3 was a molecular
switch in the cell’s metabolism, which is able to switch TNF-induced cell death from
apoptosis to necrosis [85].
Caspase-8 is recognized as a regulator of necroptosis because it can digest both RIPK1
and RIPK3. RIPK1 is composed of a N-terminal kinase domain and a C-terminal DD
domain. It is recruited to the membrane complex through interaction with other DD
containing proteins, such as the Fas receptor [86]. The cleavage of RIPK1 by caspase-8
inactives the kinase activity of RIPK1. The remaining DD domain stays in the protein
complex and facilitates the interaction between TRADD and FADD [87]. The
inhibi-tion mechanism of caspase-8 to RIPK3 is similar [88]. Therefore, caspase-8 is a negative
regulator of necrosis by removing the kinase domain of the initiator necrotic proteins
which allows the cell to choose between apoptosis and necroptosis. The processing of
RIPK1 seems to happen at an early stage, even before the maturation of caspase-8. For
example, analysis with an uncleavable procaspase-8 transgenic mouse indicates that the
non-apoptotic function was not influenced by this mutation [89]. Because caspase-8 will
change its substrate preference during maturation, RIPK1 might be a better substrate
for dimerized procaspase-8, not caspase-8. The cleavage of RIPK3 by caspase-8 does not
require high activity. The activity from caspase-8/F LIPL complex is enough to digest
RIPK3 and inhibit the necroptosis pathway [90]. Additionally, the activity of the
caspase-8/F LIPLheterodimer is lower than the caspase-8 homodimer as shown inin vitrostudies
[35, 91].
embryogenesis [92]. The key switch in this process is another substrate of caspase-8
CYLD, a RIPK1 K63 deubiquitinating enzyme [93]. Following TNF stimulation,
caspase-8 generates pro-survival signals by processing CYLD at Asp215, thereby switching cell
survival back to necroptosis. Although caspase-8 is not a component of the necroptosis
pathway, several key proteins in necroptosis are under strict regulation of caspase-8.
Therefore, caspase-8 is a very important regulator of necroptosis.
1.3.3
Non-apoptotic caspase-8 functions
The complexity of the caspase-8 enzyme comes from the evidence that its function is not
only apoptotic. Substantial evidence has been published that indicates that caspase-8
is also involved in several other cellular activities, such as embryonic development, cell
proliferation, mobility, differentiation, and NF-κB activation.
Cell development
Mouse embryos with depletion in caspase-8 perform abnormally in development, which
causes embryonic lethality at day e11.5 during gestation. Histological observations
re-vealed that heart muscle development were impaired and erythrocytes were accumulated
in caspase-8 knock-out embryos [94].In vitroculture using specific inhibitor treated mouse
embryos before implantation showed that caspase-8, but not caspase-2 and 9 is needed
for development [95]. Analysis of the substrates during development show that most of
them are related with non-apoptotic functions. Specific knock out of some substrates also
showed the same lethal phenotypes. Notably, the Casp8−/−/Rip3−/− double knock-out
mice survive and grow into adulthood [96]. This suggests that necroptosis which is
is seems immunocompetent, but keep accumulating abnormal T cells during
develop-ment. This result suggests that caspase-8 contributes to the control of homeostasis in the
immune system.
Differentiation
The prerequisite of caspase-8 activity in cell differentiation has been indicated in
sev-eral cell lines and tissues. Deletion of caspase-8 in mouse bone-marrow cells through a
conditional knock out using the Cre/loxP recombination system resulted in arresting
of monocyte differentiation into macrophages and cell death [97]. But, under the same
conditions, differentiation into dentritic cells and granulocytes was not affected. mRNA
profiles show that the mRNA of caspase-8 was down regulated in the monocytes
ac-tivation of differentiation [98]. So taken together, the differentiation of monocytes to
macrophages needs limited activity of caspase-8 to generate survival signals. Upon
stim-ulation of M-CSF, caspase-8 forms a complex with FADD, RIPK1, and the long isoform
of FLICE-inhibitory proteinF LIPL. Caspase-8 digests RIPK1 in the complex to prevent
the sustained activation of the NF-κB signaling pathway and also activates its down-stream executioner caspases [99], such as caspase-3 and caspase-6. Differential protein
profiles between monocytes and monocytes that undergo differentiation could elucidate
the caspase substrates. Mass spectrometry was used to identify the cleaved products [100].
Several proteins were identified as the downstream substrates of caspases, including
PAK-2, β-actin, and hnRNP-H, which participate in apoptosis, cytoskeleton rearrangements and differential transcriptional regulation.
In myeloid differentiation, the necessity of caspase-8 was confirmed by a
nucleophos-min (NPM) mutant that is found in myeloid leukemia. The mutated NPM specifically
apoptosis and also suppresses caspase-8 mediated myeloid differentiation [101]. Recently,
the same phenomenon was reported in the differentiation of cytotrophoblasts in forming
syncytiotrophoblasts [102, 103]. Multiple techniques show that caspase-8 is not involved
in the direct fusion of adjacent cells [104], but that caspase-8 activity is required for
preparation of trophoblasts fusion [105]. Interestingly, during these differentiation
pro-cesses, apoptotic phenomenon are always detected. For example, the phosphatidylserine
(PS) externalization is a hallmark of apoptosis, but it appears in the intercellular fusion
[106], and the cell does not die as expected. The discriminating mechanism between the
two phenomenon is still elusive. A possible hypothesis is that caspase-8 changes substrate
preference in different microenvironments.
Besides the effect of the catalytic domain of caspase-8, the DED domain, which is
removed after activation is also involved in cell differentiation at some stages of
devel-opment. The caspase-8 DED domain is accumulated during terminal differentiation of
epithelial cells. The disruption of cell differentiation caused by caspase-8 deficiency is
rescued by expressing DED domains in epithelial cells [107]. This discovery makes the
mechanism of caspase-8 regulation of cell differentiation even more complicated. More
sys-tematic and comprehensive biological research is needed to explain the effect of caspase-8
in cell differentiation.
Cell proliferation
The first evidence of a role of caspase-8 in cell proliferation was observed in studies
where caspase-8 activation was detected in TCR activated T lymphocyte proliferation
[108]. Later, various studies focused on the dual-role of caspase-8 in T cell proliferation.
of caspase-8 did not depend on Fas-FasL binding, and its activation was regulated to
prevent apoptosis. Caspase-8 is also involved in other immune cell activation
mecha-nisms. Studies on patients with immunodeficienies found an innate mutation at R248W
in caspase-8 [110]. This mutation abolished the catalytic activity of caspase-8 and
dis-tabilized the protein. As a result, dificiencies in activation of T cells, B cells and NK
cells were detected that led to the human immunodeficiency. To elucidate the function of
caspase-8 in immune cell proliferation, gene knock out techniques were used to discover
more components involved in the caspase-8 proliferation pathway. Expressing a dominant
negative FADD in T cells diminished its activation by anti-TCR antibody or exogenous
cytokines [111]. The cooperation between FADD and caspase-8 was discovered in
dou-ble knock out lymphocytes. FADD and caspase-8 form a feedback loop that limits the
outcome of autophagic signaling in T cell proliferation by preventing necrosis caused by
a hyperautophagic morphology [112]. During activation of T lymphocytes, no apoptotic
caspase-8 substrates were cleaved. The sequestering of caspase-8 was accomplished by
cF LIPL, a inhibitor of 8 [113, 114]. A lipid-raft mechanism activates of
caspase-8 by providing a platform for association of caspase-caspase-8 with the NF-κB adaptor [115]. This co-effect of caspase-8 and cF LIPL are also found in B cells [116]. Taken together,
the effect of caspase-8 in cell proliferation is very complicated. On one hand is that the
activation of caspase-8 is required for proliferation, and on the other, caspase-8 cannot
be over-activated to initiate apoptosis. The balance between the two opposite roles of
1.4
Caspase-8 is under regulation at several levels
1.4.1
Regulation by FLIP
cFLIP (cellular FLICE-inhibitory protein) was first reported to be an inhibitor of
caspase-8 [117]. The truncated form of FLIP, cF LIPS, contains two DED domains. The long
form of FLIP, cF LIPL, contains an additional C-terminal caspase-like domain in which
the catalytic Cys is replaced by Tyr. Both forms can interact with FADD and
caspase-8. cF LIPS can inhibit activation of caspase-8 in T cells by hampering dimerization
[118]. The function of cF LIPL is more controversial due to conflicting results in its
role in regulating caspase-8. On one hand, cF LIPL can block activation of caspase-8
by forming catalytically inactive heterodimer. On the other hand, the dimeric
caspase-8/cF LIPL complex has activity [119]. Upon caspase-8/cF LIPL formation, the small
levels of caspase-8 cleaved at FLIP can generate the p43 and p12 subunit. Both the
cleaved and uncleaved complex are able to bind their substrate, such as RIPK1 in a
DISC [87]. An opposing function of cF LIPL was determined by quantatively expressing
cF LIPL in vivo [120]. Expression of cF LIPL at physiological relevant levels enhanced
procaspase-8 activation and promoted apoptosis. A decrease incF LIPLlead to inhibition
of apoptosis. Only when expressed at high levels, cF LIPL can perform as an apoptotic
inhibitor. This result lead to the hypothesis that the determinant in deciding life or death
is which complex forms upon signal stimuli [7]. This hypothesis has been supported
by several phenomenon. For instance, this heterodimer complex is crucial in immune
cell proliferation. CD8+ T cell from cF LIPL transgenic cells have been shown increase
proliferation [121] by forming a complex with caspase-8. In embryo development, the
8 complex has been solved. The structure of the heterodimer is very similar to the
struc-ture of homodimer of caspase-8, with tiny differences in the dimer interface. There are
8 hydrogen bonds formed in the dimer interface in caspase-8 andcF LIPL heterodimeric
complex, compared with only 4 bonds formed in caspase-8 homodimer [123]. Loop 2’
resides in a closed conformation and provides additional binding energy. These
confor-mations are consistent with the in vitro biophysical studies which show that caspase-8
is more likely to form a heterodimer than a homodimer. In an inducible dimerization
system, although the cleavage of the inter-subunit linker is necessary for fully activation,
it is not necessary in the heterodimer. The uncleaved complex has different substrate
specificity [124]. These results suggests the differential regulation of cF LIPL. In silico
models which resemble HeLa cancer cells show the effect of the dimerization balance of
caspase-8 to its substrate, such as caspase-3 and -6 [125]. This technique might be used
to predict the consequences of a different dimeric form, which is caused by different ratio
of caspase-8 andcF LIPL.
1.4.2
Regulation by post-translational modifications
Phosphorylation is a common post-translational modification which can alter the function
and conformation of a protein. Caspase-8 is phosphorylated to regulate its activity.
Sev-eral kinases have been shown to phorphorylate caspase-8 at different sites. Biochemical
studies showed that the phosphorylation at Tyr380, which is located in the inter-subunit
linker is critical for promoting cell migration [126]. The phosphorylation of Tyr380 enables
the linker region to act as a Src homology 2 binding site providing a novel mechanism
of recruiting caspase-8 into the membrane. The association of caspase-8 with the Src
crucial for cell mobility and tumor metastasis. The phosphorylation and
dephosphory-lation of caspase-8 provides a mechanism of regudephosphory-lation of cell destination [128]. Because
RIPK1 amd RIPK3 are Ser/Thr kinases and under the regulation of caspase-8, caspase-8
is seldom able to be phosphorylated on Ser and Thr residues. However, the exemption
occures in the MAPK signaling survival pathway in neutrophils. Ser364 is
phosphory-lated by p38-MAPK which inhibits the activity of caspase-8 leading to the survival of
the cell [129]. A new site, Thr263 has been also reported to be phosphorylated by RSK2
and inhibit apoptosis [130]. This phosphoric modification mediated the ubiquitination of
caspase-8 and triggered its degradation.
The ubiquitination of caspase-8 mediated by Thr263 phosphorylation follows the
con-ventional role of the ubiquitination system. Not just caspase-8, but other caspases or
their regulators could be ubiquitinated and degradated to promote or inhibit apoptosis
[131, 132]. However, a new role of ubiquitination on caspase-8 was discovered which seems
to positively regulate caspase-8 [64]. Using mass spectrometry to analyze the components
of the DISC complex found that E3 ubiquitin ligase, CUL3, was a member of the
com-plex. Once polyubiquitinated by CUL3, caspase-8 recruited the ubiquitin-binding protein
p62 to the DISC complex [64]. The binding of p62 promoted aggregation of
polyubiqui-tinated caspase-8, keep dimeric of caspase-8 even release into cytosolic and leading to its
full activation. This mechanism provides a new mechanism for the release of caspase-8
to the cytosol. As discussed before, fully processed caspase-8 cannot maintain its dimeric
form without external facilitation. This p62-dependent binding to ubiquitin provides a
platform for maintaining dimeric caspase-8.
Caspase-8 can be sumoylated at K156 in the DED domain. This modification does
localiza-regulatory mechanism [133].
1.5
Project aims
This dissertation focuses on the effect of the dimer interface of caspase-8. Although all
cas-pases possess the same caspase fold, the behavior of initiator cascas-pases and executioner
caspase is different both in vitro and in vivo. Comparing the structure and hydrogen
bonding patterns of caspase-8 and caspase-3, we find a significant difference in the dimer
interface region. In caspase-3, the dimer interface forms an anti-parallel β-strand con-tains 4 hydrogen bonds. Residue H234 and E272 from α-helix 5 and 5’ create a network of salt-bridges across over the surface of protein. In the corresponding position, the
ini-tiator caspase-8 has different hydrogen bonds and interactions in the interface. It has two
bulky residues, Pro466 and Phe468, between the two central β-strand provided by each monomer. These two amino acid residues provide a twisted surface which increases the
difficulty of dimerization by requiring a more precise matching pattern in the
dimeriza-tion process. Mutadimeriza-tion of Phe468 to Ala has already been proved to abolish the activity
of caspase-8 [36], which suggests the importance of the dimeric pattern. Conversely, at
the corresponding position on α5 and α5’-helix, the small hydrophobic residue Thr441 and positive charged Lys473 cannot form an efficient salt-bridge. The goal of the project
presented here is to determine the proportion of the effect that the dimer interface
con-tributes to dimerization. The data obtained will compare the properties of wild type
caspase-8 and caspase-8 which contains the chimeric caspase-3 dimer interface to obtain
1.6
Figures
Figure 1.3: Constructs of caspase-8. The full length caspase-8 contains two DED do-mains in the N-terminus. Its catalytic domain is composed of the large subunit (p18) and the small subunit (p11). The five cleavage sites between the DED domain and the large subunit and the inter-subunit linker between the large and the small subunit are pointed by sticks. The bottom construct represents the short form of caspase-8 which only contains the catalytic domain. The short form of caspase-8 is soluble and can be expressed in E.coli.
Chapter 2
Materials and Methods
2.1
Materials
Dithiothreitol (DTT), acrylamide and imidazole were purchased from Acros. Sodium
chloride (NaCl), tryptone, yeast eatract, trizma base (Tris) and sodium citrate were
from Fisher. Q-Sepharose was from Amersham Biosciences. Monobasic and dibasic
phos-phate (KH2PO4,K2HPO4,´NaH2PO4,Na2HPO4), ampicillin, and molecular weight mark-ers were purchased from Sigma. IPTG(isopropyl β-D-l-thiogalactophranoside) was pur-chased from Anatrace. YM10 membranes were purpur-chased from Millipore. His-bind resin
and cell transfection kit were purchased from Novagen.
Ac-IETD-AFC(acetyl-IETD-7-amino-4-trifluoromethyl) was from Calbiochem. Mutagenesis kits were purchased from
Stratagene. All DNA restriction endonucleases were from NEB(New Engliand Biolab Inc).
Antibiotics such as penicillin/streptomycin were purchased from Invitrogen. Buffers were
filtered through either 0.45 or 0.22 µM filter membranes. The cross-linker DMP was purchased from Thermo scientific. The crysal screening kit were from Hampton research
Cell signaling; antibodies of caspase-3 and Hsp-90 were from BD transduction; Antibody
of FLAG was from Sigma. The ECL detection kit was from GE Healthcare. The cell line
HEK293A was from DR. Salvesen’s lab. DMEM medium was from corning cellgro. FBS
and trysin were from Thermo Scientific Hyclone.
2.2
Stock Solutions
Urea stock solution (10 M) was prepared in 20 mM potassium phosphate buffe (pH7.0)
as described previously [134, 135]. Molecular weight measurement was used to determine
the molarity of urea and refractive index was used to confirm it. 10 M urea stock were
made fresh and filtered before use with a 0.45 µmfilter.
2.3
Methods
2.3.1
Plasmid construction and mutagenesis
The procaspase-8 and caspase-8 genes without the N-terminal DED domain were first
mutated at adenosine 720 (the number follows full-length sequence) to cytosine. This
mutation will remove a XhoI digestion site without changing the amino acid sequence of
caspase-8. The gene was amplified by PCR from pET15b-CP8, using primers listed in
Table 1. The primers introduced a NdeI restriction site at the 5’ end and a XhoI at the
3’ end of the gene. The gel purified product was digested by NdeI and XhoI and then
ligated into the pET21b vector digested by the same enzymes.
Mutagenesis was done by the Stratagene quike change mutagenesis kit. The mutations
pcDNA3.1(-) was purchased from Invitrogen. The 8-amino acid FLAG tag was
syn-thesized by IDT with XhoI and EcoRIat 5’ end and 3’ end respectively. The separated
chains were dissolved in distilled and deionized water and heated to 95◦C, and then
cooled down very slowly to anneal. The annealed DNA was gel purified and digested
by XhoI and EcoRI simultaneously, then inserted into the digested pcDNA3.1(-). The
insertion and sequence of FLAG was confirmed by sequencing. The full length caspase-8
and procaspase-8(D5A) were ligated into pcDNA3.1(-) between XbaI and XhoI
restric-tion sites. The FLAG tag was conjugated to the 3’ end of caspase-8. The dimer-interface
mutations were added with the same method mentioned above. All the plasmids were
sequenced to confirm the mutations.
2.3.2
Protein Purification
All protein sample during purification were performed at 4◦C unless noted otherwise. All
procaspase-8 (wild type and mutated) were purified with a C-terminal-(His)6-tag from
E.coli BL21(DE3) lysS cells. The purification protocols are identical for all procaspase-8
purifications.
Cells were grown in Fernback flasks containing 1L of LB medium added with 50
µg/ml ampicilin at 37◦C. When the optical densigy of cultures reached 0.8 (absorbance measured at 600 nm), IPTG was added into the culture with a final IPTG concentration
of 0.4 mM to induce the expression of protein. The temperature was reduced to 25◦C
for 9 hours for protein expression. The cells were collected by centrifugation at 5,000
rpm for 15 minutes. After resuspended in lysis buffer (50 mM NaCl, 50 mM Tris, 50
mM imidazole, pH 7.9), the cells lysed with a French Press. The soluble protein was
step of protein purification is through a histidine affinity column. Protein with His-tag
binded to the column, was washed by lysis buffer and eluted with elution buffer (50 mM
NaCl, 500 mM imidazole, 50 mM Tris, pH 7.9). Imidazole was removed by dialyzing
the eluted protein against 50 mM Tris, 50 mM NaCl pH 7.9 overnight. The protein
was further purified by ion-exchange chromatography, the Q-sepharose column. Protein
was eluted with a salt gradient from 50-400 nM NaCl (50 mM Tris, pH 7.9). Bradford
assay was used to test which fractions contained protein. The bradford assay positive
fraction were collected, pooled, concentrated (YM 10 membrane) and dialyzed overnight
against 50 mM Tris, 50 mM NaCl pH 7.9 with 5 mM DTT added just prior to dialysis.
The concentrated protein was stored at−20◦C. The concentration of procaspase-8 were
determined using 280 of 23,380 M−1cm−1. The extinction coefficients were determined by the sequence of the protein.
The purification of caspase-8 without the N-terminal DED domain was similar to
the general protocol. After performing Q-sephrose chromatography, all the fractions were
collected and concentrated until nearly dry to enable the protein to digest itself to become
fully activated caspase-8. The membrane was washed at room temperature in 1 M Tris, pH
8.0 and supplemented with 10 mM DTT. The protein was washed off from the membrane
and dialyzed against 50 mM Tris, 50 mM NaCl with 5 mM DTT overnight and then 20
mM Tris, 10 mM DTT overnight. The self-cleavage of caspase-8 was checked by 12%
SDS-PAGE. If more than 90% of caspase-8 was cleaved into two chains, the sample was
2.3.3
Equilibrium folding studies
Equilibrium folding studies were performed as described in previously published paper
[136]. Time-course studies were first conducted to measure the unfolding time of
caspase-8. Various concentrations of protein in 20 mM potassium phosphate buffer were added
to various concentrations of urea from 0 M to 9 M. The final protein concentration was
4 µM, 2 µM and 1 µM. Emission scans were taken by excitation at both 280 nm and 295 nm while collecting emission data from 300 nm to 400 nm. The emission scan data
showed that caspase-8 was unfolded very fast, within 5 min, and the signal showed no
change after 6 M urea. Therefore, the titrator was used to perform the folding studies of
caspase-8.
The titrator used is Microlab 500 which is connected to the fluorometer (PTI).
Caspase-8 incubated in 9 M urea and 0 M urea buffer were connected to each syringe
of the titrator with equal caspase-8 concentration. The protein concentrations were 4
µM, 2 µM and 1 µM. A program was generated by PTI to direct the fluorometer to take emission of each sample and titrate protein sample into the cuvette. Caspase-8 in
0 M urea was first added into the 4 ml cuvette. After a emission scan was taken, 50 µl of caspase-8 in 9M urea was added to the cuvette. A stir bar with 280 rpm/min was
used to mix the protein sample for 1 min and a new emission scan was taken to
mea-sure the signal under this urea concentration. The unfolding of procaspase-8 is done as
urea concentration increased. These two steps were repeated for 40 times to reach a final
concentration of 6 M urea in the end. After the mixture, the urea concentration at each
circle can be calculated by the ratio of 0 M urea sample and 9 M urea sample.
ˆ
λ=
N
X
i=1 (Iiλi/
N
X
i=1
(Ii) (2.1)
The average emission wavelength versus urea concentration was plotted and analyzed.
The fluorescence data were fitted into the two-state model:
N *) U (2.2)
where N representes the native 8 monomer and U representes unfolded
caspase-8 monomer. We use [P] to represents the total molar concentration of the protein, so that:
[P] = [N] + [U] (2.3)
and the molar fraction of [N] and [U] is:
fN = [N]/[P] (2.4)
fU = [U]/[P] (2.5)
The ratio of fN and fU is equilibrium constant Keq:
Keq =fU/fN (2.6)
The Gibbs free energy is calculated by the following equation:
∆G=−RT ln(Keq) (2.7)
Under the assumption that the free energy change in each step and the concentration of
denaturant are linearly correlated [135], the free energy change in this studies is shown
as:
∆G= ∆GH2O−m
1[urea] (2.8)
where ∆GH2O stands for the free energy change in the absence of urea andm
1 stands
for the cooperativity index of the reaction. At every urea concentration tested, the
am-plitude of the fluorescence signal is assumed to be a linear combination of fractions from
each species.
Y =YNfN +YUfU (2.9)
where YN and YU represent the amplitudes of the fluorescence signals for the native
and unfold caspase-8. Then we assume that the amplitudes of both protein species are
linearly dependent on the concentration of urea:
YN =YN0 +m2[urea] (2.10)
YU =YU0 +m3[urea] (2.11)
whereYN0 andYU0 stands for the amplitudes of the signals for the native and unfolded caspase-8 in the absence of urea.
We use KaleideGraph to plot data. The equation for fitting a two-state equilibrium
((m1+m2∗m0) + (m3+m∗4∗m0)∗exp(M))/(1 +exp(M)) (2.12)
where;M = (−(m5+m6∗m0)/0.5921) (2.13)
where m0 is the urea concentration;m1 and m3 represents the y-intercepts of pre-transition and post-pre-transition; m2 and m4 stands for the slopes of pre-transition and post-transition; m5 represents the ∆GH2O and m6 is the m-value calculated from the fitting.
2.3.4
Size Exclusion Chromatography
Size exclusion chromatography can discriminate proteins through their hydrodynamic
volume and in some cases molecular weight[137]. The 200 10/300 GL column is composed
of crosslinked agarose and dextran. These two polymer can form a porous matrix. The
principle for separating protein is that protein with different size will elute through the
matrix at different rates. Larger proteins will flow faster because they cannot enter the
pores of the matrix. In the same way, smaller proteins have a longer route because they
will pass through fitted pores of the matrix. The retention time will be different because
of the variance in flow route. The column is attached to an AKTA FPLC (Amersham
Biosciences, Piscataway, NJ). The elution volumes of protein samples were determined by
taking absorbance measurement at 280 nm during the elution process. The absorbance
will be recorded by UNICORN control software and used to determine the molecular
weight of the sample.
The superdex 200 10/300GL sizing column can separate protein with molecular weight
used as standard and ran on the column. The elution volume were recorded and fitted
into a linear equation with the independent variable was the log of molecular weight of
the protein standard. The equation for weight calculation is:
y =−4.019x+ 21.45 (2.14)
where y is the elution volume and x is the log of molecular weight of the desired
protein.
2.3.5
Procaspase-8 Activity Assay
The activity of procaspase-8 (wild type and mutated) were measured using a Synergy 2
multi-mode microplate reader. The purified procaspase-8 was diluted into activity assay
buffer (20 mM Pipes (pH 7.5), 100 mM NaCl, 0.01% CHAPS, 10 mM DTT, 5% sucrose
and supplement with sodium citrate). The sodium citrate concentration is from 0M to
1M in 0.1 or 0.2 M increments. The samples were incubated at room temperature for 4
hours. The substrate Ac-IETD-AFC with a final concentrations ranging from 2 µM to 75µM were first added to a 96 wells plate. Procaspase-8 was injected into the wells with a final protein concentration of 50 nM. The fluorescence emission was measured at 505
nm after excitation at 400 nm and collected for 120 s.
The AFC references (RF) were collected by measuring the fluorescence emission of
AFC with different AFC concentrations (1 µM, 3 µM, 5 µM, 7µM, 9 µM). The slope of emission versus AFC concentration is the RF.
The slope of the fluorescence emission in the activity assay with different substrate
concentration is divided by the RF, which is calculated by the AFC reference assay. This
parame-ters of procaspase-8 are obtained by fiting the data to the Michaelis-Menten equation:
v = kcat[E0][S]
Km+ [S]
kcatis the turnover number and represents the maximum number of substrate molecules
converted to product per enzyme molecule per second. E0 is the enzyme concentration.
Km is the Michaelis constant which represents the substrate concentration when the
re-action rate is half of the maximum rate.S is the substrate concentration and we assume the substrate concentration is constant.
2.3.6
Kinetics of procaspase-8 activation
The activation of procaspase-8 is measured with Synergy 2 micro plate reader. Monomeric
procaspase-8 was diluted in activation buffer (1 M sodium citrate, 50 mM sodium
phos-phate (pH 7.5), 10 mM DTT, and 0.01% CHAPS) at various final protein concentrations
from 26.67 nM to 300 nM and incubated at room temperature. At indicated times, certain
amount of protein sample was titrated into the 96-well plate with final protein
concen-tration at 20 nM. Each well already contained the substrate, Ac-IETD-AFC with final
concentration at 50µM. This substrate concentration is high enough to saturate all the enzyme. Activity was determined at room temperature for the first 120 s of the
reac-tion to prevent the effect of substrate on caspase-8 dimerizareac-tion. The kinetic model and
M *) I *) D
m
A∗
where ’M’ represents monomeric procaspase-8, ’I’ represents intermediate dimeric
procaspase-8 which have low activity, ’D’ represents fully activated dimeric procaspase-8,
and A∗ represents procaspase-8 aggregation (no activity).
2.3.7
Chemical cross-linking
Wild-type procaspase-8 and mutated procaspase-8 were dialyzed against 50 mM sodium
phosphate buffer (pH 7.5) supplemented with 1 mM DTT in 4◦C overnight. The protein
was activated in the same phosphate buffer containing different concentration of sodium
citrate at room temperature. The concentration ranging was from 0-1 M. The final protein
concentration was 7µM. The cross-linker, dimethyl pymelimidate, was added with a final concentration from 5 to 100 µM and the samples were incubated at room temperature for 30 minutes. The protein was precipitated with 10% TCA in 4◦C and the pellet was
washed with acetone twice before analyzed via 12-18% gradient SDS-PAGE.
2.3.8
Analytical ultracentrifuge(AUC)
AUC experiments were performed on a Beckman Coulter Optima XL-I analytical
upon double-sector centrepieces and interference optics. The proteins were dialyzed at
4◦C in 50 mM sodium phosphate buffer with and without sodium citrate. The samples
were equilibrated at 24,000 rpm, and the the absorbance was measured at 280 nm. Three
concentrations were picked for each protein sample.
The sedimentation of the protein during ultracentrifuge is related to its mass [138].
The centrifuge force Fsedimentation is related to protein mass, distance to the center and
the speed.
Fsedimentation =mω2r−Vpρω2r (2.15)
where Vp stands for the partial volume and ρ stands for the density of the solution.
However, the frictional force opposeds the movement of protein:
Ff riction =vf (2.16)
where v is the migration rate of protein and f represents the frictional coefficient. After long periods of centrifugation, protein will stop migrating until the forces on it are
exactly balanced, that is when Fsedimentation = Ff riction. The mass of the protein can be
calculated by the molecular weightm. Then the particle’s volume Vp is calculated by the
equation:
Vp = ¯V m (2.17)
the ¯V is the partial specific volume. Partial specific volume is the volume change when unit gram (dry weight) of particles is dissolved in unit volume of the solute. For normal
specific volume of the proteins is contributed by each amino acid. It is calculated by their
weighted average. The partial specific volume of each amino acid is listed in Table 2.2. The
experimental data in sedimentation equilibrium assume a Boltzmann distribution[139].
Ar =cdexp{M(1−Vpρ)ω2(r2−r02)/2RT} (2.18) where Ar is the measured signal, crepresents the protein concentration at the end of
sample holder, is the molar extinction coefficient, d represents the pathlength,R is gas constant, andT is the absolute temperature. The molecular mass, M, can be calculated by fitting to this model.
If we assume procaspase-8 is in a reversible monomer-dimer self-association
equilib-rium 2M *) M2. The data can be fitted into the two-components model:
Ar =c1dexp{M(1−Vpρ)ω2(r2−r02)/2RT}+c22dexp{2M(1−Vpρ)ω2(r2−r20)/2RT} (2.19)
The relationship between monomer and dimer concentration follows mass action law
c2 =c21K12, so the equation can be rewritten as:
Ar =c1dexpM(1−Vpρ)ω2(r2−r20)/2RT +c 2
1K122dexp 2M(1−Vpρ)ω2(r2−r20)/2RT (2.20)
2.3.9
Caspase-8 crystalization
Crystal conditions screen
The purified caspase-8 with (P466I, F468V, T441H, K473E) was concentrated to 9.29
mg/ml and treated with a twofold molar excess of the reversible inhibitor (IETD-CHO)
at 4◦C for 1 h. The caspase-8-IETD complex was crystalized by hanging-drop vapour
diffusion. Commercial crystal screening kits were used to get the initial conditions for
crystalization. The complex of caspase-8-IETD were mixed with equal amounts of
reser-voir solution and incubated at 4◦C. Long rod-shape crystals grew out after a week. A
full data set was collected to a resolution of 3.32 A◦ at 100K at the SER-CAT beamline,
APS(Argonne,IL).
The optimum crystallization condition is 15% PEG6000, 5% MPD and 0.1 M MES pH
6.5. Similar conditions were generated around this condition by varying the concentration
of PEG6000 and MPD, and the pH of MES.
2.3.10
Cell culture and transfection
HEK-293A cells (ATCC:CRL-1573) were cultivated in Dulbecco’s modified Eagle’s medium
(DMEM). The medium was supplemented with 10% heat-inactivated fetal bovine serum
to make it complete. The HEK-293A cell is an adhere cell line. The frozen stock taken
from liquid nitrogen was warmed up and thaw in 37◦C water bath. After the stock was
thawed, fresh complete DMEM was added dropwise. Centrifuge at 1,000 rpm to collect
cell and remove the DMSO in the stock. Cells were gently resuspended in fresh DMEM,
then appropriate aliquots was inoculated into new flask with medium. Cells was incubated
in 37◦C incubator with 5% CO2.
conflu-ence. Remove culture medium and cells were rinsed by pre-warmed PBS. The cells were
trypsinized by trpsin-EDTA solution and the digestion was quenched by adding fresh
complete DMEM until cell layer was dispersed. The suspended cells were centrifuged
at 1000 rpm and medium were removed. Appropriate amount of fresh DMEM complete
medium was added and cells were gently resuspended by pipetting. Then proper number
of cells were inoculated into new flask with DMEM complete medium for subculture.
For transfection, the cells were split into a 24-well plate. When the confluence reached
50-80% , the cells were transfected by the Nanojuice transfection kit. The transfection
condition were optimized by varying ratio of DNA, Nanojuice core transfection reagent
and nanojuice transfection Booster. The best transfection condition for HEK293A is
determined as transfected 0.5µg of DNA per well with 0.5µlof core transfection reagent and 1 µl of transfection Booster. The core reagent and booster were mixed with 50 µl uncompleted DMEM and incubated at room temperature for 5 min. Total DNA (0.5
µg) was added into the mixture and incubated at room temperature for 15 minutes and then added into corresponding well. The negative control or the dilution vector for
transfected DNA was pcDNA3.1(-), positive control is pcDNA3.1(-)+caspase-3(V266E)
[42]. Cells were treated and harvested after 24 hours. For flow cytometry experiments,
cells were trypsinized in the well, resuspended in the culture medium, washed with cold
PBS twice and stained with Annexin V-PE and 7-AAD from the Apoptosis detection
Kit (BD pharmingen). Cells were analysed by FACS after incubation with Annexin
V-PE and 7-AAD in dark for 15 min under room temperature. The transfection efficiency
were calculated by transfecting a GFP gene into HEK293A cells. Individual experiments
were corrected by dividing the transfection efficiency and multiplying by 100 to give