Revealing signaling pathway deregulation by using gene expression signatures and regulatory motif analysis
Full text
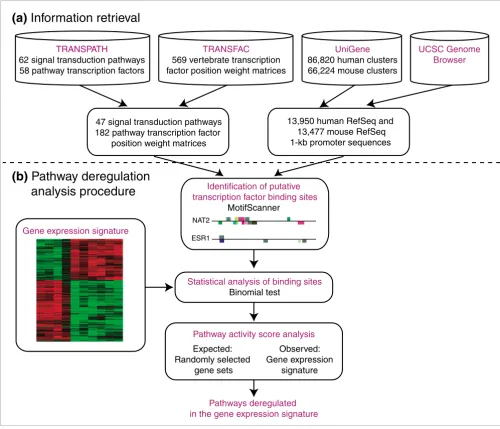
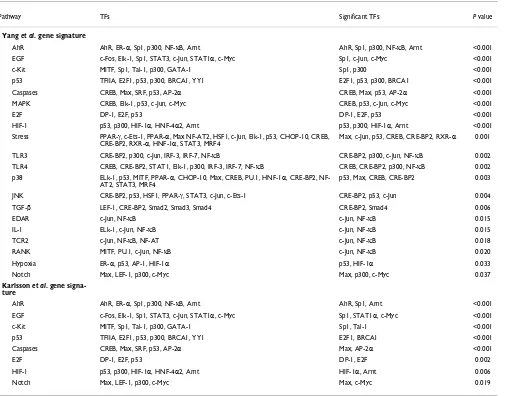
Figure


Related documents
Following Tannen’s (1994) findings it can be said that both males and females use this linguistic strategy in face-to-face communication, but the education of the speakers
However, the Swedish regulation has a broader scope (it also covers construction products intended for use in walls and ceilings in an indoor environment) and the
The proteome of BALF, mucus plugs, and epithelial cells from the lungs of elastase-exposed mice showed increased levels of all these proteins as compared with controls, except for
For the purposes of this paper, blended learning means integrating the online and face-to-face formats to create a more effective learning experience than either medium can
The extract of Carica papaya Linn and Tinospora cordifolia plant showed a significant (P < 0.05) dose-dependent of the antipyretic effect in yeast induced
In the present study, the results of meta-analyses suggest that genetic deletion of GSTM1 may contribute to increased susceptibility to NPC whereas GSTT1 polymorphism may not..
The voltage over the floating capacitor bank is controlled utilizing the excess switching vectors alongside an altered SVM conspire which stays away from
Skippers’ Meetings start promptly at 10am. Arrive early to rig your boat and get it on the beach. We would like to get the races started ASAP! Patrol boats are on the water, so
