0095-1137/03/$08.00⫹0 DOI: 10.1128/JCM.41.7.3284–3292.2003
Copyright © 2003, American Society for Microbiology. All Rights Reserved.
Nucleotide Sequence-Based Multitarget Identification
T. Vinayagamoorthy,* Kirk Mulatz, and Roger Hodkinson
Bio-ID Diagnostic, Inc., Saskatoon, Saskatchewan S7N 4N1, Canada
Received 9 January 2003/Returned for modification 21 March 2003/Accepted 10 April 2003
MULTIGEN technology (T. Vinayagamoorthy, U.S. patent 6,197,510, March 2001) is a modification of conventional sequencing technology that generates a single electropherogram consisting of short nucleotide sequences from a mixture of known DNA targets. The target sequences may be present on the same or different nucleic acid molecules. For example, when two DNA targets are sequenced, the first and second sequencing primers are annealed to their respective target sequences, and then a polymerase causes chain extension by the addition of new deoxyribose nucleotides. Since the electrophoretic separation depends on the relative molec-ular weights of the truncated molecules, the molecmolec-ular weight of the second sequencing primer was specifically designed to be higher than the combined molecular weight of the first sequencing primer plus the molecular weight of the largest truncated molecule generated from the first target sequence. Thus, the series of truncated molecules produced by the second sequencing primer will have higher molecular weights than those produced by the first sequencing primer. Hence, the truncated molecules produced by these two sequencing primers can be effectively separated in a single lane by standard gel electrophoresis in a single electropherogram without any overlapping of the nucleotide sequences. By using sequencing primers with progressively higher molecular weights, multiple short DNA sequences from a variety of targets can be determined simultaneously. We describe here the basic concept of MULTIGEN technology and three applications: detection of sexually transmitted pathogens (Neisseria gonorrhoeae,Chlamydia trachomatis, andUreaplasma urealyticum), detection of contaminants in meat samples (coliforms, fecal coliforms, andEscherichia coli O157:H7), and detection of single-nucleotide polymorphisms in the humanN-acetyltransferase (NAT1) gene (S. Fronhoffs et al., Carci-nogenesis 22:1405-1412, 2001).
DNA-based technologies such as the use of PCR and DNA probes (16, 17) have led to a wide range of genomic identifi-cation methods (1, 2, 3, 8, 12, 13, 20, 21, 32, 38, 40). However, the ultimate method for identifying a DNA target and, hence, a specific organism is considered to be the determination of a signature nucleotide sequence. The conventional chain dideoxynucleotide termination sequencing method (28) is con-sidered the “gold standard” for determining nucleotide se-quences, but it can process only one target at a time. This processing inefficiency of conventional sequencing increases the cost of screening for multiple pathogens and has therefore limited the use of this approach in routine diagnostic testing. Although attempts have been made to sequence multiple tar-gets simultaneously (4, 5, 7, 15, 24, 35), none could produce an electropherogram consisting of distinct nucleotide sequences from multiple targets. MULTIGEN (34) overcomes this prob-lem, and we describe here its basic concepts and present three examples of applications for routine diagnostic testing.
Figure 1A presents a schematic view of MULTIGEN tech-nology, showing the determination of DNA sequences from three genomic regions:␣,, and␦. Each of the three regions is amplified with corresponding pairs of primers: region ␦is amplified with primers i and i⬘, region  is amplified with primers ii and ii⬘, and region␣is amplified with primers iii and iii⬘. These specific genomic regions can be amplified simulta-neously by the multiple PCR (MPCR). As shown in Fig. 1B,
there are various options for designing the PCR primers around the segment of DNA that is of interest. Furthermore, the sequencing primer site can be located at different sites around the amplicon. Once amplicons from the ␣, , and ␦ regions are amplified, the desired regions of the amplicons can be sequenced. The determination of nucleotide sequences of known DNA targets is primarily used for microbial identifica-tion and genetic variaidentifica-tions such as single-nucleotide polymor-phisms (SNPs) wherein only short segments of the target DNA are sequenced. In order to restrict the length of the sequencing segments without a readthrough of the amplicon, a short stretch at the 3⬘end of the amplicon is sequenced. Thus, the generated DNA sequence will consist of the DNA sequence of the downstream PCR primer plus a variable number of nucle-otides from the target DNA sequence beyond the 3⬘end of the downstream PCR primer. Sequencing primer a is used to se-quence segment A on amplicon␣, sequencing primer b is used to sequence segment B on amplicon, and sequencing primer d is used to sequence segment D on amplicon␦(Fig. 1C). In order to analyze the truncated molecules generated from the different amplicon targets in a single lane, sequencing primers of various molecular weights are used. The sequencing primers are modified such that the molecular weight of sequencing primer b is greater than the combined molecular weights of sequencing primer a and segment A. The molecular weight of sequencing primer d is greater than the combined molecular weights of sequencing primer b and segment B. The sequences generated on the same gel from all three amplicons are shown schematically in Fig. 1D. The truncated molecules generated by the lower-molecular-weight sequencing primers will migrate first, followed by those generated by the larger sequencing * Corresponding author. Mailing address: Bio-ID Diagnostic, Inc.,
7, LFK Biotechnology Complex, 410 Downey Rd., Saskatoon, S7N 4N1 Saskatchewan, Canada. Phone: (306) 975-9161. Fax: (306) 938-0751. E-mail: [email protected].
3284
on May 15, 2020 by guest
http://jcm.asm.org/
primers. This prevents any overlap in the migration of the various series of truncated DNA molecules. They are then detected and recorded when they cross the path of a scanner in an automated sequencer. Therefore, by using a mixture of different sequencing primers of progressively increasing mo-lecular weights, different sets of truncated DNA molecules specific to each DNA target can be generated and detected simultaneously in a single lane by using a slab gel or in the same capillary tube by using capillary electrophoresis.
MATERIALS AND METHODS
Materials.Escherichia coliO157:H7 was supplied by the Department of Food Science, Faculty of Agriculture, University of Saskatchewan, Saskatoon, Saskatchewan, Canada. PCR-based detection kits for three high-risk subtypes of the human papillomavirus (HPV)—HPV18, HPV31, and HPV33—and three
sexually transmitted pathogens (Neisseria gonorrhoeae,Chlamydia trachomatis,
andUreaplasma urealyticum) were purchased from Maxim Biotech. These in-cluded primers and template DNA of the target organisms (Table 1). All PCR and sequencing primers (except the primers for HPV subtypes) were designed by Bio-ID Diagnostic, Inc., Saskatoon, Saskatchewan, Canada, by using Oligo 6.0 software (Life Sciences Software Resources) and synthesized by Sigma-Aldrich (Canada). Human DNA was purchased from Boehringer Mannheim GmbH. Primer modification can involve the attachment of a macromolecule with an appropriate molecular weight. One such modification includes coupling the primer to the macromolecule via a bifunctional linker molecule. The sequencing
primer is first synthesized with a C6 amino modification at the 5⬘end and is then
coupled to a carboxyl group on the linker molecule. The resulting complex is purified and coupled to the macromolecule via its single amino group.
Preparation of total DNA.Cultures ofE. coliO157:H7 were grown overnight in Luria-Bertani broth at 37°C in a shaker water bath. Total DNA from bacterial culture, as well as human genomic DNA from buccal swabs obtained from human volunteers, was extracted by using a QIAmp DNA Minikit (Qiagen). The purity and yield of the DNA was determined by using a spectrophotometer (Ultrospec 3000; Pharmacia Biotech, Cambridge, United Kingdom).
MPCR.MPCR of target amplicons was performed in a 50-l volume
contain-ing 5 l of 10⫻buffer (Bio-ID Diagnostic, Inc., and Maxim Biotech). The
corresponding thermocycling protocol is shown in Table 2. The amplified mul-tiple targets were purified by using PSIclone HTS (Princeton Separations,
Princeton, N.J.). Then, 5l of purified MPCR reaction mixture was separated on
2% agarose electrophoresis, stained with 0.1% ethidium bromide, and visualized under UV light at 254 nm on a transilluminator (FBTIV-88; Fisher Scientific) and photographed with a Polaroid photo documentation camera (FBPDC-34; Fisher Scientific).
Cycle sequencing and capillary electrophoresis.The respective amplicons were sequenced by using corresponding sequencing primers (Table 3) by cycle sequencing by using the ABI Prism BigDye terminator cycle sequencing ready reaction kit (version 3.0; PE Applied Biosystems) on a GeneAmp 2400 thermo-cycler (PE Applied Biosystems). Unincorporated dye terminators were removed by using Centricep chromatography columns (Princeton Separations). The sam-ples were then dried in a speedvac (DNA 120; ThermoSavant) and resuspended
in 20l of ABI Prism template suppression reagent. Samples were analyzed by
capillary electrophoresis by using the ABI Prism genetic analyzer 310. The
47-by-50-m uncoated capillary was filled with performance-optimized polymer
6 (acrylamide-urea polymer) and heated to 50°C. Next, 20l of the sequencing
mixture was transferred to an Eppendorf tube. Samples were drawn into the capillary by an electrokinetic injection at 2 kV for 75 s. The electrophoresis was carried out at 15 kV for 36 min.
RESULTS
To illustrate the basic concept and technical feasibility of MULTIGEN technology, experiments were performed with three HPV subtypes (HPV18, HPV31, and HPV33) to show that accurate identification of such closely related genomes could be achieved. The basic experimental model involved producing specific amplicons from plasmid clones carrying the complete genomes of HPV subtypes and then simultaneous cycle sequencing of the 3⬘-terminal end of the pooled ampli-cons by using sequencing primers of various molecular weights. The DNA sequences of the elecropherograms thus produced were BLAST searched (National Institutes of Health [NIH]) to identify homology between these respective loci of the target sequences and those in GenBank (NIH). Initially, we gener-ated three amplicons of 360, 350, and 413 bp from three sep-arate plasmid clones carrying target DNA segments (E6 re-gion) HPV18, HPV31, and HPV33, respectively (10, 26, 29). Two combinations of these amplicons were used as templates to simultaneously sequence the 3⬘ end of the amplicons. The FIG. 1. Schematic representation of novel features of MULTIGEN
technology. (A) Generation of amplicons␣,, and␦from three dif-ferent targets by MPCRs; (B) possible PCR primer sites for generating target amplicons and possible sites for sequencing primers; (C) relative molecular sizes and position of sequencing primers a, b, and d to the targets A, B, and D; (D) relative electrophoretic migration of trun-cated DNA molecular species produced from primer extension of targets A, B, and D.
on May 15, 2020 by guest
http://jcm.asm.org/
first experiment was designed to show that MULTIGEN could generate and read sequences simultaneously from two separate DNA targets (Table 4). We used two sequencing primers (HPV18 [molecular weight, 13,899] and HPV31 [molecular weight, 28,276]). The electropherogram produced two separate sequences consisting of 14 and 55 nucleotides, respectively. As determined by BLAST search, the sequences showed 100% homology with HPV18 and HPV31, respectively. As predicted, the electropherogram had a distinct “nonsignal” region be-tween the two sequences (Fig. 2A and C). In our next exper-iment, the number of targets was increased to three subtypes. Amplicons from all three HPV templates were simultaneously sequenced by using sequencing primers HPV18 (molecular weight, 13,899), HPV33 (molecular weight, 19,108), and HPV31 (molecular weight, 28,276). The electropherogram produced had three distinct sequences of 14, 19, and 55 nucle-otides accurately identifying HPV18, HPV33, and HPV31, re-spectively (Fig. 2B and D).
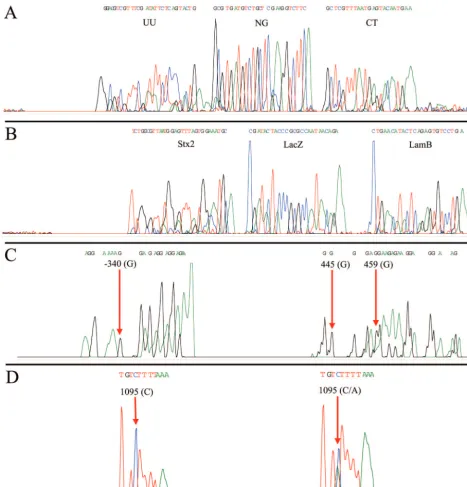
To show the utility of MULTIGEN technology in routine diagnostic testing, three applications were selected. In the first experiment we showed successful detection of three patho-gens—N. gonorrhoeae, C. trachomatis, and U. urealyticum— that are associated with sexually transmitted diseases (STDs). Signature DNA segments from all three STD targets were amplified by using MPCR, generating three amplicons of 298, 364, and 219 bp, respectively (Fig. 3A). The amplicons were simultaneously cycle sequenced by using specific sequencing primers. Figure 4A shows a single electropherogram consisting of 28 nucleotides ofU. urealyticum, followed by 25 nucleotides
ofN. gonorrhoeae, and then 23 nucleotidesC. trachomatis. A
BLAST search of the nucleotide sequence of the electrophero-gram detected 100% homology with specific DNA segments in
theureBgene (19), the CppB gene (39), and the cryptic plas-mid (31). In subsequent experiments we spiked these samples with DNA from HPV and were still able to obtain clean sig-nature sequences for all three targets with no interference. In our second experiment, signature DNA segments from the -galactosidase (lacZ) gene (14) representing coliforms, the lambda receptor (lamB) gene (6) representing fecal coliforms, and the Shiga toxin (stx2) gene (27) representing E. coli
O157:H7 were simultaneously amplified generating three am-pliconslamB(306 bp),lacZ(275 bp), andstx2(158 bp; Fig. 3B)
from the same E.coliisolate. These amplicons were simulta-neously sequenced by using specific sequencing primers. Fig-ure 4B shows a single electropherogram exhibiting all three signature nucleotide sequences of anstx2 sequence of 30
nu-cleotides, followed by alacZsequence of 25 nucleotides and a
lamB sequence of 24 nucleotides. A BLAST search of this electropherogram exhibited 100% homology with respective target gene sequences. To illustrate the specificity of MULTIGEN technology, 50 ng of the genomic DNA from the
E. coli isolate was spiked with genomic DNA from common
water protozoans: 2.5 ng ofCryptosporidium parvumand 2.5 ng
ofGiardia lamblia.Even with 10% contaminants we were able
to generate an electropherogram with all three signature nu-cleotide sequences for thelacZ,lamB, andstx2gene segments.
The sensitivity of MULTIGEN technology was estimated at two stages. The first was a sequencing step. Template titration shows that the optimal amount of double-stranded DNA per target is ca. 2 ng (12 fmol ⫽7.4⫻ 109copies) of the target
amplicon. The second stage was an MPCR step. We were able to generate all three-signature nucleotide sequences for the
lacZ,lamB, andstx2gene segments from 100 pg (0.032 fmol⫽
[image:3.603.40.545.81.335.2]1.9 ⫻ 107 copies) of the genomic the DNA. In our third
TABLE 1. PCR primers and corresponding amplicons
Application Target genome PCR primersa Amplicon size
(bp)
HPV HPV18 F, 5⬘-CACACCACAATACTATGGCGCGCT-3⬘ 360
R, 5⬘-CTGCTGGATTCAACGGTTTCTGGC-3⬘
HPV33 F, 5⬘-GCAGTAAGGTACTGCACGACTATG-3⬘ 413
R, 5⬘-CGACCCGAAATATTATGAAATCGT-3⬘
HPV31 F, 5⬘-TAAGCTCGGCATTGGAAATACCCT-3⬘ 350
R, 5⬘-CCTTCCTCCTATGTTGTGGAATCG-3⬘
Meat Stx2 F, 5⬘-CATATATCAGTGCCCGGTGTGA-3⬘ 158
R, 5⬘-GCATTTCCACTAAACTCCATTA-3⬘
LacZ F, 5⬘-AGGCATGATGCGACGCTTGT-3⬘ 275
R, 5⬘-TCTGTTATTGGCGCGGGTAG-3⬘
LamB F, 5⬘-TCTGGTCCTGGTGCCGGTCT-3⬘ 306
R, 5⬘-TCAGGACACTCTGAGTATGT-3⬘
STDs U. urealyticum F, 5⬘-CAGGAGCAATTAACTTCGCTGAAG-3⬘ 219
R, 5⬘-CAGTACTGAGAATATCGAAACGACGTC-3⬘
N. gonorrhoeae F, 5⬘-CTCTGCTTCGGCTCTCTGCTG-3⬘ 298 R, 5⬘-GAAGACCTTCGAGCAGACATCAC-3⬘
C. trachomatis F, 5⬘-GCAAGATATCGAGTATGCGTTGTTAGG-3⬘ 364 R, 5⬘-TTCATTGTACTCATTAAACGAGCGG-3⬘
NAT1 gene Amplicon 1 F, 5⬘-ATTCTCCTGCCTACATCAGAAG-3⬘ 354
R, 5⬘-AGGAAAACAAAACGAAAGC-3⬘
Amplicon 2 F, 5⬘-TTGATCAAGTTGTGAGAAGA-3⬘ 318
R, 5⬘-CTAGATACCAGAATCCATTCTCT-3⬘
aOrientation: F, forward; R, reverse.
on May 15, 2020 by guest
http://jcm.asm.org/
experiment, as an example of the ability of MULTIGEN tech-nology to identify human genetic markers, two segments of the human NAT1 (9) gene were simultaneously amplified from human genomic DNA by using MPCR primers producing two amplicons of 354 and 318 bp (Fig. 3C). These two amplicons were sequenced simultaneously by using modified sequencing
[image:4.603.43.537.79.391.2]primers. The electropherogram of the single-nucleotide poly-morphism (G/A) at locus⫺345 on the first segment and of the loci at 445 and 459 in the second segment are shown in Fig. 4C. Haploid genotyping was carried out independently on the third segment (620 bp) of the NAT1 gene for the C/A SNP at locus 1095. The homozygous C/C and the heterozygous C/A are TABLE 2. Conditions for PCR and MPCRa
Application Template/50l
Primer (amt
[pmol])/50-l
reactione
Thermocycler profile
HPV
MPCR 2.4⫻109copies of HPV18 HPV18 (10) 95° C/3-min denature
2.4⫻109copies of HPV33 HPV33 (10) (95°C/1 min, 55°C/1 min, 72°C/1 min), 35 cycles 72°C/10-min final extension; 4°C/hold
2.4⫻109copies of HPV31 HPV31 (10)
Cycle sequencing 4.8⫻1010total ampliconsb HPV18 (3.2) Step up]c(96°C/10 s, 50°C/5 s, 60°C/4 min), cycles: 25;4°C/hold
HPV33 (3.2) HPV31 (1)
Meat
MPCR 9.7⫻109copies of genomic DNA LacZ (5) 95°C/3-min denature
LamB (5) (95°C/1 min, 56.5°C/1 min, 72°C/1 min), 35 cycles 72°C/10-min final extension; 4°C/hold
Stx2 (10)
Cycle sequencing 2.7⫻1010total ampliconsb LacZ (1.6) (96°C/10 s, 50°C/5 s, 60°C/4 min), 25 cycles; 4°C/hold
LamB (1.6) Stx2 (1.6)
STD
MPCR 2.4⫻109copies of UU plasmid UU (5) 95°C/3-min denature
2.4⫻109copies of NG plasmid NG (5) (95°C/1 min, 56.5°C/1 min, 72°C/1 min), 35 cycles 72°C/10-min final extension; 4°C/hold
2.4⫻109copies of CT plasmid CT (5)
Cycle sequencing 2.2⫻1010total ampliconsb UU (1.6) (96°C/10 s, 50°C/5 s, 60°C/4 min), 25 cycles; 4°C/hold
NG (1.6) CT (1.6)
NAT1
MPCR 9.7⫻104copies of genomic DNA Amp1 (10) 95°C/3-min denature
Amp2 (10) (95°C/1 min, 54°C/1 min, 72°C/1 min), 35 cycles; 72°C/10-min final extension; 4°C/hold
Cycle sequencing 1.8⫻1010total ampliconsb Amp1 (1) Step upd(96°C/10 s, 55°C/10 s, 60°C/4 min), 25 cycles; 4°C/hold
Amp2 (1)
a
UU primer,U. urealyticumprimer; NG,N. gonorrhoeaeprimer; CT,C. trachomatisprimer; Amp1, amplicon 1; Amp2, amplicon 2.
b
Total amplicon calculation was based on the average size of the target amplicons. c
HPV33 primers were added initially; HPV18 primers were added after eight cycles, and then HPV31 primers were added after 20 cycles. d
Amplicon2 primers were run for 12 cycles, and then amplicon 1 primers were added for the remaining cycles. e
Cycle sequencing was performed with a 20-l reaction volume.
TABLE 3. Sequencing primers
Application Target genome Sequencing primera
HPV HPV18 5⬘-⌬-AATTTATTAATAAGGTGCCTGCGGTGCCAG-3⬘
HPV33 5⬘-⌬-AAAAAAAACGACATGTGGATTTAAACAAAC-3⬘
HPV31 5⬘-⌬-TATAACGTGTCAAAGACCGTTGTGTCCAGA-3⬘
Meat Stx2 5⬘-⌬-TCGTCACTCACTGGTTTCATCATA-3⬘
LacZ 5⬘-⌬-CCACGACGTTTGGTGGAATGTCTTTTGTGA-3⬘
LamB 5⬘-⌬-GCGCATCGAAAGACGGCTGGTTATTCACTG-3⬘
STDs U. urealyticum 5⬘-⌬-AACGAAGACAAAGAACGTAAAGTTGCTTAT-3⬘
N. gonorrhoeae 5⬘-⌬-CGTTTGTTGCTCTATGCTGGCGGCTTCGGT-3⬘ C. trachomatis 5⬘-⌬-CTGAAGAAAATTTGAGCAATTTCATTTTCC-3⬘
NAT1 gene Amplicon 1 5⬘-⌬-GAGAGATTCCAACTGGTATC-3⬘
Amplicon 2 5⬘-⌬-TTAATTTCTGGGAAGGATCAGCCTCAGGTG-3⬘
a⌬
, Modification to increase the primer molecular weight.
on May 15, 2020 by guest
http://jcm.asm.org/
[image:4.603.40.551.567.716.2]depicted in Fig. 4D. All of these experiments were repeated a number of times, some of them as many as 25 times, with identical results obtained with each and every repetition.
DISCUSSION
The determination of nucleic acid identity involves the bind-ing of an oligonucleotide to a specific segment of the target DNA. Some of the available methods (e.g., dot blot, microar-ray, etc.) stop at this stage, where detection is determined by relative fluorescent signals from the bound and labeled oligo-nucleotide probes, whereas others (e.g., PCR, ligase chain re-action, etc.) are processed further by polymerase-mediated reactions but are still assessed by the single fluorescent signal they produce (e.g., the size of amplicon or the release of signal quenching in real-time PCR). Features that distinguish MUL-TIGEN technology are that (i) MULMUL-TIGEN goes beyond mere target amplification and produces a distinctive and unique target sequence; that (ii) the technology involves three specific oligonucleotide primers per target, similar to nested PCR; and that (iii) as MULTIGEN generates n number of signals using all four nucleotides in a specific sequence, it increases the specificity of detection by at least 4ntimes relative
to probe- and PCR-based methods.
In order to avoid only the sequence of the downstream PCR primer showing up on the electropherogram, the sequencing primers are designed such that there are at least a few target specific nucleotides in between⫹1 of the annealing site and that of the 3⬘ downstream PCR primer. The truncated mole-cules generated during MULTIGEN cycle sequencing are less than 100 nucleotides long and therefore generate signals that are sharp, avoiding the broad signals associated with long trun-cated molecular species. We report three test models that demonstrate the potential place of MULTIGEN technology in routine diagnostic testing.
C. trachomatis, N. gonorrhea, and U. urealyticumare
com-mon causes of STDs in humans. Conventionally, these organ-isms are identified by culture and/or serological methods (25, 33). Compared to other DNA-based methods that identify only two of these organisms (i.e., C. trachomatisand/orN.
gonor-rhoeae) such as PCR (39), strand displacement (33), ligase
chain reaction (33), nucleic acid-based amplification (22), and ramification amplification (42), we show that a single MULTI-GEN test menu of STDs could include all of the important pathogens in routine clinical practices, significantly enhancing pathogen detection and thereby the level of care for patients with STDs.
Testing of food products includes tests for indicator organ-isms such as coliforms, fecal coliforms (18a), and toxin-pro-ducing pathogenic organisms such as E. coli O157:H7. Con-ventional testing includes the culture method (plate count) to determine bacterial load and acid and gas production in special growth medium (MacConkey broth or indicator media) (18), followed by confirmation by serological methods. In order to provide a test with better specificity, we show that MULTIGEN can detect the presence of coliforms, fecal coliforms, andE. coli
O157:H7 simultaneously. The speed and cost-effectiveness of MULTIGEN testing offer a significant potential contribution to-ward more effective inventory control in agribusiness.
TABLE 4. Species-specific nucleotide sequences and corresponding sequencing primers Primer (mol wt) Expected electropherogram afor: Locus and target sequence 1 Locus and target sequence 2 Locus and target sequence 3 HPV18 (13,899) HPV18 (436–450) and 5 ⬘ -AACCGTTGAATCCA-3 ⬘ HPV31 (28,276) HVP31 (445–486) and 5 ⬘ -AAGAAAAACAA AGACATTTGGATAAAAAGAAACGAT TCCACAACATAGGAGGAGG-3 ⬘ HPV18 (13,899) HPV18 (436–450) and 5 ⬘ -AACCGTTGAATCCA-3 ⬘ HPV33 (19,108) HPV33 (481–500) and 5 ⬘ -TTCATAATATTTCGGGTCG-3 ⬘ HPV31 (28,276) HVP31 (445–486) and 5 ⬘ -AAGAAAAACAA AGACATTTGGATAAAAAGAAACGAT TCCACAACATAGGAGGAGG-3 ⬘ UU (9,280) UU-UreB gene (257–284) and 5 ⬘ -GGACGTCGTTTCGATATTCTCAGTACTG-3 ⬘ NG (18,383) NG-CppB gene (3506–3530) 5 ⬘ -GCGTGATGTCTGCTCGAA GGTCTTC-3 ⬘ CT (28,048) CT-pCTT1 plasmid (453–475) and 5 ⬘ -GCTCG TTTAATGAGTACAATGAA-3 ⬘ Stx2 (7,253) Stx2 gene (492–521) and 5 ⬘ -TCTGGCGTTAATGGAGTTTAGTGGAAATGC-3 ⬘ LacZ (18,509) Lac operon (6216–6192) and 5 ⬘ -CGATACTACCCGCGCCAAT AACAGA-3 ⬘ LamB (27,921) LamB gene (824–847) and 5 ⬘ -CTGAACATAC TCAGAGTGTCCTGA-3 ⬘ NAT1 Amp1 (6,141) ⫺ 340 mutation and 5 ⬘ -TTATAAAC(G/A)T-3 ⬘ NAT1 Amp2 (30,958) 445 and 549 mutations and 5 ⬘ -CCTTGT(G/A) TCTTCC-3 ⬘ ;5 ⬘ -GTTTGAC(G/A)GA-3 ⬘ aSee definitions in Table 2, footnote a . bThe nucleotide range is given in parentheses.
on May 15, 2020 by guest
http://jcm.asm.org/
[image:5.603.52.274.71.723.2]The correlation of SNPs with human diseases (30) has gen-erated an interest in determining haploid genotypes, which include the determination of nucleotides at specific loci on both alleles. Although the determination of a nucleotide could
[image:6.603.108.481.71.432.2]be achieved by using single-nucleotide primer extension (23, 41), target-specific nucleotide probes (20), or single-nucleotide sequence analysis (11, 36), only MULTIGEN can provide both the nucleotide and the specific locus by identifying nucleotides FIG. 2. Electropherogram showing electrophoretic separation and sequences from multiple targets. (A) Electropherogram of raw data showing two distinct regions of fluorescence signals representing two stretches of sequences, (B) electropherogram of raw data showing distinct regions of fluorescence signals representing three stretches of sequences; (C) electropherogram of analyzed data showing two sequences—a 15-base nucleotide sequence generated by sequencing primer HPV18, followed immediately by a nonsignal, which is then followed by a 41-nucleotide sequence generated by sequencing primer HPV31; (D) electropherogram analyzed data showing three sequences—a 13-base nucleotide sequence generated by sequencing primer HPV18, followed immediately by a 20-nucleotide sequence generated by sequencing primer HPV33, followed immediately by a 41-nucleotide sequence generated by sequencing primer HPV31.
FIG. 3. Agarose gel electrophoresis showing amplicons from MPCRs. (A) Pathogens associated with STDs:C. trachomatis(CT; 364 bp),N. gonorrhoeae(NG; CppB gene, 298 bp), andU. urealyticum (UU;ureBgene, 219 bp). (B) Bacterial contaminants associated with meat: fecal coliforms (lamBgene, 306 bp), coliforms (lacZgene, 275 bp), andE. coliO157: H7 (stx2gene, 158 bp). (C) HumanN-acetyltransferase (NAT1) gene amplicon one (354 bp) and amplicon two (318 bp).
on May 15, 2020 by guest
http://jcm.asm.org/
[image:6.603.126.464.578.685.2]on either side of the SNP locus simultaneously at a number of SNP sites.
We have applied MULTIGEN technology here to deter-mine the nucleotide sequence from three segments (thelacZ
gene,lamBgene, andstx2gene) of the same microbial genome,
i.e.,E. coliO157:H7. This capability has applications in three areas. (i) The first is when DNA segments with desired traits are inserted in plasmid vectors to produce pharmaceuticals
[image:7.603.61.529.73.560.2]such as insulin via the -galactosidase fusion protein (37). MULTIGEN technology ensures the specific orientation of the insert in the plasmid host for proper expression and that all of the essential elements of gene expression such e.g., promoters are intact. (ii) The second application is in the posttranscrip-tional modification of eukaryotic RNA, leading to phenotypic abnormalities such as thalassemia (37). Simultaneously se-quencing a number of splicing regions of mRNA would ensure FIG. 4. Electropherogram of analyzed data showing U. urealyticum (UU; 28 nucleotides), N. gonorrhoeae (NG; 25 nucleotides), and C. trachomatis(CT; 23 nucleotides) (A);stx2gene (30 nucleotides),lacZgene (25 nucleotides), andlamBgene (24 nucleotides) (B); two segments of humanN-acetyltransferase single nucleotide polymorphism showing (G/A) SNP at locus⫺340 on the first segment and at 445 and 459 in the second segment (C); and homozygous wild type (control) showing cystine at locus 1095 with the test (human) sample showing heterozygocity (cystine/adenine).
on May 15, 2020 by guest
http://jcm.asm.org/
a proper template for translation. (iii) Finally, MULTIGEN technology can be applied to the detection of a chimeric ge-nome carrying a number of “foreign” DNA segments that could be used as biological-threat agents.
In summary, we illustrate here the scientific basis for simul-taneously obtaining multiple nucleotide sequences from viral, bacterial, and human genomic loci; the ability to subtype mi-crobes, emphasizing immediate applications in clinical testing and the food industry; human haplogenotyping; and the po-tential for determining genetic elements of “chimeric” ge-nomes that could be used as biological-threat agents. MULTIGEN technology adds a novel technical modification to the proven scientific principles of conventional sequencing and the gel electrophoresis process. MULTIGEN combines the desired characteristics of high sensitivity, high specificity, and cost-effectiveness that are paramount for critical medical decision making in routine high-volume diagnostic settings and allows the development of tests for the detection of virtually any combination of target sequences in any type of sample that contains nucleic acid material.
ACKNOWLEDGMENTS
We acknowledge Anette Kerviche of the Cancer Research Center, Saskatoon, Canada, for technical assistance with the HPV electro-pherogram, and the Department of Food Science, University of Saskatchewan, for providing bacterial cultures.
REFERENCES
1. Afshari, C. A., E. F. Nuwaysir, and J. C. Barrett.1999. Application of complementary DNA microarray technology to carcinogen identification,
toxicology, and drug safety evaluation. Cancer Res.59:4759–4760.
2. Aono, T., K. Kondo, H. Miyoshi, K. Tanaka-Taya, M. Kondo, Y. Osugi, J. Hara, S. Okada, and K. Yamanishi.1998. Monitoring of human cytomega-lovirus infections in pediatric bone marrow transplant recipients by nucleic
acid sequence-based amplification. J. Infect. Dis.178:1244–1249.
3. Bonora, S., M. C. Gutierrez, G. Di-Perri, F. Brunello, B. Allegranzi, M. Ligozzi, R. Fontana, E. Concia, and V. Vincent.1999. Comparative evalua-tion of ligaevalua-tion-mediated PCR and spoligotyping as screening methods for
genotyping ofMycobacterium tuberculiosstrains. J. Clin. Microbiol.37:3118–
3123.
4. Chee, M.1991. Enzymatic multiplex DNA sequencing.Nucleic Acids Res.
19:3301–3305.
5. Church, G. M., and S. Kieffer-Higgins.1988. Multiplex DNA sequencing.
Science240:185–188.
6. Clement, J. M., and M. Hofnung.1981. Gene sequence of the lambda
receptor, an outer membrane protein ofEscherichia coliK-12. Cell27(Pt.
2):507–514.
7. Creasey, A., L., Jr., D’Angio, T. S. Dunne, C. Kissinger, T. O’Keeffe, H. Perry-O’Keefe, L. S. Moran, M. Roskey, I. Schildkraut, and L. E. Sears. 1991. Application of a novel chemiluminescence-based DNA detection method to single-vector and multiplex DNA sequencing. BioTechniques
11:102–109.
8. Erlich, A. H.1992. PCR technology: principles and application. Oxford University Press, New York, N.Y.
9. Fronhoffs, S., T. Bruning, E. Ortiz-Pallardo, P. Brode, B. Koch, V. Harth, A. Sachinidis, H. M. Bolt, C. Herberhold, H. Vetter, and Y. Ko.2001. Real-time
PCR analysis of theN-acetyltransferase NAT allele *3,*4,*10,*11,*14 and
*17 polymorphism in squamous cell cancer of head and neck. Carcinogenesis
22:1405–1412.
10. Goldsborough, M. D., D. DiSilvestre, G. F. Temple, and A. T. Lorincz.1989. Nucleotide sequence of human papillomavirus type 31: a cervical
neoplasia-associated virus. Virology171:306–311.
11. Griffin, T. J, and L. M. Smith.2000. Single nucleotide polymorphism analysis
by MALDI-TOF mass spectrometry. Trends Biotechnol.18:77–84.
12. Hames, B. D., and S. J. Higgins.1995. Gene probe 1. Oxford University Press, New York, N.Y.
13. Hebart, H., D. Gamer, J. Loeffler, C. Mueller, C. Sinzger, G. Jahn, P. Bader, T. Klingebiel, J. Kanz, and H. Einsele.1998. Evaluation of murex CMV DNA hybrid capture assay for detection and quantitation of cytomegalovirus infection in patients following allogeneic stem cell transplantation. J. Clin.
Microbiol.36:1333–1337.
14. Hediger, M. A., D. F. Johnson, P. Nierlich, and I. Zabin.1985. DNA
se-quence of the lactose operon: the LacA gene and the transcriptional
termi-nation region. Proc. Natl. Acad. Sci. USA82:6414–6418.
15. Hubbell, E.2001. Multiplex sequencing by hybridization. J. Comput. Biol.
8:141–149.
16. Innis, M. A., D. H. Gelfand, J. J. Sninsky, and T. J. White.1990. PCR protocols. Academic Press, Inc., New York. N.Y.
17. Keller, G. H., and M. M. Manak.1989. DNA probes. MacMillan Publishers, Ltd., London, United Kingdom.
18. Koneman, W. E., S. D. Allen, V. R. Dowell, M. W. Janda, H. M. Sommers, and W. C. Winn.1988. Diagnostic microbiology. Lippincott Co., New York, N.Y.
18a.Kelly, M. T., D. J. Brenner, and J. J. Farmer III.1985.InE. H. Lennette, A. Balows, W. J. Hausler, and H. J. Shadomy (ed.), Manual of clinical micro-biology, 4th ed. American Society for Micromicro-biology, Washington, D.C. 19. Kong, F., G. James, Z. Ma, S. Gordon, W. Bin, and G. L. Gilbert.1999.
Phylogenetic analysis ofUreaplasma urealyticum: support for the
establish-ment of a new species,Ureaplasma parvum. Int. J. Syst. Bacteriol.49(Pt.
4):1879–1889.
20. Kwiatkowski, R. W., V. Lyamichev, M. de Arruda, and B. Neri.1999. Clinical, genetic, and pharmacogenetic applications of the Invader assay. Mol. Diagn.
4:353–364.
21. Lizardi, P. M., X. Huang, Z. Zhu, P. Bray-Ward, D. C. Thomas, and D. C. Ward.1998. Mutation detection and single-molecule counting using
isother-mal rolling-circle amplification. Nat. Genet.19:225–232.
22. Mahony, J. B., X. Song, S. Chong, M. Faught, T. Salonga, and J. Kapala.
2001. Evaluation of the NucliSens basic kit for detection ofChlamydia
trachomatisandNeisseria gonorrheaein genital tract specimens using nucleic
acid sequence-based amplification of 16S rRNA. J. Clin. Microbiol.39:1429–
1435.
23. Makridakis, N. M., and J. K. Reichardt.2001. Multiplex automated primer extension analysis: simultaneous genotyping of several polymorphisms.
Bio-Techniques31:1374–1380.
24. Monstein, H. J., Y. Johansson, and J. Jonasson.2000. Detection of vanco-mycin resistance genes combined with typing of enterococci by means of multiplex PCR amplification and multiple primer DNA sequencing. APMIS
108:67–73.
25. Morse, S. A. and S. K. Sarafian.1985. Sexually transmitted diseases, p.
863–876.InE. H. Lennette, A. Balows, W. J. Hausler, and H. J. Shadomy
(ed.), Manual of clinical microbiology, 4th ed. American Society for Micro-biology, Washington, D.C.
26. Pao, C. C., C. Y. Lin, J. S. Maa, C. H. Lai, S. Y. Wu, and Y. K. Soong.1990. Detection of human papilloma viruses in cervicovaginal cells using
polymer-ase chain reaction. J. Infect. Dis.161:113–115.
27. Ramachandran, V., M. A Hornitzky, K. A. Bettelheim, M. J. Walker, and S. P. Djordjevic.2001. The common Shiga toxin 2-containingEscherichia coli
serotypes and human isolates of the same serotypes possess a Stx2d toxin
type. J. Clin. Microbiol.39:1932–1937.
28. Sanger, F., S. Nicklen, and A. R. Coulson.1977. DNA sequencing with
chain-terminating inhibitors. Proc. Natl. Acad. Sci. USA74:5463–5467.
29. Seedorf, K., T. Oltersdorf, G. Krammer, and W. Rowekamp.1987. Identifi-cation of early proteins of human papilloma virus type 16 (HPV16) and
HPV18 in cervical carcinoma cells. EMBO J.6:139–144.
30. Shastry, B. S.2002. SNP alleles in human disease and evolution. J. Hum.
Genet.47:561–566.
31. Sriprakash, K. S., and E. S. Macavoy.1987. Characterization and sequence
of a plasmid from the trachoma biovar ofChlamydia trachomatis. Plasmid
18:205–214.
32. Tanke, H. J., J. Wiegant, R. P. van Gijlswijk, V. Bezrookove, H. Pattenier, R. J. Heetebrij, E. G. Talman, A. K. Raap, and J. Vrolijk.1999. New strategy for multi-colour fluorescence in-situ hybridization—COBRA: combined
bi-nary ratio labeling. Eur. J. Hum.Genet.7:2–11.
33. Van Dyck, E., M. Leven, L. Pattyn, V. Damme, and M. Laga.2001. Detection ofChlamydia trachomatisandNeisseria gonorrhoeaeby enzyme immunoas-say, culture, and three nucleic acid amplification tests. J. Clin. Microbiol.
39:1751–1756.
34. Vinayagamoorthy, T.March 2001. Multi-loci genome analysis. U.S. patent 6,197,510.
35. Wang, G., M. S. Rahman, M. Z. Humayun, and D. E. Taylor.1999. Multiplex sequence analysis demonstrates the competitive growth advantage of the
A-to-G mutants of clarithromycin-resistantHelicobacter pylori. Antimicrob.
Agents Chemother.43:683–685.
36. Wasson, J., G. Skolnick, L. Love-Gregory, and M. A. Permutt.2002. Assess-ing allele frequencies of sAssess-ingle nucleotide polymorphism in DNA pools by
pyrosequencing technonology. BioTechniques32:1144–1152.
37. Watson, D. J., M. Gilman, J. Witkowski, and M. Zoller.1997. Recombinant DNA, 2nd ed.W. H. Freeman & Co., New York, N.Y.
38. Winkler, M. A., J. Uher, and S. Cepa.1999. Direct analysis and identification ofHelicobacterandCampylobacterspecies by MALDI-TOF mass
spectrom-etry. Anal. Chem.71:3416–3419.
39. Wong, K. C., B. S. Ho, S. I. Egglestone, and W. H. Lewis.1995. Duplex PCR
system for simultaneous detection ofNeisseria gonorrhoeaeandChlamydia
trachomatisin clinical specimens. J. Clin. Pathol.48:101–104.
on May 15, 2020 by guest
http://jcm.asm.org/
40. Yager, T. D., L. Baron, R. Batra, A. Bouevitch, D. Chan, K. Chan, S. Darasch, R. Gilchrist, A. Izmailov, J. M. Lacroix, K. Marchelleta, J. Ren-frew, D. Rush-low, E. Steinbach, C. Ton, P. Waterhouse, H. Zaleski, J. M. Dunn, and J. Stevens.1999. High performance DNA sequencing, and the detection of mutations and polymorphisms, on the Clipper sequencer.
Elec-trophoresis20:1280–1300.
41. Ye, F., M. S. Li, J. D. Taylor, Q. Nguyen, H. M. Colton, W. M. Casey, M.
Wagner, M. P. Weiner, and J. Chen.2001. Fluorescent microsphere-based readout technology for multiplexed human single nucleotide polymorphism
analysis and bacterial identification. Hum. Mutat.17:305–316.
42. Zhang, W., M. Cohenford, B. Lentricha, H. D. Isenberg, E. Simon, H. Li, H. Yi, and D. Y. Zhang.2002. Detection ofChlamydia trachomatisby isothermal ramification amplification method: a feasibility study. J. Clin. Microbiol.
40:128–132.