0095-1137/11/$12.00 doi:10.1128/JCM.00276-11
Copyright © 2011, American Society for Microbiology. All Rights Reserved.
Development of a Highly Sensitive Genus-Specific Quantitative
Reverse Transcriptase Real-Time PCR Assay for Detection
and Quantitation of
Plasmodium
by Amplifying RNA
and DNA of the 18S rRNA Genes
䌤
Edwin Kamau,
1* LaDonna S. Tolbert,
2Luke Kortepeter,
1Michael Pratt,
1Nancy Nyakoe,
3Linda Muringo,
3Bernard Ogutu,
3John N. Waitumbi,
3and Christian F. Ockenhouse
1Division of Malaria Vaccine Development, United States Military Malaria Vaccine Program, Walter Reed Army Institute of Research, Silver Spring, Maryland1; Division of Entomology, Molecular Diagnostics, Walter Reed Army Institute of
Research, Silver Spring, Maryland2; and Walter Reed Project, Kenya Medical Research Institute, Kisumu, Kenya3
Received 8 February 2011/Returned for modification 22 March 2011/Accepted 31 May 2011
A highly sensitive genus-specific quantitative reverse transcriptase real-time PCR (qRT-PCR) assay for detection ofPlasmodiumhas been developed. The assay amplifies total nucleic acids (RNA and DNA) of the 18S rRNA genes with a limit of detection of 0.002 parasite/l using cultured synchronized ring stage 3D7 parasites. Parasite densities as low as 0.000362 parasite/l were detected when analyzing clinical samples. Analysis of clinical samples showed that detection of 18S rRNA genes from total nucleic acids increased the analytical sensitivity of the assay by more than 1 log unit compared to DNA only. When clinical samples with no parasites present by microscopy were analyzed by qRT-PCR, 90% (117 of 130) were positive for the presence of
Plasmodiumnucleic acids. Quantification of clinical samples by qRT-PCR using total nucleic acid versus DNA was compared to microscopy. There was a significantly greater correlation of parasite density to microscopy when DNA alone was used than with total nucleic acid. We conclude that analysis of total nucleic acids by qRT-PCR is a suitable assay for detection of low parasite levels in patients with early-stage malaria and/or submicroscopic infections and could greatly benefit malaria diagnosis, intervention trials, and malaria control and elimination efforts.
Malaria remains one of the most devastating infectious dis-eases in the world. After many years of neglect, various phil-anthropic agencies, such as Roll Back Malaria (http://www.rbm .who.int) and the Gates Foundation, have now committed to its eventual eradication (7). The success of these new initiatives hinges in part on the use of effective diagnostic and surveil-lance methods (11). However, despite the revolutionary gains from molecular approaches in diagnosis of malaria, micros-copy remains the gold standard for malaria diagnosis, clinical trial efficacy evaluation, and epidemiological surveys, despite its shortcomings (15, 16).
In expert hands, microscopy has a detection limit of 10 to 50 parasites/l (9, 12), but the average microscopist has a detec-tion limit of about 100 parasites/l, thereby limiting the use of microscopy in cases of low parasite burden (2). Studies have shown that even at submicroscopic infections, mosquitoes do get infected and can potentially transmit malaria (13, 19). Therefore, as we move to the era of malaria control and elim-ination, highly sensitive methods with high throughput capa-bilities will be critical in parasite detection and surveillance. Such methods will be important in quantifying the extent of
submicroscopic infections and giving a better insight into the dynamics of malaria transmission.
Molecular techniques for detection of specificPlasmodium
nucleic-acid sequences have enabled measurement of infec-tions that are 1 log unit lower than with microscopy or antigen detection tests (18, 20). For example, PCR assays allow explicit identification of malarial species and can be easily adapted for high-throughput application. Quantitative real-time PCR (qPCR) especially has improved the application of PCR be-cause the assay is fast, has very low risk of contamination, and is highly sensitive, specific, and quantitative. These qualities are not only ideal for diagnosis and clinical trial efficacy eval-uation, but will increasingly be needed for use in epidemiolog-ical surveys and to evaluate the success of malaria control and elimination campaigns. The superiority of PCR to microscopy as a diagnostic tool for detection of Plasmodium has been extensively described. For example, in a recent systematic re-view of surveys of populations in regions of endemicity in whichP. falciparumprevalence was measured by both micros-copy and PCR-based techniques, the prevalence of infection measured by microscopy was shown to be on average 50.8% of that measured by PCR (17).
Most PCR assays target DNA of thePlasmodiummulticopy 18S rRNA genes, which, due to their high copy numbers and mosaic of conserved and variable regions, provide an ideal molecular target for malaria parasite genus and species iden-tification and quaniden-tification. However, even slight genetic vari-ation within 18S rRNA gene sequences of the same species has been problematic. The Plasmodium genome lacks the long * Corresponding author. Mailing address: Division of Malaria
Vac-cine Development, Center for Molecular Diagnostics and Genomic Studies, Walter Reed Army Institute of Research, 503 Robert Grant Ave., Silver Spring, MD. Phone: (301) 319-7572. Fax: (301) 319-7358. E-mail: [email protected].
䌤Published ahead of print on 8 June 2011.
2946
on May 16, 2020 by guest
http://jcm.asm.org/
tandemly repeated arrays of rRNA genes found in other eu-karyotes. Instead, it contains several single 18S-5.8S-28S rRNA units distributed on different chromosomes, with the sequence encoded by the rRNA gene in one unit differing from the sequences of the corresponding rRNAs in the other units (8). Additionally, the expression of each rRNA unit is developmen-tally regulated, with different sets of rRNAs being expressed at different stages of the parasite life cycle (10, 21). As such, it is important to consider all these factors when designing a nu-cleic-acid sequence-based assay that targets 18S genes.
In this study, we describe the development of a highly sen-sitive genus-specific quantitative reverse transcriptase real-time PCR (qRT-PCR) assay for detection ofPlasmodiumand show that amplification of total nucleic acids (RNA and DNA) of the 18S rRNA genes significantly increases the analytical sensitivity of the assay. We also compare the quantification of clinical samples in detection ofPlasmodiumby qRT-PCR and microscopy.
MATERIALS AND METHODS
Samples.Samples used in this study were obtained from a Phase IIb pediatric clinical trial conducted between March 2005 and April 2006 at the KEMRI/ Walter Reed Project, Kombewa Clinic, in the Kombewa Division of Kisumu District, Nyanza Province, Western Kenya. The study was approved by the Ethical Review Committee of the Kenya Medical Research Institute, Nairobi, Kenya, and the Walter Reed Army Institute of Research Institutional Review Board, Silver Spring, MD.
The details of this study have been described elsewhere (23). Briefly, EDTA-treated blood samples were collected from study participants at enrollment (day 0) and 1 month after administration of the third and final vaccination. In addi-tion, blood was also drawn during unscheduled clinical visits from children who were sick and suspected to have malaria. For assessment of malaria, a peripheral blood smear was obtained from subjects who presented to the Walter Reed Project’s Kombewa Clinic with fever or a history of fever within 48 h or an illness that the attending doctor suspected might be due to malaria infection. After Giemsa staining, thin and thick blood smear slides from each sample were
independently examined by three expert microscopists for detection of
Plasmo-diumand counts where applicable. All malaria microscopists were fully trained
and were required to pass a competency and proficiency test prior to reading
slides for the study. Detection of asexual parasitemia of⬎0 parasites/l resulted
in the diagnosis of and treatment for malaria. The parasite density presented in this study is the average density obtained by the three independent (blinded from each other’s results) microscopists. Two hundred microliters of blood was
ali-quoted and stored at⫺20°C until it was required. Genomic nucleic acid was
extracted from whole blood using the QIAamp DNA Blood Mini Kit (Qiagen, Valencia, CA) as recommended by the manufacturer. Extracted nucleic acids
(NA) were stored at⫺20°C until they were required.
Primer and probe design.Primer and probe sets were based on 18S rRNA sequences deposited in GenBank and were designed using the Web-based soft-ware Primer3 v.0.4.0 (http://frodo.wi.mit.edu/primer3/) and/or Primer Express
Software (Applied Biosystem, Foster City, CA). ThePlasmodiumgenus primers
and probe were designed to amplify all units of rRNA distributed in all the chromosomes: 1, 5, 7, 11, and 13. They were also designed to amplify the two
types ofPlasmodium18S rRNA genes, the S type, expressed primarily in the
mosquito vector, and the A type, expressed primarily in the human host (8, 22). The regions of sequences selected were highly conserved and found only in the
genusPlasmodium. The sequence of the forward primer was 5⬘-GCTCTTTCT
TGATTTCTTGGATG-3⬘, and that of the reverse primer was 5⬘-AGCAGGTT
AAGATCTCGTTCG-3⬘. The probe sequence was 5⬘-ATGGCCGTTTTTAGT
TCGTG-3⬘, labeled with 5⬘FAM (6-carboxyfluorescein) and 3⬘TAMRA
(6-carboxytetramethyl-rhodamine) as the reporter and quencher, respectively. For theP. falciparumspecies-specific primers and probe, we used previously pub-lished sequences but used VIC instead of FAM as the reporter dye (18, 20).
qRT-PCR and qPCR.Amplification and real-time measurements were per-formed in the Applied Biosystems 7500 analytical PCR system with the following thermal profile for qPCR: 10 min at 95°C, 40 cycles of 15 s at 95°C, and 1 min at 60°C. For qRT-PCR, a 30-min cycle at 50°C was added as the initial step for the
reverse transcription process. For the reaction, 1l of template was added to 9
l of reaction master mix containing 1⫻QuantiTect Probe RT-PCR Master Mix
(Qiagen), 0.4M each primer, 0.2 M probe, and 4 mM MgCl2. For the
qRT-PCR assay, QuantiTect RT Mix (a blend of Omniscript and Sensiscript Reverse enzymes) was added to the reaction master mix as recommended by the
manufacturer at a rate of 1l per 100l of the reaction master mix.
Standard curves.For standard curves, cultured highly synchronized ring stage 3D7 parasites were used in order to emulate infected human blood samples. The percent parasitemia of the ring stage was determined by flow cytometry and
microscopy. To determine the number of parasites/l in culture material, we
multiplied the percent parasitemia by the number of red blood cells (RBCs)/l,
which were counted by Coulter analysis (Coulter AC䡠T 5 diff CP; Beckman
Coulter, Inc., Miami, FL). The limit of detection (LOD) for the PCR assays was established by creating a standard curve using cultured synchronized ring stage 3D7 parasites that were serially diluted using uninfected whole blood prior to total NA extraction. When analyzing and quantifying clinical samples, each 96-well plate was run with the standard 3D7 NA, which was serially 5-fold diluted
from 20,000 parasites to 0.256 parasite/l. Total nucleic acid (RNA and DNA)
was extracted using a QIAamp DNA Blood Mini Kit with 200l of blood (or
cultured material) and eluted in 200l of water. For each experiment, we used
1l of NA template, which is equivalent to 1l of blood from a patient or
cultured material.
Statistical analysis.For statistical analysis, a two-tailed pairedttest in Graph-Pad prism was used.
RESULTS
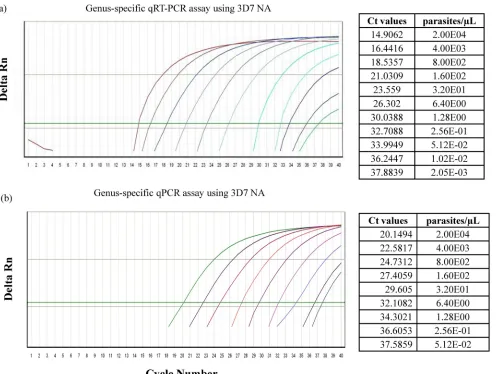
Comparison of limits of detection between qRT-PCR and qPCR assays. Standard 3D7 NA was used to establish the LOD, which was set as the lowest NA concentration at the threshold cycle number (CT) at which the normalized reporter dye emission rose above background noise. For the genus-specific qRT-PCR assay, the LOD was determined to be 0.002 parasite/l, and the LOD was 0.0512 parasite/l for the qPCR assay (Fig. 1a and b). For theP. falciparum species-specific assay, the LOD was determined to be 1.22 parasites/l for qRT-PCR and 2.44 parasites/l for qPCR (data not shown). We assessed the reproducibility of the qRT-PCR and qPCR genus-specific assays with respect to both intra- and interop-erator variability on replicate samples conducted on different days. The qRT-PCR assay was found to be more sensitive over a wide dynamic range of known parasite densities than the qPCR assay. Both assays were highly reproducible, with a mean coefficient of variation of 3% between different opera-tors performing assays on different days (Fig. 2). Next, we randomly picked a clinical sample that had been established to beP. falciparumpositive by microscopy and assessed the LOD for both genus-specific andP. falciparumspecies-specific assays by serially diluting the sample. The LOD for the genus-specific assay was established to be 0.00661 parasite/l for qRT-PCR and 0.0297 parasite/l for qPCR (Fig. 3a and b). For theP. falciparumspecies-specific assay, the LOD was established to be 1.82 parasites/l for qRT-PCR and 3.41 parasites/l for qPCR (data not shown). We tested the specificities of the assays by ensuring the assays did not amplify human NA.
Establishing assay sensitivity by inclusion of a reverse transcriptase step in clinical samples.We then compared the performance of qRT-PCR and qPCR in 603 clinical samples using genus-specific orP. falciparumspecies-specific assays in a pairedttest. There was a significant difference in the perfor-mance of qRT-PCR and qPCR for both genus-specific andP. falciparum-specific assays. For the genus-specific assay, theCT
values for qRT-PCR and qPCR were significantly different from each other (P⬍0.0001), with means⫾standard errors of the mean (SEM) of 17.69⫾ 0.2393 and 22.44⫾ 0.2373,
re-VOL. 49, 2011 TOTAL NUCLEIC ACID PCR ASSAY FOR MALARIA DIAGNOSIS 2947
on May 16, 2020 by guest
http://jcm.asm.org/
spectively. The difference between the meanCTvalues for the qRT-PCR and qPCR assays was 4.757⫾ 0.3370. For theP. falciparumspecies-specific assay, theCTvalues for qRT-PCR and qPCR were significantly different from each other (P ⬍
0.0001), with means⫾SEM of 25.27 ⫾ 0.2564 and 27.12⫾ 0.2343, respectively. The difference between the meanCT val-ues for qRT-PCR and qPCR was 1.756⫾0.3713. To show how inclusion of the RT step in the qPCR assay increased the sensitivity of the assay, the difference in theCT(⌬CT) for each clinical sample between the qRT-PCR assay and the qPCR assay was plotted against the parasite density as determined by a thick blood smear (Fig. 4). Over 5-log-unit differences in parasite density, the inclusion of the reverse transcriptase en-zyme in the qPCR assay increased the sensitivity of the assay (samples with netCTvalues of⬎0).
[image:3.585.49.547.69.443.2]Comparison of quantification by microscopy to qRT-PCR quantification.We analyzed clinical samples that had no par-asites based on microscopy using genus-specific qRT-PCR as-says. Of the 130 samples analyzed, 117 (90%) were positive by
FIG. 1. Amplification plot showing LODs for detection ofPlasmodium. To establish the LOD, standard 3D7 NA was serially 5-fold diluted from 2.00E4 to 4.10E⫺4 parasite/l, and then qRT-PCR or qPCR was run using a genus-specific assay. (a and b) Amplification plots showing LODs by qRT-PCR using standard 3D7 NA. The lowest amplification for qRT-PCR was 2.05E⫺03 parasite/l (a), and for qPCR it was 5.23E⫺02 parasite/l (b). Delta Rn, magnitude of normalized fluorescence.
FIG. 2. Reproducibility of the genus-specific assay. Data are from serially diluted standard 3D7 NA assayed on different days by different operators, represented by different colors on the graph. The data show that the assay is highly reproducible.
on May 16, 2020 by guest
http://jcm.asm.org/
[image:3.585.302.541.498.687.2]qRT-PCR, with a mean CT value of 19.60 and lowest and highestCTvalues of 11.46 and 39.41. TheseCTvalues corre-spond to quantitative values of 4.65 ⫻ 104 parasites/l and
0.000362 parasite/l, respectively. We next determined whether inclusion of the reverse transcriptase enzyme in the qPCR assay affected the quantification of the parasites in the blood over a range of parasite densities (42 to 1.17E6 parasites/
l). From 466 clinical samples, we correlated the parasite den-sity as determined by microscopy with both qRT-PCR and qPCR genus-specific assays (Fig. 5) and measured the statisti-cal significance of each assay for either all samples or samples whose parasite densities were stratified into subgroups. There was a statistically significant correlation between the parasite density measured by microscopy and either the qRT-PCR (Fig. 5a) or qPCR (Fig. 5b) molecular assay. However, the qPCR assay outperformed the qRT-PCR assay for each subgroup examined. The correlation was weakest at low parasite
densi-ties in both assays, with increasing divergence of the 95% confidence intervals as the parasite density decreased.
Diluting clinical samples extends the qRT-PCR dynamic range.We observed that at high parasite densities, quantitative PCR reached a plateau, limiting the dynamic range of the qRT-PCR. We hypothesized that the dynamic range of the qRT-PCR can be extended by further diluting the clinical samples. As such, we randomly picked 95 samples with CT
values of⬍18 and performed a 10-fold serial dilution of the NA to 10⫺4. The diluted samples were analyzed by
genus-specific qRT-PCR assay. Before dilution, the mean parasite equivalent as determined by the qRT-PCR assay was 2.09E4 parasites/l, but after dilution, the mean parasite equivalent increased to 4.33E5 parasites/l. Interestingly, the mean asite density of these samples by microscopy was 2.41E5 par-asites/l, clearly showing that dilution of extracted NA corre-lates well with microscopy at high parasite densities. These FIG. 3. Amplification plot showing LODs by qRT-PCR using a clinical sample that was serially 5-fold diluted. The lowest amplification for qRT-PCR was 6.61E⫺03 parasite/l (a) and for qPCR was 2.97E⫺02 parasite/l (b).
VOL. 49, 2011 TOTAL NUCLEIC ACID PCR ASSAY FOR MALARIA DIAGNOSIS 2949
on May 16, 2020 by guest
http://jcm.asm.org/
data represent more than 1 log unit increase in the number of parasites detected by qRT-PCR after diluting the clinical sam-ples, proving our hypothesis to be true.
DISCUSSION
The detection of parasites in subjects subclinically infected with eitherP. falciparumorP. vivaxmalaria is a stated goal of FIG. 4. Addition of the reverse transcriptase enzyme to the qPCR assay increases sensitivity. TheCTvalues of clinical samples were determined
using qRT-PCR and qPCR. The difference inCT(⌬CT) for each clinical sample was determined and plotted against the parasite density as
determined by a thick blood smear. An increase in the sensitivity of the assay by addition of the RT step was seen in all log groups.
FIG. 5. Inclusion of the reverse transcriptase enzyme in the qPCR assay affects the quantification of parasites. The log parasite density of clinical samples determined by microscopy was compared to correspondingCTvalues obtained by qRT-PCR or qPCR. There was a statistically
significant correlation between parasite densities measured by microscopy and both PCR assays, with qPCR outperforming qRT-PCR.
on May 16, 2020 by guest
http://jcm.asm.org/
the malaria vaccine and drug development programs at the Walter Reed Army Institute of Research because of the in-creasing reliance on the human malaria challenge model to assess the efficacy of malaria vaccines or to evaluate new an-timalarial drugs. Because the threshold for fever and clinical disease for both P. falciparum and P. vivax malaria is ⬍10 parasites/l blood, new and reliable diagnostic tests that are ultrasensitive and specific for malaria are needed to replace the Giemsa-stained thick blood smear, the current gold stan-dard test. It has been proposed that concomitant use of a molecular-based assay for detecting Plasmodium parasites would be an excellent safeguard against possible false-negative results, as determined by expert microscopists (3). Here, we have described a highly sensitive qRT-PCR assay for detection of the genusPlasmodium. The assay detects both rRNA and DNA, taking advantage of the high copy numbers of rRNA present per genome in a parasite. The assay has an LOD of 0.002 parasites/l, which is one of the lowest LODs reported thus far for detection ofPlasmodium. During the analysis of the clinical samples in this study, we detected the presence of parasite NA in samples that were below the LOD as estab-lished using the standard P. falciparum 3D7 strain. For in-stance, on two separate occasions, the assay detected two sam-ples that had 0.000362 and 0.0004 parasite/l, which to our knowledge is the lowest parasite density reported thus far in detection ofPlasmodiumby PCR.
Under ideal conditions, during PCR amplification, the am-plicon amount doubles every cycle (i.e., increases by 1 log2), and one ⌬CT unit thus corresponds to a 2-fold increase in analytical sensitivity (4). Here, we have shown that introduc-tion of the RT step in the genus-specific assay improved the mean analytical sensitivity of the assay approximately 10-fold. We have also shown that introduction of the RT step improved the analytical sensitivity of the P. falciparumspecies-specific assay by more than 3-fold. Several PCR assays for detection of the genus Plasmodium have been described previously. For example, Elsayed et al., (6), using molecular beacon probes, developed an 18S rRNA-based assay that could detect 0.004 parasite/l blood but required two separate PCRs. However, careful consideration of the results reported is critical, as there are many variables that affect the sensitivity of PCR assays. The sensitivity of PCR assays in detection ofPlasmodiumat the genus or species level is commonly described in para-sites/l or percent parasitemia. While many laboratories might follow similar protocols, factors such as the level of parasite synchronization of cultured samples, dilution ranges, and methods of nucleic acid extraction and molecular assay detec-tion (real-time PCR, molecular beacon, nucleic acid sequence-based amplification [NASBA], or gel electrophoresis) all con-tribute to differences in sensitivity, making cross-platform and cross-study comparisons difficult. As we move toward consid-ering PCR assays the gold standard for malaria diagnosis and surveillance, not only in clinical research, but also in future monitoring and evaluation efforts in malaria control and elim-ination campaigns, stringent standardization of such details will be required. The sensitivity of any new molecular-based diagnostic test must be compared to the current gold standard, microscopy of thick blood smears. The difficulties associated with malaria diagnosis by microscopy are numerous, and
ac-curacy depends on the microscopists’ ability, training, experi-ence, and motivation and the availability of laboratory re-sources. Microscopy can miss a substantial proportion ofP. falciparuminfections, in populations where malaria is endemic or nonendemic. Malaria microscopy is a highly perishable skill requiring continued training, practice, and testing of micros-copists to maintain high-level proficiency (16). There is wide variation among “expert” microscopists in assessing parasite density and distinguishing different parasite species. The Wal-ter Reed experience in Africa and Southeast Asia has demon-strated the need for uniform training, quality assurance, and standardized reporting methods to minimize errors that occur with microscopy (5, 15). Literature analysis shows that there are frequent discrepancies and discordant results between mi-croscopy and PCR techniques. In a recent review by Berry et al. (3), it was noted that greater than 17% of thin blood smears examined by microscopy were corrected after being checked by PCR. Additionally, the rate of misdiagnosis varied from 20% to 50% forP. malariae,P. ovale, andP. vivax. In mixed infec-tions, almost all microscopy results were inaccurate. Using the genus-specific assay, we found 90% of microscopy-negative samples to be positive by genus-specific qRT-PCR assay. This is an unusually high number compared to what has been pre-viously reported, especially considering that three independent qualified microscopists evaluated each smear. The large dis-crepancy between qRT-PCR and microscopy can be attributed to the high sensitivity of our genus-specific qRT-PCR assay, the presence ofPlasmodiumparasites not recognizable by mi-croscopy, the presence ofPlasmodiumnucleic acid circulating in subjects/patients, poor staining procedures, etc.
Some of the important parameters guiding malaria treat-ment are detection of the presence/absence of the parasite, identification of infecting species, and determination of the level of parasitemia. As demonstrated in this study, each assay has strengths and weaknesses, and applying a uniform “one size fits all” approach is both naïve and potentially misleading. The biggest difference was found at the lowest or the highest densities, with the density in qRT-PCR reaching a plateau sooner than in microscopy. Since qRT-PCR is much more sensitive than microscopy, we hypothesized that qRT-PCR was more reliable than microscopy at lower parasite densities but not at higher parasite densities. We showed this to be true when we diluted NA from the clinical samples and conse-quently obtained higher parasite densities by qRT-PCR. When samples were grouped based on individual log densities, the group with a log density of 3 was quantitatively more congruent for both qRT-PCR and microscopy than groups with higher log densities. In this group, genus-specific qRT-PCR had a mean
CTvalue of 18.75. As such, we propose that if a clinical sample is quantitatively analyzed by PCR and has aCTvalue below 18.00 (this number may vary depending on the sensitivity of the assay), the sample must be diluted further in order to obtain a more reliable quantification. In addition, diluting the sample might improve the sensitivity of the assay by diluting out PCR inhibitors that might be in the sample. Although most extrac-tion kits claim that purified NA is free of protein, nucleases, and other contaminants or inhibitors, PCR-inhibitory compo-nents in blood, such as hemoglobin and lactoferrin, are always of concern. Inclusion of amplification facilitators, such as a
VOL. 49, 2011 TOTAL NUCLEIC ACID PCR ASSAY FOR MALARIA DIAGNOSIS 2951
on May 16, 2020 by guest
http://jcm.asm.org/
single-stranded DNA binding protein or bovine serum albu-min, in the PCR mixture might prove to be helpful (1).
The Malaria Eradication Research Agenda (malERA) (http://malera.tropika.net/) states its objective as follows. “Im-proved and new tools and strategies for monitoring, evalua-tion, and surveillance are needed to track program interven-tion coverage, impact on cases and transmission, and progress toward elimination/eradication. New survey tools may help to measure transmission more simply and directly, thus enabling detection of ‘hot spots’ requiring additional resource alloca-tion.” As we move to the next phase of malaria control, we embrace the objectives of malERA and believe that additional tools, including ultrasensitive molecular assays, are needed to assess pockets of residual malaria prevalence and transmission. We believe that such molecular tools will undoubtedly require assays that are inexpensive, easy to use with a variety of de-tection platforms, and need minimal training. We propose that the assays we describe here can fulfill the Target Product Profiles (TPP) for two separate indications, one for quality control and quality assurance programs in malaria diagnosis (i.e., reference centers) and the second for surveillance in malaria elimination and eradication campaigns. The qRT-PCR assay is a good fit for the modified TPP for the eradication agenda: the assay is highly sensitive, reliable and accurate; compatible with most PCR systems found in resource-con-strained environments; and, importantly, cost-effective through reduction in the volume of the PCR mixture, thereby reducing the cost of goods.
In handling of clinical samples, NA extraction and storage of the samples should be considered critical bottlenecks for stan-dardization of PCR as a gold standard for malaria diagnosis. We propose standardization of NA extraction procedures (methods) and protocol across the board as an initial step toward standardization of PCR as a gold standard for malaria diagnosis. Since the QIAamp DNA Blood Mini Kit is one of the most widely used extraction kits, we propose adopting use of this kit as a standard procedure for extraction of NA from whole blood for malaria diagnosis. Additionally, we propose that a publically available resource, such as ATCC, the Malaria Research and Reference Reagent Resource Center (MR4), or the WHO, should prepare and supply standard NA, such as 3D7, to be used worldwide by all researchers and in clinical settings for quantitation and as-sessment of PCR sensitivity.
A radical changeover from traditional malaria microscopy to molecular PCR-based assays will require careful thought and consideration regarding the impact that such a change would have on the clinical development pathway of malaria vaccines or drugs that are assessed in human challenge models. At the Walter Reed Army Institute of Research, we have used stan-dard malaria microscopy by qualified expert malaria microsco-pists who undergo proficiency training and examination before each clinical trial as the definitive method that determines whether a subject will initiate treatment after the identification of two unambiguous malaria parasites in Giemsa-stained thick blood smears. This approach has been validated and used successfully in over 1,000 human subjects in approximately 57 human clinical challenge trials to date (2011). Importantly, the use of expert microscopists who detect the appearance of ma-laria parasites in the peripheral circulation prior to the onset of
symptoms consistent with clinical malaria is an important ob-jective in our studies. Therefore, we are keen to consider alternative diagnostic assays that can detect the presence of parasites earlier than microscopy. Our eagerness to employ such an assay will be tempered by the potential for PCR contamination of products that may inadvertently give a false-positive result for someone who actually has no ma-laria parasites, which could lead to a false conclusion as to the effectiveness of a malaria vaccine or drug intervention. Nevertheless, with stringent quality assurance programs, segregation plans for sample preparation and assay detec-tion, and endorsement by regulatory agencies, we believe the time has come to transition from microscopy to molec-ular-based PCR assays.
In conclusion, we have described a highly sensitive quanti-tative real-time PCR assay for detecting Plasmodiumat the genus level and have shown that adding an RT step signifi-cantly increases the sensitivity of real-time PCR. We have shown that at high parasite density, the quantitative PCR assay reaches a limit of quantification that can be extended by fur-ther diluting the sample. Our genus-specific qRT-PCR assay has proven to be extremely useful in detection ofPlasmodium
while maintaining the same robustness and sensitivity. The assay is currently being used in our institute in support of human clinical trials for detection of bothP. falciparumandP. vivax. The assay is also being used in animal studies for detec-tion ofP. berghei, P. yoelii, and P. knowlesi. In addition, the assay was recently successfully used in detection of Plasmo-dium from mosquitoes collected in the field for surveillance studies.
ACKNOWLEDGMENT
We acknowledge Kathy Moch for supplying cultured highly synchro-nized ring stage 3D7 parasites.
REFERENCES
1.Al-Soud, W. A., and P. Radstrom.2001. Purification and characterization of
PCR-inhibitory components in blood cells. J. Clin. Microbiol.39:485–493.
2.Anonymous.1988. Malaria diagnosis: memorandum from a W.H.O. meeting.
Bull. World Health Organ.66:575–594.
3.Berry, A., F. Benoit-Vical, R. Fabre, S. Cassaing, and J. Magnaval.2008.
PCR-based methods to the diagnosis of imported malaria. Parasite15:484–
488.
4.Cnops, L., J. Jacobs, and M. Van Esbroeck.2010. Validation of a four-primer real-time PCR as a diagnostic tool for single and mixed Plasmodium infections. Clin. Microbiol. Infect. doi:10.1111/j.1469 0691.2010.03344.x. 5.Durrheim, D. N., P. J. Becker, and K. Billinghurst.1997. Diagnostic
dis-agreement—the lessons learnt from malaria diagnosis in Mpumalanga. S.
Afr. Med. J.87:1016.
6.Elsayed, S., K. Plewes, D. Church, B. Chow, and K. Zhang.2006. Use of molecular beacon probes for real-time PCR detection of Plasmodium fal-ciparum and other Plasmodium species in peripheral blood specimens.
J. Clin. Microbiol.44:622–624.
7.Galinski, M. R., and J. W. Barnwell.2008. Plasmodium vivax: who cares?
Malar J.7(Suppl. 1):S9.
8.Gardner, M. J., et al.2002. Genome sequence of the human malaria parasite
Plasmodium falciparum. Nature419:498–511.
9.Guerin, P. J., et al. 2002. Malaria: current status of control, diagnosis, treatment, and a proposed agenda for research and development. Lancet
Infect. Dis.2:564–573.
10.Li, J., R. A. Wirtz, G. A. McConkey, J. Sattabongkot, and T. F. McCutchan.
1994. Transition of Plasmodium vivax ribosome types corresponds to
sporo-zoite differentiation in the mosquito. Mol. Biochem. Parasitol.65:283–289.
11.Mehlotra, R. K., et al.2010. Molecular-based assay for simultaneous detec-tion of four Plasmodium spp. and Wuchereria bancrofti infecdetec-tions. Am. J.
Trop. Med. Hyg.82:1030–1033.
12.Moody, A.2002. Rapid diagnostic tests for malaria parasites. Clin. Microbiol.
Rev.15:66–78.
13.Muirhead-Thomson, R. C.1954. Low gametocyte thresholds of infection of
on May 16, 2020 by guest
http://jcm.asm.org/
Anopheles with Plasmodium falciparum; a significant factor in malaria
epi-demiology. Br. Med. J.4853:68–70.
14. Reference deleted.
15.Ohrt, C., Purnomo, M. A. Sutamihardja, D. Tang, and K. C. Kain.2002. Impact of microscopy error on estimates of protective efficacy in
malaria-prevention trials. J. Infect. Dis.186:540–546.
16.Ohrt, C., et al.2007. Establishing a malaria diagnostics centre of excellence
in Kisumu, Kenya. Malar. J.6:79. doi:10.1186/1475-2875-6-79.
17.Okell, L. C., A. C. Ghani, E. Lyons, and C. J. Drakeley.2009. Submicroscopic infection in Plasmodium falciparum-endemic populations: a systematic
re-view and meta-analysis. J. Infect. Dis.200:1509–1517.
18.Perandin, F., et al.2004. Development of a real-time PCR assay for detec-tion of Plasmodium falciparum, Plasmodium vivax, and Plasmodium ovale
for routine clinical diagnosis. J. Clin. Microbiol.42:1214–1219.
19.Schneider, P., et al.2007. Submicroscopic Plasmodium falciparum gameto-cyte densities frequently result in mosquito infection. Am. J. Trop. Med.
Hyg.76:470–474.
20.Veron, V. S., S. Simon, and B. Carme.2009. Multiplex real-time PCR de-tection of P. falciparum, P. vivax and P. malariae in human blood samples.
Exp. Parasitol.121:346–351.
21.Waters, A. P.1994. The rRNA genes of Plasmodium. Adv. Parasitol.34:33–79. 22.Waters, A. P., C. Syin, and T. F. McCutchan.1989. Developmental
regula-tion of stage-specific ribosome popularegula-tions in Plasmodium. Nature342:438–
440.
23.Withers, M. R., et al.2006. Safety and reactogenicity of an MSP-1 malaria vaccine candidate: a randomized phase Ib dose-escalation trial in Kenyan
children. PLoS Clin. Trials1:e32.
VOL. 49, 2011 TOTAL NUCLEIC ACID PCR ASSAY FOR MALARIA DIAGNOSIS 2953