Past and present of sediment and carbon biogeochemical cycling models
Full text
Figure

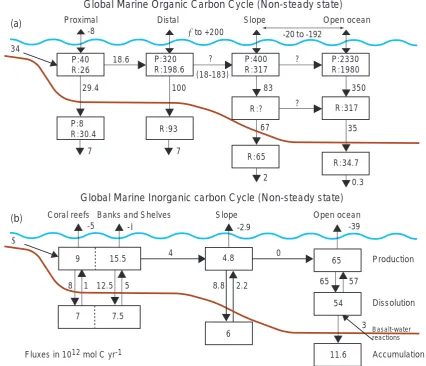
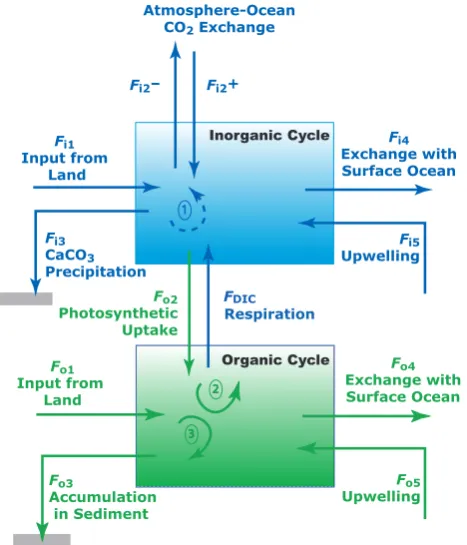
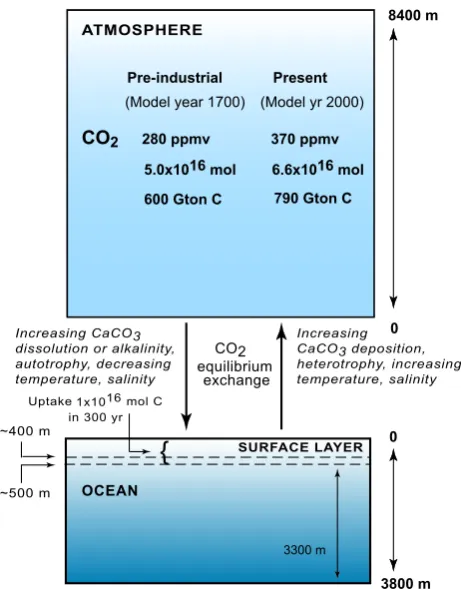
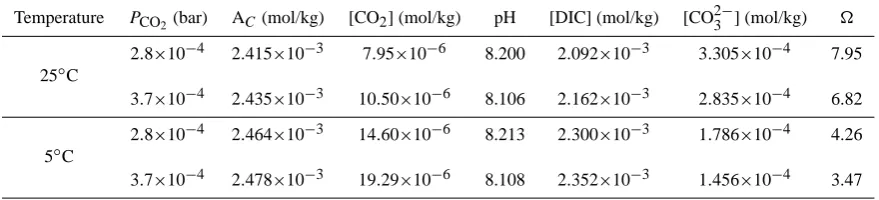



Related documents
Worksheets Worksheets for nutrition staff, nutrition services and materials, health indicators and program participant data, participant feedback, determining nutrition
frequent business travelers and reveals what innovative travel and expense management program managers are doing to modernize their roles, engage travelers, optimize programs,
We found four patients in whom both alleles were mutated; three of these patients had the same homozygous mutation (c.154delA; p.N52fsX80), a single base pair deletion that inserts
The effectiveness of self help technology for emotional symptoms in the present analysis is similar to the analysis of 8 studies in adults with depression and anxiety in pri- mary
Study endpoints were clinicians ’ assessments of dry eye signs (ocular surface staining intensity, TBUT, and Schir- mer score) as well as OSDI overall and Symptoms, Vis- ual
Because the long-term renal outcomes of AAV pa- tients with persistent hematuria during clinical remission were heterogeneous, it is clinically pertinent to differen- tiate the
1 Equal = Fixed effect meta-analysis with equal weights; 2 Patients = Fixed effect meta-analysis with number of patients as weights; 3 Events = Fixed effect meta-analysis with number
In the past 12 years, the absolute number of Chinese ar- ticles showed a more than 6-fold increase (from 99 to 605 between 2000 and 2011) in publications in international
