R E S E A R C H
Open Access
An inverse metabolic engineering approach
for the design of an improved host platform
for over-expression of recombinant
proteins in
Escherichia coli
Chaitali Ghosh, Rashmi Gupta and Krishna Jyoti Mukherjee
*Abstract
Background:A useful goal for metabolic engineering would be to generate non-growing but metabolically active quiescent cells which would divert the metabolic fluxes towards product formation rather than growth. However, for products like recombinant proteins, which are intricately coupled to the growth process it is difficult to identify the genes that need to be knocked-out/knocked-in to get this desired phenotype. To circumvent this we adopted an inverse metabolic engineering strategy which would screen for the desired phenotype and thus help in the identification of genetic targets which need to be modified to get overproducers of recombinant protein. Such quiescent cells would obviate the need for high cell density cultures and increase the operational life span of bioprocesses.
Results:A novel strategy for generating a library, consisting of randomly down regulated metabolic pathways in E. coliwas designed by cloning small genomic DNA fragments in expression vectors. Some of these DNA fragments got inserted in the reverse orientation thereby generating anti-sense RNA upon induction. These anti-sense
fragments would hybridize to the sense mRNA of specific genes leading to gene‘silencing’. This library was first screened for slow growth phenotype and subsequently for enhanced over-expression ability. Using Green Fluorescent Protein (GFP) as a reporter protein on second plasmid, we were able to identify metabolic blocks which led to significant increase in expression levels. Thus down-regulating theribBgene (3, 4 dihydroxy-2-butanone-4-phosphate synthase) led to a 7 fold increase in specific product yields while down regulating the genekdpD(histidine kinase) led to 3.2 fold increase in specific yields.
Conclusion:We have designed a high throughput screening approach which is a useful tool in the repertoire of reverse metabolic engineering strategies for the generation of improved hosts for recombinant protein expression.
Keywords:Recombinant protein, Inverse metabolic engineering,Escherichia coli, Improved host platform
Background
Improvement of metabolic phenotype through directed genetic modifications is the main goal of metabolic gineering [1-4]. The classical approach of metabolic en-gineering requires a detailed knowledge of enzyme kinetics, the system network, and intermediate pools involved, and on this basis, a genetic manipulation is proposed for some presumed benefits. Recent progress in molecular genetics methods makes it possible to
knockout or over-express targeted genes in most microor-ganisms [5-9]. A key question in metabolic engineering is how to identify gene targets that have direct or indirect impact on a particular phenotype of interest [10].
For the over-production of metabolites the issue is comparatively straight forward since only the regulatory blocks and bottlenecks in the specific pathway involved in product synthesis need to be removed. Secondly, the supply of precursor metabolites to the pathway need to be enhanced thereby improving the metabolic flux in the pathways. However, for products like recombinant
* Correspondence:[email protected]
School of Biotechnology, Jawaharlal Nehru University, New Delhi, India
proteins there is a vast range of precursors along with the transcriptional/translational mechanism all of which are coupled to the growth process. Many metabolic engineering approaches have therefore attempted to enhance both growth and recombinant protein produc-tion inE. coli[11,12].One approach has been to supple-ment the genes which get down-regulated due to the stress associated with recombinant protein expression [13]. Similarly key factors which has impact on growth and expression yield, like glycolysis and Tri Carboxylic Acid (TCA) cycle enzymes, ATPase’s, RNA polymerases etc. have been co-expressed [14-16].
A novel alternative strategy to design overproducers of the recombinant product would be to make these precur-sors available specifically for product rather than biomass formation. We therefore decided to design a strategy to screen for metabolically active but non-growing quiescent cells which have been earlier shown to be more effective for recombinant protein production [17].
The concept of inverse metabolic engineering involves first to identify the desired phenotype, then to determine environmental or genetic conditions that confer this pheno-type, and finally to alter the phenotype of the selected host by genetic manipulation [18,19]. In this study, we adopted this approach where an E. coli genomic library was screened to obtain quiescent cells which might be better producers of recombinant proteins. For this, we randomly and partially down-regulated the biosynthetic pathways in the cell, in order to identify those blockages which help in diverting the metabolic flux away from growth. Cells with slow growth phenotype or growth stoppage were then screened for enhanced protein expression capability. An additional advantage of non-growing, quiescent cells is that they can increase the operational life span of bio-processes and improve process economics by decoupling product for-mation from cell growth [20].
In this study anE. coligenomic library was prepared in a pRSET A vector having a strong promoter as well as in pBAD33 having a comparatively weaker promoter because even a small down regulation of some genes can have significant effects. Screening of the genomic library by dif-ferent approaches (slow growth or enhanced GFP fluores-cence) led to the identification of clones with different down-regulated genes which blocked growth and simultan-eously improved recombinant protein expression.
Results and discussion
Identification of metabolic pathways which lead to 'no growth’or slow-growth phenotype through genomic library screening
We constructed a library consisting of a set of clones with metabolic pathways which were ‘knocked-down’ rather than‘knocked-out’, allowing us to include the role of essential genes. In our study the strategy adopted was
to clone small fragments of genomic DNA (~ 200–800 bases), the idea being that some of these DNA fragments would get inserted in an opposite orientation (compared to the coding strand) in the expression vector. On induction, the RNA produced would be complimentary to the mRNA (of a functional protein) which would then hybridize leading to (partial) silencing of the gene. In eukaryotic systems an ideal antisense could be as short as a 22–25 bp fragment (as then a dicer molecule can act, leading to RNAi). But in prokaryotic systems, RNAi is absent and not many studies have been done on the antisense mechanism. So by choosing a 200–800 bp fragment we attempted to ensure that at least partial gene silencing would take place.
The concept of inverse metabolic engineering was used to screen this library for the desired ‘no-growth’ phenotype and then to identify the genetic factors that confer this phenotype. We were interested in a ‘no-growth’or ‘slow-growth’phenotype where the meta-bolic activity was undiminished; the rationale being such clones would divert the metabolic flux away from bio-mass formation and towards product synthesis.
Construction of the antisense library in pRSET A vector Genomic DNA fragments of sizes ranging from 200– 800 bp (as we required only a part of the gene which would act in an antisense fashion to block transcription) were isolated after an optimized partial digestion of the
E. coli genomic DNA and subsequently ligated into a high copy number vector (pRSET A) with a strong T7 promoter. The constructed shotgun library in pRSET A was transformed intoE. coliBL21 pLysS strain.
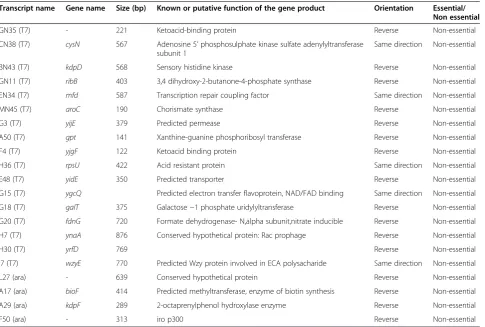
These clones were sequenced and used to do a BLAST search against the E. coli genome database. The lists of identified genes are given in Table 1. The details of the genes were collected from the PEC (Profiling of E. coli Chromosome) and Ecocyc database.
These 17 clones were co-transformed with a second plasmid pBAD33-GFP, where the GFP gene was cloned under the arabinose promoter and had a compatible p15 ‘ori’ with pUC ‘ori’ of pRSET A. This GFP reporter gene was used to check whether the antisense expression (while
blocking growth) also helped in the overexpression of re-combinant protein. All these experiments were done in LB media. For all the 17 clones one flask was induced only with 0.02% of arabinose (control), while the second test flask was co-induced with both IPTG and arabinose, to check the effect of the antisense transcript on GFP expres-sion. A third flask for all the co-transformants was kept uninduced to observe the effect if any, of leaky expression (2ndcontrol). The library contained large number of ran-dom inserts in pRSET A (which did not show any affect) which essentially served as a negative control.
Figure 2 (A,B,C) shows the effect of co-expression of clone GN11 having the anti-sense of ribB gene (rib-3, 4 dihydroxy-2-butanone-4-phosphate synthase). In Figure 2B the uninduced culture had a basal level GFP expression of 5 to 7 AU while GFP expression in the ara induced control culture increased linearly from 6 to 39 AU in 8 hours post induction. In the test system the fluorescence of the co-induced culture overtook the control flask after 2 hours to give a final fluorescence of 53 AU demonstrating the positive effect of antisense induction on GFP expression. Since the final biomass concentrations obtained in the test culture was signifi-cantly lower, the specific product yield was much higher
at 347 AU/g DCW, a 7 fold increase over the control culture (47 AU/g DCW) (Figure 2C).
Figure 2 (D,E,F) shows the co-expression of the clone EN34 coding for mutation frequency decline (mfd) protein. In Figure 2E the uninduced flask showed a basal level expression of 16.9 AU throughout the cultivation. The control flask showed a linear increase from 10 AU to 54 AU in 8 hours. However, while the expression was ini-tially low in the co-induced culture, it overtook the control flask after 3 hours and gave a final GFP fluorescence of 72 AU. The specific product yield of arabinose induced GFP was found to be 88 AU/gm DCW in the 8thhour post induction in the control culture whereas in the arabinose and IPTG induced culture it was calculated to be 378 AU/ gm DCW which was 4 fold higher (Figure 2F).
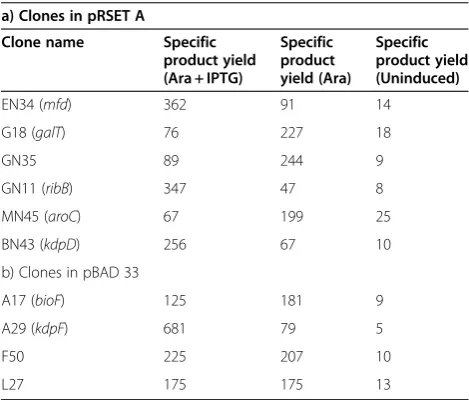
Similar studies were conducted with the rest of the 15 clones. However, not all the clones gave such a dramatic increase in the specific product yield though we did ob-serve a significant increase in specific product yield in some cases which are listed in Table 2.
Construction of the antisense library in pBAD33
In the previous screening strategy we over-expressed the transcripts under the strong T7 promoter, which has Table 1 List of transcripts whose blockage leads to growth stoppage
Transcript name Gene name Size (bp) Known or putative function of the gene product Orientation Essential/ Non essential
GN35 (T7) - 221 Ketoacid-binding protein Reverse Non-essential
CN38 (T7) cysN 567 Adenosine 5’phosphosulphate kinase sulfate adenylyltransferase subunit 1
Same direction Non-essential
BN43 (T7) kdpD 568 Sensory histidine kinase Reverse Non-essential
GN11 (T7) ribB 403 3,4 dihydroxy-2-butanone-4-phosphate synthase Reverse Non-essential
EN34 (T7) mfd 587 Transcription repair coupling factor Same direction Non-essential
MN45 (T7) aroC 190 Chorismate synthase Reverse Non-essential
G3 (T7) yijE 379 Predicted permease Reverse Non-essential
A50 (T7) gpt 141 Xanthine-guanine phosphoribosyl transferase Reverse Non-essential
F4 (T7) yjgF 122 Ketoacid binding protein Reverse Non-essential
H36 (T7) rpsU 422 Acid resistant protein Same direction Non-essential
E48 (T7) yidE 350 Predicted transporter Reverse Non-essential
G15 (T7) ygcQ Predicted electron transfer flavoprotein, NAD/FAD binding Same direction Non-essential
G18 (T7) galT 375 Galactose−1 phosphate uridylyltransferase Reverse Non-essential
G20 (T7) fdnG 720 Formate dehydrogenase- N,alpha subunit,nitrate inducible Reverse Non-essential
H7 (T7) ynaA 876 Conserved hypothetical protein: Rac prophage Reverse Non-essential
H30 (T7) yrfD 769 Reverse Non-essential
I7 (T7) wzyE 770 Predicted Wzy protein involved in ECA polysacharide Same direction Non-essential
L27 (ara) - 639 Conserved hypothetical protein Reverse Non-essential
A17 (ara) bioF 414 Predicted methyltransferase, enzyme of biotin synthesis Reverse Non-essential
A29 (ara) kdpF 289 2-octaprenylphenol hydroxylase enzyme Reverse Non-essential
certain disadvantages. Firstly, IPTG itself is partially toxic to cells and also the expression of T7 RNA polymerase leads to growth retardation [21,22]. This would mask the true growth retardation due to the anti-sense effect of the tran-scripts. Secondly, since the anti-sense works at the RNA
level we should use only a transcriptional vector and not an expression vector to rule out the possibility that the toxicity of a randomly expressed polypeptide is responsible for growth retardation. Thirdly, hyper expression of the anti-sense RNA seems to be a clumsy approach to block and/or down-regulate gene expression (even though this was pri-marily a screening strategy for identifying potential targets for manipulation). We therefore chose a much better regu-lated transcriptional vector for the construction of a second library which would identify targets where even a partial down-regulation would lead to diversion of metabolic flux towards product synthesis.
The genomic library was prepared under the ara pro-moter in pBAD33 (a transcriptional vector), which is a tightly regulated promoter. Moreover as we were not constrained to use the BL21 (DE3) strain we chose
E. coli DH5α since it has superior transformation effi-ciency and is much more stable strain for retaining plas-mids. A dual plasmid system was used to directly screen for the desired phenotype. Competent DH5α cells pre-transformed with a plasmid pNER31 (having GFP under the lac promoter) was used for co-transformation with the genomic library. This allowed us to directly screen for the over-expressing phenotype. Upon co-induction of the transcripts, we screened for colonies exhibiting in-tense green color of GFP (under UV) compared to the control colonies in the plate. More than 30,000
A
B
C
D
E
F
Figure 2Time profiles of growth, Fluorescence spectra and Specific product yields of GN11 (ribB) in A, B, and C and EN34 (mfd) in D, E, and F respectively in LB media.(▲) uninduced; (■) induced with arabinose; (♦) induced with arabinose and IPTG.
Table 2 Specific product yield (AU/gm DCW) of various clones under three different conditions, uninduced, induced with arabinose and induced with arabinose and IPTG both (6 hrs post induction)
a) Clones in pRSET A
Clone name Specific product yield (Ara + IPTG)
Specific product yield (Ara)
Specific product yield (Uninduced)
EN34 (mfd) 362 91 14
G18 (galT) 76 227 18
GN35 89 244 9
GN11 (ribB) 347 47 8
MN45 (aroC) 67 199 25
BN43 (kdpD) 256 67 10
b) Clones in pBAD 33
A17 (bioF) 125 181 9
A29 (kdpF) 681 79 5
F50 225 207 10
transformants of the library was screened. Finally 4 clones (Table 1), exhibiting higher GFP expression was selected for further studies. Shake flask studies on these clones were done in different media LB, TB, M9 along with appropriate controls.
We first needed to factor out the effect of GFP expres-sion on growth retardation by comparing the growth of the uninduced culture with the IPTG induced culture. A very slight decline in growth was observed post induc-tion in LB media which is typical of GFP induced cul-tures. However when the second, anti-sense gene carrying plasmid, was induced by arabinose a significant drop in growth was observed. The growth profile of the co-induced culture was compared with the control culture where only GFP was induced. We observed that the growth declined sharply in all the co-induced cultures (Figure 3).
Comparing the product profile of kdpF (2-octaprenyl-phenol hydroxylase enzyme) antisense expressing clone A29, we observed that, the specific product yield of the
co-induced culture was 8.4 fold higher than the control culture which was induced with IPTG only (Figure 4). Co-expression studies were also done with rest of the 3 clones (data not shown).
One of the open questions in the quest for improved host platforms is how targets are to be identified which confer the desired phenotype, given the lack of a compre-hensive kinetic model for E. coli and the absence of detailed information for the regulatory mechanism, which operate inside the cells. There are therefore several approaches for the modifications of the E. coli host to achieve the desired goal [23-25]. In our case the design strategy was essentially a high throughput screen which helped us select better producers. It is interesting to note that all the selected genes were non-essential and also not directly linked to either growth or product formation.
For example, the selected clone GN11 contained an insert of 403 bp of theribBgene (3, 4 dihydroxy-2-buta-none-4-phosphate synthase) in the reverse direction.
A
B
C D
ribB serves as the biosynthetic precursor for the xylene ring of riboflavin [26,27]. It is reported thatribBmutant
E. coli leads to slow growth but remains metabolically active which corroborates our studies [28]. However the discovery that blocking the generibBcan help divert the metabolic flux towards recombinant protein production was both non obvious and counter intuitive. Similarly, clone BN43 coded for a protein KdpD (sensory histidine kinase) which is expressed during osmotic shock and is otherwise nonessential. Clearly in this case there is no direct link to the gene function and the observed chan-nelization of metabolic flux towards recombinant pro-tein production. In the case of clone MN45 which contained part of the antisense aroC (chorismate syn-thase gene, which catalyzes the formation of chorismate in aromatic amino acid biosynthesis) its blockage should ideally lead to a decrease in the biosynthetic pathways involved in growth. However, an exact estimate of the growth inhibition is difficult given that complex nitrogen sources are present in the medium which can supply the growing cell with the desired amino acids. The fact that blocking chorismate synthase led to a complete growth stoppage was interesting and points to the criticality of even single metabolic reactions in growth. All these results underscore the fact that there is no direct link at the pathway level between gene function and observed phenotype which could possibly be the result of a com-plex regulatory response within the cell.
Conclusion
In a novel approach towards the design of an ideal host platform for the over-expression of recombinant pro-teins, two different high throughput screening strategies were designed to identify genes whose down-regulation would lead to a slow growth phenotype and also possibly divert the metabolic flux towards over-expression of re-combinant proteins. The leads obtained did not rely on a priori knowledge of the regulated and interconnected nature of the E. coli’s metabolic network. At a practical
level we were able to obtain very useful targets for gene knock-out/knock-down which would enable the design of better hosts for protein expression. It would be inter-esting to study the combined and synergistic effect of these identified blocks. At a more fundamental level a detailed analysis of the phenotypic affects of these meta-bolic blocks can lead to a better understanding of the regulatory mechanisms in theE. colinetwork.
Materials and methods Growth media and antibiotics
Media and bulk chemicals were purchased from local manufacturers, Himedia, Qualigens, Difco and MERCK. Media used were Luria Broth [LB] (bactotryptone 10 g/l, yeast extract 24 g/l and NaCl 15 g/l), Terrific Broth [TB] (YE 24 g/l, tryptone 12 g/l, glucose 0.5% in which 2.31 g/l KH2PO4and 12.54 g/l K2HPO4were added after
autoclav-ing separately), and minimal media [M9] (NaCl 0.5 g/l, NH4Cl 1 g/l, K2HPO4 3 g/l, 1 M CaCl2 0.1 ml/l, 1 M
MgSO42 ml/l and 20% glucose 10 ml/l). Antibiotics used
were ampicillin 100μg/ml (1X), kanamycin 50μg/ml (1X) and chloramphenicol 30μg/ml (1X).
Bacterial strains and plasmids
The T7 based expression vector pRSET A (ampicillin, Invitrogen) which had pUC‘ori’, was used in these stud-ies. pBAD33 (chloramphenicol, Beckwith) [29] having p15‘ori’witharapromoter and compatible with pRSET A, was used for expression of GFP gene (used as a re-porter protein) and the construction of second library. The plasmids were maintained and expressed in E. coli
DH5α cells. E. coli BL21 (DE3) from stratagene (USA) and E. coli BL21 (DE3) pLysS were used for expression studies. Strain JM109 (Promega, USA) was used for the genomic library preparation. pNER31 (ampicillin) having GFP gene under ‘lac’ promoter and induced by IPTG, was gifted by Summers, Cambridge, UK and pET14b-GFP (ampicillin) where pET14b-GFP gene was cloned under T7 promoter, was gifted by J. Singh, ICGEB, India.
A
B
C
Construction of genomic library in vectors pRSET A and pBAD33
Genomic DNA was isolated from JM109 by the Hexade-cyltrimethylammonium bromide (CTAB) method. This was partially digested with eitherSauIIIA orNlaIII both four base cutters. The expression vectors pRSET A and pBAD33 were digested withBglII andSphI which gener-ate compatible cohesive ends with SauIIIA and NlaIII respectively. These linearised vector fragments were Calf Intestinal Alkaline Phosphatase (CIAP) treated to pre-vent self ligation and ligated with the digested and gel eluted genomic fragments of size ranging from 200 to 800 bp. The ligation mixture was used to transform electrocompetent BL21(DE3) pLysS and DH5α cells respectively and plates were incubated at 37°C over-night. Approximately 8000 and 30,000 colonies were obtained in each library respectively. The quality of the library that was constructed was checked by pick-ing a few colonies and checkpick-ing for the insert size by colony PCR.
Screening of the library by two different approaches A preliminary screening of the pRSET A vector library was done on LB agar plates. Replica plating was done onto two different plates. One plate containing IPTG was considered as induced and the second plate without IPTG was consid-ered as control. Slow growing colonies upon induction were identified and there growth profile was determined by performing shake flask studies in different media viz. LB, TB and M9.
For the second library under pBAD33, a co-transformation protocol was followed where electrocompetent cells of DH5α already containing the pNER31 plasmid (with GFP) was prepared by glycerol wash method and electroporated with the ligation mixture containing the genomic library. Primary screening was done on LB agar plates by induction of both the plasmids with IPTG (1 mM final concentration) and arabinose (0.02%, final concentration). Colonies with enhanced fluorescence were picked, and the genomic frag-ments cloned in the second plasmid (containing the library) which were responsible for enhanced fluorescence, was determined by sequencing. The growth and product profiles were determined for the selected clones by shake flask stud-ies which were done in M9, TB and LB media.
Selected clones which showed enhanced fluorescence upon induction were grown overnight with shaking at 37°C in 3 ml LB tubes. Secondary inoculation was done by adding 100μl of overnight grown culture in 10 ml of LB, TB and M9 medium in 100 ml flasks. After 1.5 hours when the OD600 of 0.3-0.5 was attained, cultures
were induced by adding 1 mM IPTG (final concentra-tion). After induction, the OD600was monitored at
regu-lar intervals.
For measuring the metabolic activity of non growing cells in minimal media, 200μl of samples were taken at regular intervals post induction. These samples were centrifuged at 13,200 rpm for 2 minutes and the super-natant was stored at 4°C. Glucose levels were estimated by the 3, 5-Dinitrosalicylic acid (DNSA) method.
The expression of GFP was monitored by Varian (Carry Eclipse) Fluorescence Spectrophotometer with an excitation wavelength of 488 nm and emission wave-length of 514 nm.
The specific yield was calculated using the formula Fluorescence/OD = AU/gm DCW (the dry cell weight factor is 0.3 gm/L/1OD600).
Abbreviations
GFP: Green fluorescent protein; CTAB: Hexadecyltrimethylammonium bromide; DNSA: 3, 5-Dinitrosalicylic acid; CIAP: Calf Intestinal alkaline phosphatase; IPTG: Isopropylβ-D-1-thiogalactopyranoside.
Competing interests
The authors declare that they have no competing interests.
Authors’contributions
CG and KJM designed the experiments and wrote the manuscript. CG performed all the experiments. RG performed few experiments. All authors read and approved the final manuscript.
Acknowledgement
CG gratefully acknowledges CSIR fellowship, Govt. of India. The authors thank Dr. Beckwith (HMS, Boston), Dr. Summers (Cambridge, UK) and Dr. J. Singh (ICGEB, India) for gifting the plasmids pBAD33, pNER31 and pET14b-GFP respectively.
Received: 18 January 2012 Accepted: 11 June 2012 Published: 3 July 2012
References
1. Bailey JE:Towards a science of metabolic engineering.Science1991,
252:1668–1675.
2. Stephanopoulos G, Sinskey AJ:Metabolic engineering methodologies and future prospects.Trends Biotechnol1993,11:392–396.
3. Stephanopoulos G:Metabolic fluxes and metabolic engineering.
Metab Eng1999,1:1–11.
4. Stephanopoulos G, Kelleher J:How to make a superior cell.Science2001,
292:2024–2026.
5. Baudin A, Ozier-Kalogeropoulos O, Denouel A, Lacroute F, Cullin C:
A simple and efficient method for direct gene deletion inSaccharomyces
cerevisiae.Nucleic Acids Res1993,21:3329–3330.
6. Datsenko KA, Wanner BL:One step inactivation of chromosomal genes in
Escherichia coliK-12 using PCR products.Proc Natl Acad Sci USA2000,
97:6640–6645.
7. Lutz R, Bujard H:Independent and tight regulation of transcriptional units inEscherichia colivia the LacR/O, the TetR/O and AraC/I-1-I-2 regulatory elements.Nucleic Acids Res1997,25:1203–1210.
8. Mumberg D, Muller R, Funk M:Yeast vectors for the controlled expression of heterologous proteins in different genetic backgrounds.Gene1995,
156:119–122.
9. Voss I, Steinbuchel A:Application of a KDPG-aldolase gene-dependent addiction system for enhanced production of cyanophycin inRalstonia
eutrophastrain H16.Metab Eng2006,8:66–78.
10. Yong J, Stephanopolous G:Multi-dimensional gene target search for improving lycopene biosynthesis inEscherichia coli.Metab Eng2007,
9:337–347.
12. Chou CP:Engineering cell physiology to enhance recombinant protein production inEscherichia coli.Appl Microbiol Biotechnol2007,76:521–532. 13. Choi JH, Lee SJ, Lee SJ, Lee SY:Enhanced production of insulin-like
growth factor I fusion protein inEscherichia coliby coexpression of the down-regulated genes identified by transcriptome profiling.Appl Environ Microbiol2003,69:4737–4742.
14. March JC, Eiteman MA, Altman E:Expression of an anaplerotic enzyme, pyruvate carboxylase, improves recombinant protein production in
Escherichia coli.Appl Environ Microbiol2002,68:5620–5624.
15. Weikert C, Canonaco F, Sauer U, Bailey JE:Co-overexpression of RspAB improves recombinant protein production inEscherichia coli.Metab Eng 2000,2:293–299.
16. Lütke-Eversloh T, Stephanopoulos G:Combinatorial pathway analysis for improved L-tyrosine production inEscherichia coli: identification of enzymatic bottlenecks by systematic gene overexpression.Metab Eng 2008,10:69–77.
17. Mukherjee KJ, Rowe DC, Watkins NA, Summers DK:Studies of single-chain antibody expression in quiescentEscherichia coli.Appl Environ Microbiol 2004,70:3005–3012.
18. Bailey JA, Sburlati A, Hatzimanikatis V, Lee K, Renner W, Tsai P:Inverse metabolic engineering: a strategy for directed genetic engineering of useful phenotypes.Biotechnol Bioeng1996,52:109–121.
19. Delgado J, Liao JC:Inverse flux analysis for reduction of acetate excretion inEscherichia coli.Biotechnol Prog1997,13:361–367.
20. Sonderegger M, Schümperli M, Sauer U:Selection of quiescentEscherichia
coliwith high metabolic activity.Metab Eng2005,7:4–9.
21. Srivastava P, Bhattacharaya P, Pandey G, Mukherjee KJ:Overexpression and purification of recombinant human interferon alpha2b inEscherichia coli.
Protein Expr Purif2005,41(2):313–322.
22. Banack T, Clauson N, Ogbaa N, Villar J, Oliver D, Firshein W:Overexpression of the Hda DnaA-related protein inEscherichia coliinhibits
multiplication, affects membrane permeability, and induces the SOS response.J Bacteriol2005,187(24):8507–8510.
23. Chin JW, Cirino PC:Strain engineering strategies for improving whole-cell biocatalysis: engineering Escherichiacolito overproduce xylitol as an example.Meth Mol Biol2011,743:185–203.
24. Pandhal J, Ow SY, Noirel J, Wright PC:Improving N-glycosylation efficiency inEscherichia coliusing shotgun proteomics, metabolic network analysis, and selective reaction monitoring.Biotechnol Bioeng2011,108(4):902–912. 25. Waegeman H, De Lausnay S, Beauprez J, Maertens J, De Mey M, Soetaert W:
Increasing recombinant protein production inEscherichia coliK12 through metabolic engineering.N Biotechnol2011, [Epub ahead of print]. 26. Bacher A:Biosynthesis of flavins, Chemistry and biochemistry of
flavoenzymes, Volume 1. Boca Raton, Fla: CRC press; 1991:215–259. 27. Bacher A, Eisenreich K, Kis K, Ladenstein R, Richter G, Scheuring J, Weinkauf
S:Biosynthesis of flavins. InBioorganic chemistry frontiers, Volume 3. Edited by Dugas H, Schmidtchen FP. Berlin, Germany Springer; 1993:147–192. 28. Raina S, Mabey L, Georgopoulos C:TheEscherichia coli htrPgene product
is essential for bacterial growth at high temperatures: mapping, cloning, sequencing, and transcriptional regulation of htrP.J Bacteriol1991,
173:5999–6008.
29. Guzman LM, Belin D, Carson MJ, Beckwith J:Tight regulation, modulation, and high-level expression by vectors containing the arabinose PBAD promoter.J Bacteriol1995,177:4121–4130.
doi:10.1186/1475-2859-11-93
Cite this article as:Ghoshet al.:An inverse metabolic engineering
approach for the design of an improved host platform for
over-expression of recombinant proteins inEscherichia coli.Microbial Cell
Factories201211:93.
Submit your next manuscript to BioMed Central and take full advantage of:
• Convenient online submission
• Thorough peer review
• No space constraints or color figure charges
• Immediate publication on acceptance
• Inclusion in PubMed, CAS, Scopus and Google Scholar
• Research which is freely available for redistribution