Sphingosine 1-phosphate receptor 3 and RhoA signaling mediate inflammatory gene expression in astrocytes
Full text
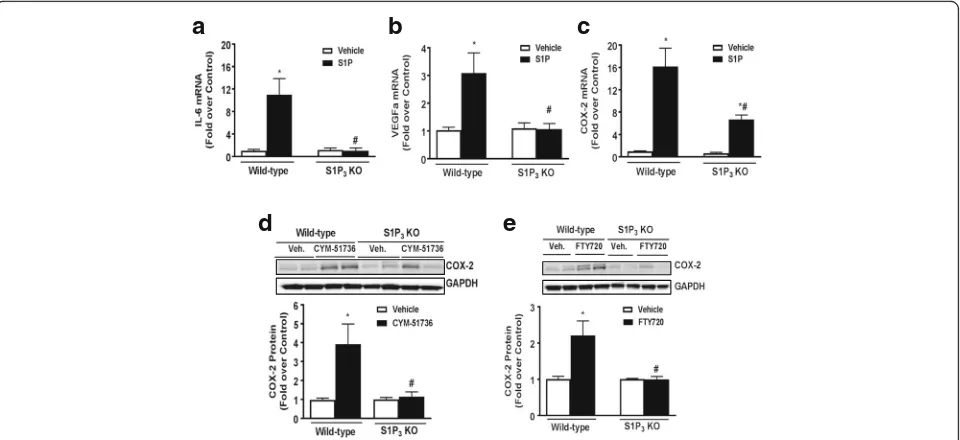
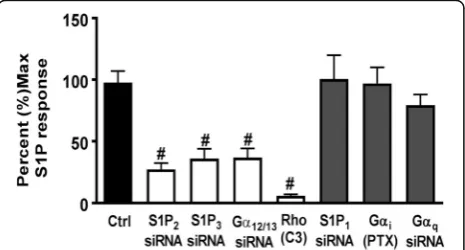
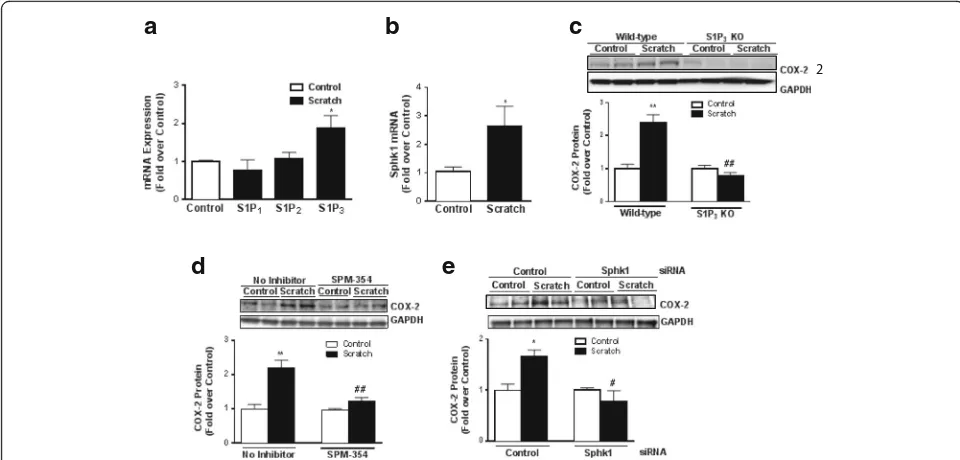
Figure

Related documents
The direct effect is relatively straightforward: a higher foreign ownership share for given values of γ i reduces all agents’ ownership of domestic firms and thus naturally affects
market environment we also study a two-lottery individual decision problem and one- market entry games with ambiguous and risky information.. For these two
Dept of Cardiovascular Critical Care, Boston Children’s Hospital Associate Professor Cardiology (Pediatrics), Harvard Medical School. Pietro
As this study reveals that there is in fact, a rise in the food production, change in cropping patterns and rise in incomes through wage employments and reduction in the
Knowledge and Management of Post Partum Haemorrhage Among Skilled Birth Attendants in Primary Health Centres of Jos North LGA, Plateau State*. Lydia Babatunde Bulndi 1 ,
We showed that a more conservative definition of QFT-Plus positivity, based on double-positive antigen tube results (TB1 and TB2), significantly reduces the positivity rate to 0.6%
Martin, supra note 209, at 7; see also K AHN ET AL ., supra note 112, at 6-29 (“If the employee indicates a belief that the appraisal is inaccurate, the allegations should be
Among the trees known to be graceful are caterpillars, trees with at most 4-end vertices, trees with diameter at most 5, trees with at most 35 vertices,
