Copyright © 1997, American Society for Microbiology
Rapid Identification and Fingerprinting of Candida krusei by
PCR-Based Amplification of the Species-Specific
Repetitive Polymorphic Sequence CKRS-1
ARNAUD CARLOTTI,* FOAD CHAIB, ANDRE´E COUBLE, NICOLAS BOURGEOIS,VIRGINIE BLANCHARD,ANDJEAN VILLARD
Laboratoire de Mycologie Fondamentale et Applique´e aux Biotechnologies Industrielles, Faculte´ de Pharmacie, Universite´ Claude Bernard Lyon-I, 69373 Lyon cedex 08, France
Received 30 October 1996/Returned for modification 10 February 1997/Accepted 3 March 1997
A PCR method was developed to identify and fingerprint Candida krusei isolates simply and rapidly. The primer pair Arno1 and Arno2 was designed to amplify the polymorphic species-specific repetitive sequence CKRS-1 (C. krusei repeated sequence 1) that we identified in the nontranscribed intergenic regions (IGRs) of rRNA genes in C. krusei LMCK31. The specificity, sensitivity, reproducibility, and fingerprinting ability of the PCR assay were evaluated. Amplification products were obtained from all 131 C. krusei isolates studied. No other yeast species of medical importance (n526), including species similar to C. krusei, species of pathogenic filamentous fungi, or a variety of pathogenic bacteria, yielded a PCR product with these primers. This PCR assay allowed for the identification of C. krusei in less than 6 h. The PCR assay was sensitive enough to detect as little as 10 to 100 fg of C. krusei-purified DNA and proved to be reproducible. Since amplification products varied both in number and in molecular weight according to the strains, PCR patterns allowed strains to be distinguished. To ascertain the epidemiological usefulness of this PCR fingerprinting, the patterns of the 131 isolates were compared. A total of 95 types which corresponded to 95 independent strains were delineated (discriminatory power 5 1 with n 5 95). Comparison of the results of PCR fingerprinting and those of fingerprinting with the CkF1,2 probe showed that they concurred. In addition, this work yields insights into the mechanisms involved in generating polymorphisms in the IGRs of C. krusei. Since this method is simpler and faster than established identification and genotyping methods of this important pathogenic species, it is a critical improvement for clinical microbiology laboratories relevant not only to diagnosis but also to epide-miology.
Candida krusei is an important emerging pathogenic yeast, particularly in immunocompromised patients (13, 23, 41). In addition to its known lack of susceptibility to fluconazole (11, 30), a relatively decreased susceptibility of C. krusei to several other antifungal agents has recently been documented (3). Little is known about the epidemiology of infection with this species, but the potential for nosocomial transmission must be considered (3). Hence, improvements in the methods available for diagnosis and epidemiological studies of C. krusei are crit-ical to clincrit-ical laboratories. For instance, they may allow for the earlier detection and identification of C. krusei, which would permit the earlier initiation of adapted antifungal ther-apy.
The PCR (26) is the most rapid and sensitive technique for detecting a specific DNA sequence. Several reports indicate that PCR might be a useful test for either the early detection of Candida species and their rapid identification to the species level (4, 10, 12, 14–16, 19, 20, 24, 25, 27–29, 32, 34, 36, 37, 40) or the fingerprinting of isolates (22, 32, 35, 36). Most of these PCR assays have been devoted to Candida albicans and a few have included C. krusei (4, 12, 20, 27, 40). In addition, none of them combined both specificity and fingerprinting ability in one simple assay.
We recently developed the species-specific CkF1,2 DNA
probe which proved to be useful for both identification (7) and fingerprinting (5) of C. krusei. We have shown by sequence analysis of the two fragments (fragments F1 and F2) that constitute CkF1,2 that they represent polymorphic forms of the nontranscribed intergenic region (IGR) of rRNA genes and that they contained a repeated sequence, CKRS-1 (C. krusei repeated sequence 1), which is made up of a variable number of tandemly repeated sequences of about 165 bp (re-peated eight times in F1 and seven times in F2) (8). We believe that this sequence is the primary basis for the polymorphic CkF1,2 fingerprint pattern and the fingerprinting differences between strains.
In an attempt to combine both identification and finger-printing of C. krusei, we developed, on the basis of the nucle-otide sequence of CkF1,2, a PCR assay targeting the CKRS-1 region. We report here on the species specificity, sensitivity, reproducibility, and fingerprinting ability of this simple and rapid assay. We also compared PCR to our previous scheme of typing C. krusei isolates (5).
MATERIALS AND METHODS
Strains.In total, 169 strains of yeast, filamentous fungi, and bacteria were examined (Table 1). The 30 reference yeast strains belonged to 26 different species, according to Kreger-van Rij (17). The type strain of the species C. krusei, CBS 573 (Centraal Bureau voor Schimmelcultures, Delft, The Netherlands), was included, as was C. krusei LMCK31 (equivalent of K31), from which the CkF1,2 DNA probe was originally selected (5, 7, 8). A total of 131 strains of C. krusei
from clinical (n5129) or environmental (n52) sources, labelled K1 to K147,
were also studied. Species phenotypically related to C. krusei (i.e., Candida
valida) or difficult to distinguish from C. krusei by means of miniaturized
iden-tification systems (e.g., Candida glabrata, Candida inconspicua, Candida lambica,
* Corresponding author. Mailing address: LMFABI, Faculte´ de Pharmacie, Universite´ Lyon I, 8, avenue Rockefeller, 69373 Lyon cedex 08, France. Phone: (33) 78 77 72 76. Fax: (33) 78 77 72 12. E-mail: [email protected].
1337
on May 15, 2020 by guest
http://jcm.asm.org/
Candida norvegensis, Candida rugosa, Candida zeylanoides, Yarrowia lipolytica
[formerly Candida lipolytica], and Zygosaccharomyces rouxii) were also included (Table 1). Representative pathogenic filamentous fungi and bacterial strains were included as well (Table 1). Fungal strains were grown in YM medium (glucose, 1%; yeast extract, 0.3%; malt extract, 0.3%; Bacto Peptone, 0.5%) at 30°C. When the strains were not purchased from international collections, their identifications were done by conventional methods (2, 17) and were further verified with the API 32C system (API bioMerieux, Marcy l’Etoile, France). Yeasts were maintained at 4°C on YM agar slants. Bacterial strains were grown as described previously (6).
DNA extraction.Yeast whole-cell DNA was extracted as described previously (5). DNAs from filamentous fungi were extracted as described by Bainbridge et al. (1). Bacterial DNAs were extracted as described elsewhere (6). DNA
con-centrations and A260/A280ratios were determined spectrophotometrically with an
Ultrospec UV monitor (LKB, Cambridge, England).
Precautions against contamination of PCR mixtures.To avoid possible con-tamination of samples we used the universal precautions suggested by Kwok and Higushi (18). Cross-contamination by aerosols was reduced by using separated areas for PCR preparation and PCR product analysis. UV irradiation was used for microcentrifuge tubes, racks, surfaces of laboratory benches, and instruments by using UV lamps on the benchtop. We included appropriate negative controls, which contained all of the reagents except the template DNA during the PCR
with each set used for amplification. Careful laboratory procedures (autoclaving of buffers and distilled water used for PCRs, aliquoting of reagents, use of aerosol-resistant pipette tips, frequent changing of gloves, premixing of reagents, and addition of DNA as the last step) were used. In all experiments reported herein, the negative controls tested negative.
Oligonucleotide primers for the PCR and probe.From the nucleotide se-quences of the F1 and F2 fragments of the CkF1,2 DNA probe we selected two primers which flanked the species-specific variable region CKRS-1 (1,256 bp long in F1) that is located in the nontranscribed spacer 2 of the intergenic region of rRNA genes of C. krusei (Fig. 1). We have established that four major polymor-phic forms of IGRs existed in strain LMCK31, and F1 and F2 are the largest of the four (8). The Dataminder software (a gift from K. Usdin, National Institutes of Health, Bethesda, Md.) was used for primer selection on a Macintosh Centris 660AV computer (Apple, Cupertino, Calif.). The primers were purchased from
Gibco BRL (Gibco BRL, Gaithersburg, Md.). The primers are Arno1 (59-GGC
CAACACATACATACCTT-39) and Arno2 (59-GGTAGGATACTAACCACA
GC-39) (approximate positions 2,148 to 2,167 and 3,970 to 3,989, respectively, on
the F1 fragment of CkF1,2). The primers spanned a 1,841-bp-long fragment in F1 and a 1,652-bp-long fragment in F2.
To ensure the identity of the amplified product and for assay sensitivity, Southern blot hybridization analyses were performed with the F1 fragment of the CkF1,2 DNA probe (100 ng) labelled directly with horseradish peroxidase by using the ECL kit (Amersham, Les Ulis, France) and detected by enhanced
chemiluminescence, as described previously (5, 7). The phagelDNA digested
with HindIII was used as a standard and was labelled under the same conditions.
PCR conditions.Sequences were amplified with a thermal cycler (model
Croc-odile II; Appligene, Illkirsh, France). Each PCR assay was performed in 50ml of
a reaction mixture containing 50 mM KCl; 10 mM Tris-HCl (pH 8.3); 1.5 mM
MgCl2; 0.001% (wt/vol) gelatin; 0.01% Triton X-100; 100mM (each) dATP,
dCTP, dGTP, and dTTP (all from Appligene); 0.2mM (each) primer; 25ml of
template DNA (about 2.5 to 10 ng); and 1.25 U of Taq DNA polymerase
(Appligene). Samples were overlaid with 20ml of mineral oil prior to PCR
amplification. For PCR amplification initial denaturation was for 4 min at 92°C, followed by 32 cycles of annealing for 0.5 min at 55°C, extension for 2 min at 72°C, and denaturation for 0.5 min at 92°C. After the last cycle, final extension was performed at 72°C for 10 min. PCR amplification was determined to be optimum under these conditions, but no change in the PCR amplification pat-terns occurred when annealing temperatures were between 45 and 60°C.
Analysis of the amplified DNA.Portions (10ml) of the PCR products were analyzed on a 1% agarose gel (type II medium EEO; Sigma Chemical Co., St. Louis, Mo.). Electrophoresis was conducted in Tris-borate-EDTA buffer (0.089 M Tris, 0.089 M boric acid, 0.02 M EDTA [pH 8.1]) at 80 V for 1 to 2 h. Either
the phagelDNA digested with HindIII or the molecular weight marker Raoul
I (all from Appligene) was used as the standard. The gels were stained with ethidium bromide (0.5 mg/ml) and were photographed with 300-nm transillumi-nation through an orange filter and with Polaroid type 55 film. For Southern hybridization analysis, the separated amplified products were transferred to a positively charged nylon membrane (Appligene) by Southern blotting (33) under a vacuum and were then baked at 80°C for 15 min. Hybridization, prehybridiza-FIG. 1. Positions of the oligonucleotide primers Arno1 and Arno2 used for PCR identification and fingerprinting of C. krusei. A schematic physical map of the F1 fragment of CkF1,2 probe is shown. F1 represents a polymorphic form of
the IGRs of rRNA genes of LMCK31. It encompasses the 39end of the 25S
rRNA gene, NTS-1 region, a 5S rRNA gene, and an NTS-2 region. White boxes indicate repetitive sequences (a, b, c, and CKRS-1) in F1. The structure of CKRS-1, which is made up of a variable number of repeated elements (kre) in the IGRs is expanded. The last element, kre-7, is a truncated element. b, bases. Abbreviations for restriction enzymes: E, EcoRI; H, HinfI; N, NsiI; S, SmaI; Ev,
[image:2.612.320.555.72.224.2]EcoRV; St, StyI. TABLE 1. Yeasts, filamentous fungi, and bacterial strains screened
by PCR with primer pair Arno1 and Arno2
Species Strain designationa
Yeasts
Candida albicans ...ATCC 2091 Candida albicans serotype A ...LML 1a Candida albicans serotype B ...LML 1b Candida boidinii ...34 F 1 Candida famata ...CBS 1795 T Candida glabrata...CBS 138 T Candida guilliermondii ...CBS 6021 T Candida humicola ...CBS 2839 T Candida inconspicua ...CBS 180 T Candida kefyr...CBS 607 T
Candida krusei ...CBS 573 T, K1-K147 (131)b
Candida lambica...CBS 1876 T Candida lusitaniae...CBS 6936 T Candida norvegensis ...LML Q243 Candida parapsilosis...CBS 604 T Candida rugosa ...SIPHV 823 Candida tropicalis...CBS 94 T
Candida valida...CBS 638 T, CBS 635 (2) Candida zeylanoides ...IPP 207
Cryptococcus neoformans...CBS 132 T Geotrichum candidum...CBS 109.12 Kluyveromyces marxianus...CBS 712 T Rhodotorula glutinis...CBS 20 T Rhodotorula rubra ...CBS 17 T Saccharomyces cerevisiae ...CBS 1171 T Trichosporon cutaneum...IPP 654
Yarrowia lipolytica ...CBS 6124 T, LML 1051 (2) Zygosaccharomyces rouxii ...LML Z1
Filamentous fungi
Aspergillus fumigatus ...LML AR27 Aspergillus flavus...LML AF7 Mucor mucedo ...LML M4
Bacteria
Escherichia coli...JM109 Nocardia asteroides...IPP 1750-88 Rhodococcus sp. ...LML R1 Staphylococcus aureus...LML S1
aAbbreviations: ATCC, American Type Culture Collection, Rockville, Md.;
CBS, Centraal Bureau Voor Schimmelcultures, Delft, The Netherlands; IPP, Institut Pasteur, Paris, France; LML, Laboratoire de Mycologie de la Faculte´ de Pharmacie, Lyon, France. The numbers in parentheses are the total number of
C. krusei strains studied.
bMultiple isolates (n553) from 17 patients, single isolates (n576) from 76
patients, and unrelated strains (n52) from the environment.
on May 15, 2020 by guest
http://jcm.asm.org/
tion, washing, and chemiluminescence detection were performed as described elsewhere (5, 7).
Comparison of PCR amplification patterns and CkF1,2 hybridization pat-terns.In order to compare PCR amplification patterns and CkF1,2 hybridization
patterns, the same set of strains (n510) was analyzed both by PCR and by
Southern hybridization analysis (SHA) with CkF1,2 after HinfI digestion of genomic DNA as described previously (5).
Reproducibility of PCR amplification.The reproducibility of the PCR patterns
was ensured first by incorporating DNA from CBS 573Tas the positive reference
into each assay. Second, all the PCR amplification patterns were established at least twice for each template DNA. Third, the PCR amplification patterns of five selected strains were compared, within a gel and between gels, when using DNA
extracted from either different cultures (n55) or the same culture (n55). In
each case the amplification products for a given strain exhibited the same pat-tern, regardless of the DNA extract or the culture.
RESULTS
Specificity of the PCR.PCR with primer pair Arno1 and Arno2 was specific for the species C. krusei. PCR products were found with all 131 C. krusei strains studied but not with the other yeast species, filamentous fungi, or bacteria exam-ined. Figure 2 provides an example of the species specificity of the PCR assay. While no amplification signal was observed with DNAs of the medically most important yeast species or those species resembling C. krusei, amplification products were observed with C. krusei CBS 573Tand LMCK31. Amplification
products varied both in number and in molecular weight ac-cording to the strain, which allowed for clear discrimination. Two major amplified products of about 1,480 and 1,320 bp were observed for CBS 573T. Four major amplification
prod-ucts of about 1,840, 1,660, 1,500, and 1,350 bp were observed for LMCK31. The first two of these four products corre-sponded exactly to the expected lengths deduced from nucle-otide sequence data for the F1 and F2 fragments of CkF1,2 which corresponded to the two largest polymorphic forms of the IGR of LMCK31, as shown previously (5, 7, 8). These results suggest that four major polymorphic forms of CKRS-1
and the flanking region delimited with the primers exist in LMCK31 and that two major polymorphic forms exist in CBS 573T. These forms varied in size both between patterns and
within a pattern by about 160 to 180 bp. Other examples of the amplification patterns obtained with different C. krusei strains are presented in Fig. 3 to 7.
Sensitivity of the PCR.To determine the sensitivity of the PCR, three series of 104, 103, 102, 10, and 1 fg of C. krusei
LMCK31 DNA were amplified. In all three tests 100 to 10 fg of DNA, the DNA equivalent of about 0.3 to 3 yeast cells accord-ing to Riggsby et al. (31), or roughly 30 to 300 copies of the IGR, could be detected by agarose gel electrophoresis (Fig. 3A). In addition to the four major bands observed previously, three minor ones were seen when 10 pg of template DNA was used (Fig. 3A).
The results obtained by agarose gel electrophoresis were confirmed by Southern blot hybridization with the peroxidase-labelled fragment F1 of probe CkF1,2. With this probe a high hybridization signal was still observed with PCR products from 10 fg of template DNA (Fig. 3B). In addition, the hybridization signals obtained with the minor bands were still observable when 100 fg of template DNA was used for the PCR assay (Fig. 3B).
Reproducibility of the PCR.To determine the reproducibil-ity of the PCR assay, DNA was extracted from a single strain from either different cultures or the same culture and was used as a template so that the pattern of amplification could be compared. A total of five different strains were thus studied. The pattern for a given strain was always the same, regardless of the DNA extract or the cultures. For example, Fig. 4 pre-sents the results obtained with strains LMCK31 and K3. The same pattern shown in Fig. 2, with four major amplified
frag-FIG. 2. Specificity of the PCR with primer pair Arno1 and Arno2. PCR was done with about 2.5 to 5 ng of genomic DNA template from various yeast species. Amplification products were separated by electrophoresis on a 1% (wt/vol) agarose gel at 100 V for 2 h, stained with ethidium bromide, and vizualized by UV irradiation. The molecular size standard was a Raoul molecular size marker. Molecular sizes are indicated (in base pairs) to the right. Molecular sizes of amplified products are indicated in boldface type.
FIG. 3. Sensitivity of the PCR with primer pair Arno1 and Arno2. Purified DNA from C. krusei LMCK31 was serially diluted in water (from 10 pg to 10 ag in 25ml) and was used as a template for the PCR assay. Amplification products were separated by electrophoresis and vizualized as described in legend to Fig. 2(A) prior to being blotted onto a positively charged nylon membrane. The blot was probed with about 100 ng of peroxidase-labelled fragment F1 of the CkF1,2 DNA probe, and hybridization was revealed by chemiluminescence (B). (A) A phagelDNA HindIII digest was used as the standard. Numbers to the left are in base pairs.
on May 15, 2020 by guest
http://jcm.asm.org/
[image:3.612.361.510.71.306.2]ments and three minor ones, was always observed for strain LMCK31, regardless of the culture or the extraction (Fig. 4). However, a decreased intensity of amplification products was observed on one occasion (lane LMCK31 A). This suggested a decreased yield in the corresponding DNA amplification. A unique pattern was also observed with strain K3 when DNA templates were from different cultures and extractions (Fig. 4). Moreover, the amplification pattern of strain CBS 573Twas
established more than 15 times and was always the same, even if variations in intensities were seen on rare occasions. In addition, duplicates of amplification patterns were always the same for all the strains examined.
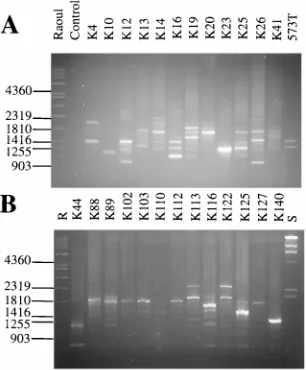
Fingerprinting with the PCR.To assess the fingerprinting ability and the discriminatory power of the PCR assay, a total of 131 isolates of C. krusei from our collection, most of which have previously been typed by SHA with the CkF1,2 probe (5, 7; unpublished data), were examined. They represented either related strains (n553) (multiple isolates from a single patient [n517]) or unrelated strains (n5 78) from either different patients (n576) or the environment (n52). A large variety of PCR patterns was observed for the isolates. A total of 95 patterns that corresponded to the 95 (17 178) independent strains or patients were observed. The patterns for multiple isolates from a single patient were the same regardless of the anatomic site or date of isolation. For instance in Fig. 5, for the 12 isolates three amplification patterns which corresponded to isolates from each of the three patients were observed. The patterns contained between one and four major amplification products. The two isolates from patient P1 were isolated 87 days apart from samples of the mouth and urine. They exhib-ited the same pattern. The four isolates from patient P2 were isolated during a period of 9 days from cultures of samples of blood (two times), the sinus, or the mouth. The six isolates from patient 3 were obtained during a period of 191 days in samples from either the throat (four times), the nose, or a bronchoalveolar aspiration. They all exhibited the same
pat-tern. Hence, the maintenance of the same pattern over a 6-month period was evident for isolates from patient P3.
[image:4.612.104.256.67.267.2]In contrast, epidemiologically unrelated isolates of C. krusei had distinctly different amplification profiles. For instance, the amplification patterns of 26 isolates are presented in Fig. 6. Distinctly different patterns that included between one and eight major amplification products which varied in size and number according to the strain were observed (Fig. 6). All the different patterns were easily differentiated from each other. The patterns for all strains except strains K102, K103, and K112, which were isolated 45 days apart from either stools or cultures of blood from a single patient, were different. For the
FIG. 4. Reproducibility of PCR with primer pair Arno1 and Arno2. Shown here is an example of the amplification patterns obtained with DNAs of strain LMCK31 extracted from either the same culture (I) or different cultures (II) and DNAs of strain K3 extracted from different cultures (III) used as templates. Electrophoresis and vizualization conditions were the same as described in the legend to Fig. 2. Raoul, molecular size marker. Numbers to the left and right are in base pairs.
FIG. 5. Example of PCR fingerprinting of C. krusei-related isolates. The amplification patterns obtained by PCR with Arno1 and Arno2 of purified DNA templates from multiple isolates from three patients (patients P1, P2, and P3) are shown. Conditions were as described in the legend to Fig. 2. Standard,lDNA
HindIII digest; Raoul, molecular size marker. Numbers to the right are in base
[image:4.612.362.511.68.273.2]pairs.
FIG. 6. Example of PCR fingerprinting of unrelated and related (K102, K103, and K112) isolates. The amplification patterns obtained with Arno1 and Arno2 are shown. Conditions were the same as described in the legend to Fig. 2. S, standardlDNA HindIII digest. R: Raoul, molecular size marker. Numbers to the left are in base pairs.
on May 15, 2020 by guest
http://jcm.asm.org/
[image:4.612.360.513.498.683.2]131 isolates, results of typing by PCR and typing by SHA with the CkF1,2 probe agreed in stating the identity or nonidentity of strains (data not shown). PCR with primer pair Arno1 and Arno2 showed a 100% typeability, 100% reproducibility, and a discriminatory power (D) of 1 (for n595 independent strains). PCR results compared with those of SHA with CkF1,2.We believed that polymorphism in the CKRS-1 region targeted by PCR is the primary basis for the polymorphic CkF1,2 finger-printing pattern and fingerfinger-printing differences between strains. To test this hypothesis we compared PCR patterns and CkF1,2 Southern hybridization patterns of HinfI-digested genomic DNA for 10 selected strains. Generally, PCR patterns and hybridization patterns contained the same number of major bands (Fig. 7). For example, for CBS 573T, two fragments of
1,480 and 1,320 bp were observed in the PCR patterns and two HinfI restriction fragments of 2,950 and 2,780 bp hybridized with CkF1,2. In both cases the size difference between the two fragments within a pattern was about 160 to 170 bp. Similarly, for strain K65, two fragments of 1,430 and 1,120 bp were observed in PCR patterns and two major HinfI restriction fragments of 3,458 and 2,580 bp hybridized with CkF1,2. How-ever, the size differences between the two fragments were about 320 bp (23160 bp) and 900 bp, respectively. Relative differences in size between PCR fragments and hybridizing fragments were consistently either about 1,450 bp (for exam-ple, 2,950 and 1,480 bp or 2,780 and 1,320 bp for CBS 573Tand
2,577 and 1,120 bp for K65) or 2,000 bp (for example, 3,458 and 1,430 bp for K65) for all 10 C. krusei strains shown in Fig. 7 and the other C. krusei strains studied (data not shown). Within a single pattern the two values could be encountered (for example, in strain K65). In LMCK31 patterns, we found the differences between both amplified products and CkF1,2-hybridizing fragments to be about 160 to 180 bp and relative differences between fragments in PCR patterns and CkF1,2 hybridization patterns to be about 2,000 bp (data not shown). The expected variations deduced from nucleotide sequence data for F1 and F2 were 189 and about 2,000 bp, respectively, if CKRS-1 was the major source of variation. This suggested that CKRS-1 is the primary basis for differences within a
pat-tern and between patpat-terns, but that a deletion of about 600 bp outside that region could be involved in the polymorphisms revealed by probe CkF1,2.
DISCUSSION
The findings of this study demonstrate that PCR amplifica-tion of the species-specific CKRS-1 repetitive polymorphic se-quence of the intergenic region of rRNA genes in C. krusei can be used effectively both to identify and to fingerprint isolates rapidly and reliably in one simple sensitive assay. It is clear that the PCR assay with primers Arno1 and Arno2 is species spe-cific and sensitive. Several PCR-based methods of detection and identification of Candida species have been reported. Overall, they use either (i) detection of specific genes by using species-specific primers or (ii) analysis of the amplification products obtained with universal primers by either restriction endonuclease analysis (REA; PCR-REA or PCR-restriction fragment length polymorphism analysis) (15, 20, 40) or analysis with species-specific oligonucleotide probes (12, 27, 34, 40). Recently, sequential tandem PCR of both conserved and vari-able regions of the transcribed large-subunit RNA gene has also been described (14), as has single-strand conformational polymorphism analysis (38), for the identification of Candida spp.
[image:5.612.60.296.70.232.2]Few of these techniques have been applied to C. krusei (4, 12, 20, 27, 40), and generally, they involved small numbers of strains. Moreover, our approach presents some advantages over those methods that have been described to date. While species-specific sequences of genes that encode actin (16), heat shock proteins (10), and cytochrome P-450 lanosterol-a -de-methylase (L1A1) (4) have been extensively used for PCR-based identification of C. albicans, except for L1A1, the lack of species-specific nucleotide sequence information for these C. krusei genes prevents the development of C. krusei-specific primers. As we developed and sequenced the first species-specific fingerprinting probe for C. krusei, CkF1,2, we were able to design two species-specific primers for the identifica-tion of that species by PCR amplificaidentifica-tion of the CKRS-1 re-gion. We chose that sequence because we thought that it rep-resented a target of choice (7) for both the identification and typing of C. krusei strains because it is a species-specific, poly-morphic, and repeated region. This potential is confirmed here. Presumptive identification of C. krusei by PCR-REA has been reported (20). Our PCR assay is less complicated than PCR-REA and allows for the accurate and reliable identifica-tion of C. krusei. PCR-based identificaidentifica-tion of Candida species at the species level by using both constant and variable regions of either the small-subunit 18S rRNA gene (27) or the large-subunit 25S rRNA gene (14) or the internal transcribed spacer regions of rRNA genes (12, 40) have been reported. The method described by Niesters et al. (27) required hybridization with species-specific probes, while the method described by Haynes et al. (14) did not. The latter required a tandem PCR with both universal and specific primers sequentially, adding to the time required to perform PCR-based tests and to the risk of sample contamination due to additional sample manipula-tion. Fujita et al. (12) developed a microtitration plate enzyme immunoassay to detect Candida species, including C. krusei, with species-specific probes derived from the internal tran-scribed spacer region of fungal ribosomal DNA. While this approach proved to be rapid and sensitive with other Candida species, the investigators reported a relatively poor reactivity of their C. krusei digoxigenin-labeled probe. Recently, rapid iden-tification of Candida species by DNA fingerprinting with PCR with arbitrary primers has also been described (34). The
FIG. 7. Comparison of PCR amplification patterns and Southern hybridiza-tion patterns with CkF1,2 of genomic DNA from 10 selected strains. (A) Am-plification patterns obtained with primer pair Arno1 and Arno2. (B) Genomic DNAs from the strains were digested with HinfI, and restriction fragments were separated by electrophoresis on a 0.8% agarose gel at 40 V for 18 h prior to being blotted to a positively charged nylon membrane. The blot was probed with about 100 ng of peroxidase-labelled CkF1,2 probe, and hybridization was revealed by chemiluminescence. Standard,lDNA HindIII digest; Raoul, molecular size marker. Number to the left of each panel are in base pairs.
on May 15, 2020 by guest
http://jcm.asm.org/
method required highly complex pattern comparison, and only one C. krusei strain was evaluated. Therefore, PCR with prim-ers Arno1 and Arno2 is simpler and faster than previously described methods of identification (identification of C. krusei is achieved in less than 6 h when using the rapid DNA extrac-tion procedure). In addiextrac-tion to reliable identificaextrac-tion of the species, this assay allows for the simultaneous typing of the isolates. Moreover, it has been evaluated with a significant number of well-characterized strains. This is also the first dem-onstration of the potential of IGRs containing variable num-bers of tandemly repeated sequences for PCR-based identifi-cation and fingerprinting of a Candida species.
There are no data on the sensitivity of the different assays cited above with regard to the identification of C. krusei. The sensitivity of detection reported here for C. krusei is higher than those generally reported so far for C. albicans, which are in the range of 1 to 50 pg of purified DNA (9, 10, 21). However, the sensitivity of 10 to 100 fg recorded here is of the same order as either the one reported by Kan (16) by using the actin gene (25 fg) and32P-labeled oligonucleotide probe as the target or
that reported by Polanco et al. (28) (125 fg) by using the 18S rRNA gene as the target. The high sensitivity recorded with PCR of the CKRS-1 region can certainly be explained by the repetitive nature of this target sequence, which is repeated as much as the rRNA cistron. Warner (39) reported that, depend-ing on the strain, there are approximately 100 to 220 copies of the rRNA gene repeat array in Saccharomyces cerevisiae. The sensitivity of detection of our PCR assay (10 to 100 fg) is also higher than that of dot blot hybridization analysis with the CkF1,2 DNA probe, which is 60 to 120 ng (7). This represents about a 1,000-fold increase in sensitivity.
For fingerprinting C. krusei isolates, we developed the first typing scheme that proved useful for epidemiological studies by using HinfI restriction of genomic DNA and Southern hy-bridization with CkF1,2 (5, 7). It showed a 100% typeability, a good reproducibility, and a high discriminatory power (D . 0.98). The usefulness of this typing scheme has recently been confirmed by others (3). Fingerprinting by PCR amplification of the CKRS-1 region gave the same results with regard to typeability, reproducibility, and discriminatory power (D5 1 with 95 independent strains) as our original typing scheme, as expected, but it is much simpler to perform and is faster and it is available to every laboratory, contrary to hybridization-based techniques. Despite the large number of strains studied, this typing method identified epidemiologically unrelated isolates as being different, yet it was still able to identify multiple isolates from the same patient as being the same strain. It allowed us to demonstrate for the first time the maintenance of a single strain in one patient over a 6-month period and at different anatomic sites. The data collected confirmed and extended our previous conclusions that have also recently been confirmed by others (3). First, each patient was infected or colonized with his or her own distinct strain; second, multiple isolates from the same patient were identical, whatever the date or anatomic site of isolation; and third, there was no evidence of nosocomial transmission (but this may be due simply to the patient population studied). This clinical epide-miological evidence supports an endogenous origin for the colonizing and infecting C. krusei isolates. PCR methods for fingerprinting other Candida strains have been reported (22, 32, 36). These methods rely on amplification of variable re-gions flanked by conserved rere-gions in which the primers are either (i) designed or (ii) random (35). Random amplification of polymorphic DNA presents well-known drawbacks, includ-ing lack of reproducibility and difficulty in pattern comparison.
To the best of our knowledge, none of the PCR-based methods have been successfully applied to C. krusei.
Comparison of typing of the CKRS-1 region by PCR and SHA with CkF1,2 confirms and extends our previous results (5, 7, 8). First, this comparison clearly demonstrates that the poly-morphism within a strain and differences in fingerprints be-tween strains are mainly due to variations in lengths occurring in the CKRS-1 region. We have shown by sequence compari-son that in strain K31 these variations in length are due to duplication or deletion of the C. krusei repeated subelement (kre) of about 164 to 165 bp that generates the polymorphic forms of the IGR (8). The size differences of multiples of about 160 to 180 bp recorded for the different strains studied by PCR of the CKRS-1 region strengthen our previous conclusions. In addition, the present results suggest that differences between the fingerprints of different strains involved (i) a variable num-ber of the polymorphic forms of IGR according to the strain, (ii) a variation in the overall length of the CKRS-1 region and hence variable numbers of kre elements, and (iii) the fact that, to account for the high degree of variability observed in these numerous strains, either the truncated element observed in K31 or other kre elements could be of various sizes, depending on the strain, to explain the overall size variation of the CKRS-1 region between strains. We also have shown that two types of relative length differences between CkF1,2-hybridizing fragments and PCR patterns account for yet another source of variability within and between patterns. This work is critical to understanding both the mechanisms of variability in C. krusei strains and how fingerprints evolve between strains. More gen-erally, this work yields insights to assess the value of variable repetitive sequences as epidemiological markers.
In conclusion, PCR amplification of the CKRS-1 region provides a sensitive, rapid, and reliable method for the simul-taneous identification and discrimination of C. krusei isolates. The method is faster and simpler than the available identifi-cation methods, especially conventional identifiidentifi-cation meth-ods. We believe that this work is a crucial improvement in the means of identification of this important pathogenic yeast, which possesses marked antifungal resistance. This is critical to the clinical microbiology laboratory because it improves both the rapidity of identification of isolates (it reduces the time required for identification to about 6 h) and it facilitates large epidemiological studies. The effectiveness and simplicity of the method make it available to almost all laboratories. Finally, this work confirms and extends our previous findings regarding the mechanisms involved in generating fingerprinting differ-ences. The direct detection by our PCR assay of C. krusei in biological samples is in progress.
ACKNOWLEDGMENTS
We are indebted to Christopher Kvaal for reading the manuscript. For providing the strains we thank the microbiology laboratories of S. Bretagne, V. Lavarde, B. Druel, G. Barbe´, M. Chaumarat, G. Carre´, and G. Zambardi.
REFERENCES
1. Bainbridge, B. W., C. L. Spreadbury, F. G. Scalise, and J. Cohen. 1990. Improved methods for the preparation of high molecular weight DNA from large and small scale cultures of filamentous fungi. FEMS Microbiol. Lett.
66:113–118.
2. Barnett, A. A., R. W. Payne, and D. Yarrow. 1990. Yeasts: characteristics and identification, 2nd ed. Cambridge University Press, New York, N.Y. 3. Berrouane, Y. F., R. J. Hollis, and M. A. Pfaller. 1996. Strain variation
among and antifungal susceptibilities of isolates of Candida krusei. J. Clin. Microbiol. 34:1856–1858.
4. Burgener-Kairuz, P., J. P. Zuber, P. Jaunin, T. G. Buchman, J. Bille, and M.
Rossier.1994. Rapid detection and identification of Candida albicans and
Torulopsis (Candida) glabrata in clinical specimens by species-specific nested
on May 15, 2020 by guest
http://jcm.asm.org/
PCR amplification of a cytochrome P-450 lanosterol-a-demethylase (L1A1) gene fragment. J. Clin. Microbiol. 32:1902–1907.
5. Carlotti, A., R. Grillot, A. Couble, and J. Villard. 1994. Typing of Candida
krusei clinical isolates by restriction endonuclease analysis and hybridization
with CkF1,2 DNA probe. J. Clin. Microbiol. 32:1691–1699.
6. Carlotti, A., and G. Funke. 1994. Rapid distinction of Brevibacterium species by restriction analysis of rDNA generated by polymerase chain reaction. Syst. Appl. Microbiol. 17:380–386.
7. Carlotti, A., A. Couble, J. Domingo, K. Miroy, and J. Villard. 1996. Species-specific identification of Candida krusei by hybridization with the CkF1,2 DNA probe. J. Clin. Microbiol. 34:1726–1731.
8. Carlotti, A., T. Srikantha, K. Schro¨ppel, C. Kvaal, J. Villard, and D. R. Soll. A novel repeat sequence (CKRS-1) containing a tandemly repeated subele-ment (kre) accounts for differences between Candida krusei strains finger-printed with the probe CkF1,2. Curr. Genet., in press.
9. Chryssanthou, E., B. Andersson, B. Petrini, S. Lofdahl, and J. Tollemar. 1994. Detection of Candida albicans DNA in serum by polymerase chain reaction. Scand. J. Infect. Dis. 26:479–485.
10. Crampin, A. C., and R. C. Matthews. 1993. Application of the polymerase chain reaction to the diagnosis of candidosis by amplification of an HSP 90 gene fragment. J. Med. Microbiol. 39:233–238.
11. Fisher, M. A., S. Shen, J. Haddad, and W. F. Tarry. 1989. Comparison of in vivo activity of fluconazole with that of amphotericin B against Candida
tropicalis, Candida glabrata, and Candida krusei. Antimicrob. Agents
Che-mother. 35:1443–1446.
12. Fujita, S.-I., B. A. Lasker, T. J. Lott, E. Reiss, and C. J. Morrison. 1995. Microtitration plate enzyme immunoassay to detect PCR-amplified DNA from Candida species in blood. J. Clin. Microbiol. 33:962–967.
13. Goldman, M., J. C. Pottage, and D. C. Weaver. 1993. Candida krusei funge-mia: report of 4 cases and review of the literature. Medicine (Baltimore)
72:143–150.
14. Haynes, K. A., T. J. Westerneng, J. W. Fell, and W. Moens. 1995. Rapid detection and identification of pathogenic fungi by polymerase chain reac-tion amplificareac-tion of large subunit ribosomal DNA. J. Med. Vet. Mycol.
33:319–325.
15. Hopfer, R. L., P. Walden, S. Setterquist, and W. E. Highsmith. 1993. De-tection and differentiation of fungi in clinical specimens using polymerase chain reaction (PCR) amplification and restriction enzyme analysis. J. Med. Vet. Mycol. 31:65–75.
16. Kan, V. L. 1993. Polymerase chain reaction for the diagnosis of candidemia. J. Infect. Dis. 168:779–783.
17. Kreger-van Rij, N. J. W. 1984. The yeasts. A taxonomic study, 3rd ed. Elsevier Science Publishers B.V., Amsterdam, The Netherlands. 18. Kwok, S., and R. Higushi. 1989. Avoiding false positives with PCR. Nature
(London) 339:237–238.
19. Lehmann, P. F., D. Lin, and B. A. Lasker. 1992. Genotypic identification and characterization of species and strains within the genus Candida by using random amplified polymorphic DNA. J. Clin. Microbiol. 30:3249–3254. 20. Maiwald, M., R. Kappe, and H.-G. Sonntag. 1994. Rapid presumptive
iden-tification of medically relevant yeasts to the species level by polymerase chain reaction and restriction enzyme analysis. J. Med. Vet. Mycol. 32:115–122. 21. Makimura, K., S. Y. Murayama, and H. Yamaguchi. 1994. Detection of a
wide range of medically important fungi by the polymerase chain reaction. J. Med. Microbiol. 40:358–364.
22. McCullough, M. J., B. C. Ross, and P. C. Reade. 1995. Genetic differentia-tion of Candida albicans strains by mixed-linker polymerase chain reacdifferentia-tion. J. Med. Vet. Mycol. 33:77–80.
23. Mcquillen, D. P., B. S. Zingman, F. Meunier, and S. M. Levitz. 1992. Invasive infections due to Candida krusei—report of ten cases of fungemia that include three cases of endophthalmitis. Clin. Infect. Dis. 14:472–478. 24. Miyakawa, Y., T. Mabuchi, K. Kagaya, and Y. Fukazawa. 1992. Isolation and
characterization of a species-specific DNA fragment for detection of
Can-dida albicans by polymerase chain reaction. J. Clin. Microbiol. 30:894–900.
25. Miyakawa, Y., and T. Mabuchi. 1994. Characterization of a species-specific DNA fragment originating from the Candida albicans mitochondrial ge-nome. J. Med. Vet. Mycol. 32:71–75.
26. Mullis, K. B., and F. A. Faloona. 1987. Specific synthesis of DNA in vitro via a polymerase-catalyzed chain reaction. Methods Enzymol. 155:335–350. 27. Niesters, H. G. M., W. H. F. Goessens, J. F. M. Meis, and W. G. V. Quint.
1993. Rapid, polymerase chain reaction-based identification assays for
Can-dida species. J. Clin. Microbiol. 31:904–910.
28. Polanco, A. M., J. L. Rodriguez-Tudela, and J. V. Martinez-Suarez. 1995. Detection of pathogenic fungi in human blood by the polymerase chain reaction. Eur. J. Clin. Microbiol. Infect. Dis. 14:618–621.
29. Rand, K. H., H. Houck, and M. Wolff. 1994. Detection of candidemia by polymerase chain reaction. Mol. Cell. Probes 8:215–221.
30. Rex, J. H., M. A. Pfaller, A. L. Barry, P. W. Nelson, and C. D. Webb. 1995. Antifungal susceptibility testing of isolates from a randomized, multicenter trial of fluconazole versus amphotericin B as treatment of nonneutropenic patients with candidemia. Antimicrob. Agents Chemother. 39:40–44. 31. Riggsby, W. S., L. J. Torres-Bauza, J. W. Wills, and T. M. Townes. 1982.
DNA content, kinetic complexity, and the ploidy question in Candida
albi-cans. Mol. Cell. Biol. 2:853–862.
32. Scho¨nian, G., O. Meusel, H. Tietz, W. Meyer, Y. Gra¨ser, I. Tausch, W.
Presber, and T. G. Mitchell.1993. Identification of clinical strains of Candida
albicans by DNA fingerprinting with the polymerase chain reaction. Mycoses
36:171–179.
33. Southern, E. M. 1975. Detection of specific sequences among DNA frag-ments separated by electrophoresis. J. Mol. Biol. 98:503–517.
34. Thanos, M., G. Scho¨nian, W. Meyer, C. Schweynoch, Y. Gra¨ser, T. G.
Mitchell, W. Presber, and H.-J. Tietz.1996. Rapid identification of Candida species by DNA fingerprinting with PCR. J. Clin. Microbiol. 34:615–621. 35. van Belkum, A. 1994. DNA fingerprinting of medically important
microor-ganisms by use of PCR. Clin. Microbiol. Rev. 7:174–184.
36. van Belkum, A., W. Melchers, B. E. de Pauw, S. Scherer, W. Quint, and J. F.
Meis.1994. Genotypic characterization of sequential Candida albicans iso-lates from fluconazole-treated neutropenic patients. J. Infect. Dis. 69:1062– 1070.
37. van Deventer, A. J. M., W. H. F. Goessens, A. van Belkum, H. J. A. van Vliet,
E. W. M. van Etten, and H. A. Verbrugh.1995. Improved detection of
Candida albicans by PCR in blood of neutropenic mice with systemic
can-didiasis. J. Clin. Microbiol. 33:625–628.
38. Walsh, T. J., A. Francesconi, M. Kasai, and S. J. Chanock. 1995. PCR and single-strand conformational polymorphism for recognition of medically im-portant opportunistic fungi. J. Clin. Microbiol. 33:3216–3220.
39. Warner, J. R. 1989. Synthesis of ribosomes in Saccharomyces cerevisiae. Microbiol. Rev. 53:256–271.
40. Williams, D. W., M. J. Wilson, M. A. O. Lewis, and J. C. Potts. 1995. Identification of Candida species by PCR and restriction fragment length polymorphism analysis of intergenic spacer regions of ribosomal DNA. J. Clin. Microbiol. 33:2476–2479.
41. Wingard, J. R. 1995. Importance of Candida species other than C. albicans as pathogens in oncology patients. Clin. Infect. Dis. 20:115–125.