0095-1137/10/$12.00 doi:10.1128/JCM.00362-10
Copyright © 2010, American Society for Microbiology. All Rights Reserved.
Simultaneous Sequence Analysis of the 16S rRNA and
rpoB
Genes by
Use of RipSeq Software To Identify
Mycobacterium
Species
䌤
Keith E. Simmon,
1* Øyvind Kommedal,
2,3Øystein Saebo,
3Bjarte Karlsen,
3and Cathy A. Petti
1,4Associated Regional and University Pathologists (ARUP) Institute for Clinical and Experimental Pathology, Salt Lake City, Utah1;
Section for Microbiology and Immunology, The Gade Institute, University of Bergen, Bergen, Norway2; iSentio, Bergen, Norway3;
and Departments of Medicine and Pathology, University of Utah School of Medicine, Salt Lake City, Utah4
Received 22 February 2010/Returned for modification 9 June 2010/Accepted 24 June 2010
The 16S rRNA gene is commonly used to identify Mycobacterium spp., but alternative DNA targets can
provide better resolution to the species level. We evaluated a novel system that enables simultaneous ampli-fication, sequencing, and analysis of two different DNA targets in a single tube to identify clinical isolates of Mycobacterium spp. For 139 clinical isolates, we found that the 16S rRNA gene alone identified 67 (48%) isolates as single species, 68 (49%) isolates to the complex or group level, and 4 (3%) isolates to the genus level
only. TherpoBgene alone identified 117 (84%) isolates as single species, 10 (7%) isolates to the complex or
group level, and 12 (8%) isolates to the genus level only. Combining the separate analyses for sequencing of two gene targets, 119 (86%) isolates were identified as single species and 10 (7%) isolates were identified to the complex or group level. Seven (5%) isolates were identified as novel species within established groups, and 3 (2%) were identified to the genus level only. Dual-locus identification identified 110 (79%) isolates as single species and 22 (16%) isolates to the complex or group level. Six (4%) were identified as novel species within established groups, and 1 (1%) was identified to the genus level only. Identifications were more accurate when
both the 16S rRNA andrpoBgenes were screened, and reliance on a single gene target was suboptimal. RipSeq
dual-locus software provides an accurate alternative method for laboratories using two different gene targets for microorganism identification.
Accurate identification of Mycobacteriumspecies is impor-tant because the clinical relevance and susceptibility patterns differ among species and impact clinical decision-making. Rou-tine molecular identification ofMycobacteriumspecies is often hindered by the lack of genetic heterogeneity within the 16S rRNA gene. Other DNA targets have been used to resolve taxonomic uncertainties, and studies have demonstrated their use for identification in clinical laboratories (1–3, 5, 7, 11, 16). Recently, more scrutiny has been applied to these targets, particularly on their relative values for describing new species, and data have suggested that reliance on a single DNA target for identification may have limitations (14, 15, 20). Routine implementation of alternative targets for identification is lim-ited by cost, longer turnaround times, a lack of references in databases, a lack of consensus on the optimal target or region within a DNA target (1, 10), and uncertainty about expected intraspecies variability (14, 18).
We hypothesized that concurrent sequencing of the 16S rRNA gene and an alternative DNA target could provide bet-ter discrimination for identification ofMycobacteriumspp., and we sought a method that would not significantly impact work-flow, cost, or analysis time. RipSeq software (iSentio, Bergen, Norway) offers a solution for laboratories that use two genetic loci for identification by enabling simultaneous amplification, sequencing, and analysis of two different DNA targets in a
single tube. The RipSeq dual-locus identification system has an algorithm that analyzes mixed electropherograms obtained by sequencing of two different gene targets, searches two different databases, each representing one of the amplified targets, and provides a ranked list of best-match results for each target gene. Using clinical isolates, we evaluated the added value of using two DNA targets for identification of
Myco-bacterium sp. by standard laboratory protocols performed
according to CLSI guidelines (6). We also assessed the ac-curacy of the RipSeq dual-locus identification system for identification ofMycobacteriumspp. by comparing the iden-tity scores of consensus sequences obtained by sequencing of two gene targets individually to those obtained by the simultaneous dual-locus method.
MATERIALS AND METHODS
In April 2008 and August 2008, we prospectively collected consecutive clinical isolates that were identified at ARUP Laboratories asMycobacteriumspp. by partial 16S rRNA gene sequencing. Template DNA was prepared by a 15-min boil of isolate suspensions in PrepMan Ultra sample preparation reagent (Ap-plied Biosystems, Foster City, CA). Amplification was performed in a 25-l reaction mixture containing 1⫻FastStart PCR master mix (Roche Diagnostics Corporation, Alameda, CA), a 0.2M concentration of each primer, and 2l of template. Primers for amplification of the 16S rRNA andrpoB(2) genes were 16S-5F (5⬘-TTGGAGAGTTTGATCCTGGCTC-3⬘), 16S-534R (5⬘-TACCGCG GCTGCTGGCAC-3⬘),rpoB-mycoF (5⬘-GGCAAGGTCACCCCGAAGGG-3⬘), and rpoB-mycoR (5⬘-AGCGGCTGCTGGGTGATCATC-3⬘). PCR mixtures were amplified by an initial hold at 95°C for 10 min and then 35 cycles of denaturing at 95°C for 30 s, annealing at 55°C for 30 s, and extension at 72°C for 1 min 30 s. The reaction ended with a final extension at 72°C for 2 min and a hold at 4°C. A single amplification reaction mix was prepared for each isolate and contained primers for both the 16S rRNA andrpoBgenes. Gel electrophoresis was used to confirm the amplification of both targets (16S rRNA gene product,
⬃500 bp; andrpoBproduct,⬃750 bp). PCR products were purified using Am-pure beads (Agencourt Bioscience Corp., Beverly, MA) per the manufacturer’s
* Corresponding author. Mailing address: Associated Regional and University Pathologists (ARUP) Institute for Clinical and Experimen-tal Pathology, 500 Chipeta Way, Salt Lake City, UT 84108. Phone: (801) 583-2787, ext. 2035. Fax: (801) 584-5109. E-mail: keith.simmon @aruplab.com.
䌤Published ahead of print on 7 July 2010.
3231
on May 16, 2020 by guest
http://jcm.asm.org/
instructions and were sequenced with the following primers or primer combina-tions: 16S-5F, 16S-534R,rpoB-mycoF,rpoB-mycoR, 16S-5F plusrpoB-mycoF, and 16S-534R plusrpoB-mycoR. Sequencing was done using Big Dye Terminator reagents on a model 3730xl sequencer (Applied Biosystems) by a standard automated sequencer protocol.
Editing of sequences generated with a single primer was done by manual inspection, using Contig Express in the Vector NTI suite (Invitrogen, Carls-bad, CA). Edited sequences were aligned in MEGA 4.0 (19). All sequences were trimmed to the same size for comparison. Bootstrapped neighbor-joining trees were constructed using the Jukes-Cantor parameter. Dual-locus electropherograms were uploaded and bulk analyzed directly in the RipSeq software.
Identification of isolates by use of individual gene targets.Isolate identifica-tions based on the 16S rRNA gene were based on CLSI guidelines (6), which state that 100% identity to a reference sequence is required for species identi-fication. Identification with therpoBgene was performed using criteria outlined by Adekambi et al., who recommended an identity of 98.0 to 100% for species identification (1, 2). GenBank reference sequences were used for identification for both the 16S rRNA andrpoBgenes.
Analysis of sequences with RipSeq software.All samples were initially run with standard settings. The basis for the dual-locus module is the RipSeq algorithm, which has been described previously (12, 13). The dual-locus module includes the RipSeq base-calling procedure and a modified alignment strategy. The basic principles of the search are to divide the mixed electropherograms into smaller pieces and to construct every possible combination of bases (words) within each piece. Each single word is subsequently compared to the corresponding area of the reference sequences in the database. Before a RipSeq search can be per-formed, three parameters are set. The first two are the 5⬘-end trimming and 3⬘-end trimming of the electropherograms. The third is theyaxis cutoff. Ayaxis cutoff is used to avoid base calling of low signals from the baseline of the electropherograms, as these would significantly reduce the specificity of the analysis.
Because the RipSeq dual-locus system is intended for use on cultured isolates and the ratio between the different targets is constant, the signal peak height and technical quality of the electropherograms should be similar every time. This observation enables establishment of standard parameter settings and bulk anal-ysis of multiple samples. For each sample, the program will generate a consensus answer based on the forward and reverse sequences for both gene targets. The consensus answer will be generated only when there is agreement within all results. If one of the results is not in agreement or if one of the targets is not detected, a warning will be displayed, and the available results for that sample must be evaluated manually.
Interpretation by the RipSeq algorithm.A RipSeq interpretation scheme set identification to the species level by use of the 16S rRNA gene to 99.5 to 100% and identification to the species level by use of therpoBgene to 97.8 to 100%. When anrpoB sequence matched a reference sequence with an identity of
⬎99.0%, the algorithm weighted therpoBresult with a higher priority. When an identity score obtained using therpoBgene reference did not meet species guidelines, the database was interrogated. When the database lacked anrpoB gene reference for the species, 16S rRNA gene identification was used. When a correspondingrpoBreference was in the database but below the cutoff value, we classified the isolate as aMycobacteriumspecies closely related to its correspond-ing 16S rRNA gene identity.
The quality and accuracy of base calling for the 16S rRNA andrpoBgenes were determined by inferring a consensus sequence based on the variance in identity scores toward the top reference for the forward and reverse sequenc-ing reactions. The identity scores toward the reference were compared for the dual-locus method and the consensus sequences of individual gene targets.
RESULTS
16S rRNA gene identification.Alignment of 462 bp of the
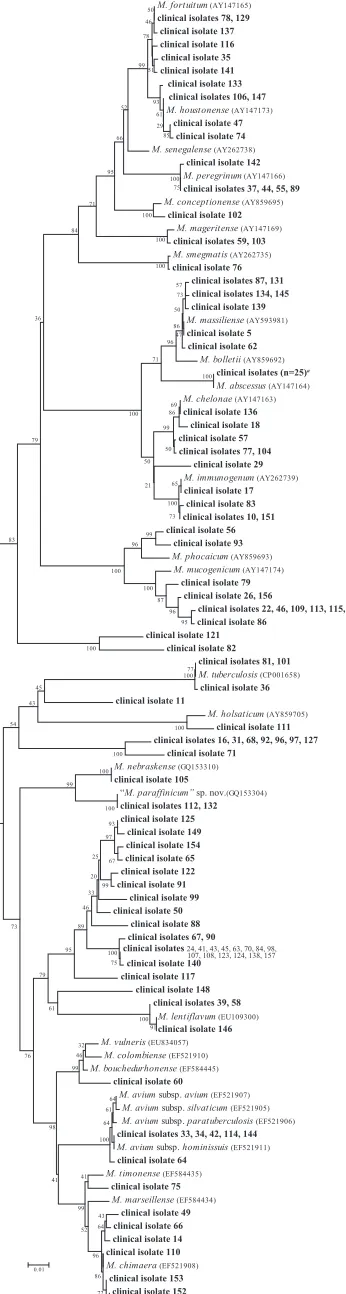
16S rRNA genes of all 139 isolates gave 28 (20%) unique DNA sequences, with 72 (16%) variable positions over the aligned area (Fig. 1). The largest cluster of isolates sharing an identical sequence belonged to theMycobacterium chelonae/M. absces-suscomplex (n⫽38), and this cluster represented the majority of clinical isolates.M. gordonaewas the second largest group and comprised three sequence variants, with 17, 10, and 1 isolate of the variants. The difference between these three
variants was 2 to 4 bp, and each had 100% identity to a GenBank reference annotated as M. gordonae. Overall, 135 (97%) isolates shared 100% identity, 2 shared 99.8% identity, and 1 isolate each shared 99.6% and 99.4% identities to a GenBank reference sequence. Using CLSI guidelines, 67 (48%) isolates were identified as single species, 68 (49%) iso-lates were identified to the complex or group level, and 4 (3%) were identified to the genus level only (Table 1).
rpoBgene identification. Alignment of 684 bp of the rpoB
gene gave 70 (50%) unique DNA sequences with 260 (38%) variable positions. Among the 38 M. chelonae/M. abscessus
complex isolates with identical 16S rRNA gene sequences, 14 differentrpoBgene sequences were obtained. The largest clus-ter of isolates sharing identicalrpoBsequences belonged toM.
abscessus(n⫽25) (Fig. 2). Twenty-eight isolates identified as
M. gordonae by 16S rRNA gene sequencing contained the
largest variability in therpoBgene sequence. Overall, 65 (47%) isolates shared 100% identity, 63 (45%) shared 98.0 to 99.9% identity, and 11 (8%) shared⬍98.0% identity to a GenBank reference sequence. Using guidelines set forth by Adekambi et al. (1, 2), 117 (84%) isolates were identified as single species, 10 (7%) isolates were identified to the complex or group level, and 12 (8%) were identified to the genus level only (Table 1).
Comparison of 16S rRNA andrpoBgenes.With integration
of therpoBgene, 50 additional organisms were identified to the species level compared with identification by use of the 16S rRNA gene alone (117 [84%] versus 67 [48%] isolates). Most notably, therpoBgene distinguished between members of the
M. chelonae/M. abscessuscomplex,M. kansasiifromM. gastri,
M. mucogeniumfrom M. phocaicum, and M. conceptionense
fromM. senegalense. Four isolates that were identified to the
genus level only by both targets probably represented unde-scribed species in theMycobacteriumgenus. For an additional 6 isolates, 16S rRNA gene results indicated species-level de-scriptions, although therpoB sequence data may suggest un-described species. Five of these isolates were related to M.
gordonae, and one was a member of the M. chelonae/M.
ab-scessuscomplex. A lack of agreement was also observed
be-tween 16S rRNA andrpoBgene identifications for organisms within the M. fortuitum group and the M. avium complex. Genetic heterogeneity was observed among 28 clinical isolates in the M. gordonae group, with 3 unique 16S rRNA gene sequences and 14 unique rpoB sequences. Compared to all other groups, this group exhibited the mostrpoBgene heter-ogeneity, and 5 isolates shared⬍98% identity to a GenBank reference sequence. Combining the separate analyses of the sequences of two gene targets, 119 (86%) isolates were iden-tified as single species and 10 (7%) isolates were ideniden-tified to the complex or group level. Seven (5%) were identified as novel species within established groups, and 3 (2%) were iden-tified to the genus level only (Table 1).
Dual-locus identification with 16S rRNA andrpoBgenes by
use of RipSeq software.The dual-locus algorithm was able to
identify closely related 16S rRNA gene reference sequences for 138 of 139 samples without manual inspection of the mixed electropherograms or database scrutiny. For one sample, the reverse sequence was not successfully matched to the 16S rRNA gene reference, and manual inspection of the electro-pherograms was required. Comparison of the percent identi-ties between the forward and reverse sequences showed an
on May 16, 2020 by guest
http://jcm.asm.org/
average percent identity variance of 0.2%; the lowest variance was 0.0%, and the highest was 0.5%, with a median of 0.2%.
The dual-locus algorithm was able to identify closely related
rpoBgene reference sequences for 134 of 139 samples without manual inspection of the electropherograms or database scru-tiny. Of the 5 failures, 4 were due to a lack of reference sequences in GenBank to place in therpoBgene database. All 4 samples were identified using 16S rRNA gene identities. The remaining failure was due to lower peaks in the electrophero-gram for therpoBgene than in that for the 16S rRNA gene. Comparison of the percent identities between the forward and reverse sequences of successful samples showed an average percent identity variance of 0.3%; the lowest variance was 0.0%, and the highest variance was 3.0%, with a median vari-ance of 0.2%. When we reviewed the isolate with the highest variance, we found that this was due to very poor technical quality in the nonmixed 3⬘ end of the forward electrophero-gram. After reanalysis, the variance for this particular isolate was reduced to 0.1%, and the maximum variance for any iso-late was 1.7%.
With the dual-locus algorithm, 110 (79%) isolates were iden-tified as single species, and 22 (16%) isolates were ideniden-tified to the complex or group level. Six (4%) were identified as novel species within established groups, and 1 (1%) was identified to the genus level only (Table 1). Comparison of the percent identities for the averaged forward and reverse sequence re-actions from the RipSeq algorithm to those for the consensus sequences of individual reactions showed average percent identity variances of 0.1% (range, 0.0 to 0.6%; median, 0.1%) and 0.3% (range, 0.0 to 1.7%; median, 0.2%) for the 16S rRNA andrpoBgenes, respectively.
DISCUSSION
[image:3.585.53.274.60.714.2]In this study, we examined identifications obtained by use of individual gene targets and then compared them to those ob-tained by a novel method for sequencing two loci in a single reaction tube. Regardless of method, identifications were more accurate when both the 16S rRNA and rpoB genes were
[image:3.585.302.543.89.187.2]FIG. 1. Neighbor-joining tree of 462-bp region of 16S rRNA genes of 139 clinical isolates and reference strains. Branch support is re-corded at nodes, as a percentage of 100 bootstrap iterations. Footnote symbolaindicates clinical isolates 1, 5, 8, 13, 18, 21, 25, 27, 28, 29, 40, 51, 52, 53, 54, 57, 61, 62, 69, 73, 77, 85, 87, 95, 100, 104, 118, 119, 128, 130, 131, 134, 135, 136, 139, 145, 150, and 155.
TABLE 1. Comparison of identification with 16S rRNA gene,rpoB, or both and identification with dual-locus module in RipSeq
Identification level
No. (%) of isolates
16S rRNA gene sequencing
rpoBgene sequencing
Combined analysis of 16S rRNA and
rpoBgenes
RipSeq dual-locus
method
Species 67 (48) 117 (84) 119 (86) 110 (79) Group or complex 68 (49) 10 (7) 10 (7) 22 (16)
Genus 4 (3) 12 (8) 3 (2) 1 (1)
Possible novel species
7 (5) 6 (4)
on May 16, 2020 by guest
http://jcm.asm.org/
screened. Although the rpoB gene target alone provided a larger number of species-level identifications than the 16S rRNA gene, the 16S rRNA gene allowed us to evaluate isolates nearingrpoB cutoff guidelines. For example, using the rpoB
gene, 12 isolates could not be identified further than the genus level, compared with only 4 isolates identified by use of the 16S rRNA gene. Results from 16S rRNA gene sequencing also improved our ability to interpret rpoB gene sequence data. Additionally, the 16S rRNA gene allowed us to classify isolates in the correct phylogenetic context in the absence of species descriptions orrpoBgene references. Based on these observa-tions, we believe that reliance on either the 16S rRNA orrpoB
gene alone is not the optimal approach for definitive identifi-cation ofMycobacteriumspecies.
In addition to demonstrating the importance of two DNA targets for species-level identification, we evaluated an alter-native to multitarget DNA analysis by using the RipSeq dual-locus module. Previously, the RipSeq algorithm was shown to resolve mixed electropherograms for a single gene (12, 13). In this study, we demonstrated that this software provides a reli-able algorithm to accurately identifyMycobacteriumspecies by discriminating two gene targets in a single sample, without a significant loss of fidelity. Performance of dual-locus sequenc-ing in a ssequenc-ingle reaction tube, as opposed to individual reaction tubes for the two gene targets, had the added advantages of limiting overall reagent costs and labor and improving turn-around times for reporting results to the species level. The editing of individual sequences and their final analyses were the most time-consuming steps, as it took about 10 min to assemble, edit, and export a consensus sequence and to per-form a BLAST search for each sequence. We calculated that these steps would involve approximately 2 to 3 days of work for 139 isolates. The automation in the RipSeq application al-lowed for the identification of the 139 isolates in roughly 4 h, including evaluation of a few suboptimal electropherograms. Even with some manual adjustment using the bulk run feature, analysis of samples was significantly faster than that by the conventional individual sequencing method.
[image:4.585.74.247.69.716.2]One of the limitations of RipSeq software is an inability to construct consensus sequences. Mixed samples require sepa-rate analyses of the forward and reverse electropherograms. Also, the base-calling step cannot differentiate which base comes from a particular template; instead, base calling results in a string of DNA letters containing a large number of IUPAC codes (e.g., Y⫽T and C) that are used directly in the subse-quent matching with the reference databases. As a conse-quence, the actual template sequences cannot be derived di-rectly from the dual-locus base-calling method. In cases of new variants or novel species, the RipSeq identity scores will indi-cate them, but confirmation and further investigations will require individual sequencing of the respective targets. Addi-tionally, for dual-locus sequences, sequence quality currently
FIG. 2. Neighbor-joining tree of 684-bp region ofrpoBgenes of 139 clinical isolates and reference strains. Branch support is recorded at nodes, as a percentage of 100 bootstrap iterations. Footnote symbola indicates clinical isolates 1, 8, 13, 21, 25, 27, 28, 40, 51, 52, 53, 54, 61, 69, 73, 85, 95, 100, 118, 119, 128, 130, 135, 150, and 155.
on May 16, 2020 by guest
http://jcm.asm.org/
cannot be assessed using traditional phred scores (8). As an alternative, we assessed the quality of sequences based on the identity scores obtained from each forward and reverse se-quence and measured the overall identity variance. When the analysis algorithm was followed, the average variances were 0.2% and 0.3% for the 16S rRNA andrpoBgenes, respectively. More variance was observed with the rpoB gene, and this observation can be explained partly by the larger proportion of nonperfect matches in the database for therpoBgene.
We selected the 16S rRNA and rpoB genes because they have been well-recognized and accepted targets for identifica-tion ofMycobacteriumspp. (1–3, 9). Prior investigations have utilized type or well-characterized strains to evaluate the per-formances of alternative targets for microorganism identifica-tion and, similarly, used well-characterized strains and/or type strains as references. We believe that the strengths of our study are that we (i) used only clinical isolates and (ii) did not limit the reference set to only type strains for identification, instead using all available reference sequences in GenBank. For the 16S rRNA gene, a type strain sequence was available for each species identified in this study. Not surprisingly, type strain sequences were much more limited for the region of therpoB
gene and were not available forM. arupense,M. kansasii, M.
gastri, M. gordonae, and M. szulgai. Although the 16S rRNA
andrpoBgenes were robust targets, we found a lack of inter-species variability in therpoBgene for both theM. fortuitum
and M. aviumcomplex isolates, particularly forM. fortuitum
andM. houstonenseorM. intracellulareandM. chimaera. Prior
studies revealed that there is a lack of consensus about the phylogeny of these complexes, and without the use of multiple DNA targets, definitive species identifications cannot be de-termined reliably (4, 17).
Identifications were more accurate when both the 16S rRNA andrpoBgenes were screened, and reliance on a single gene target was suboptimal. The RipSeq dual-locus software pro-vides an accurate, timesaving, and cost-effective alternative method for laboratories that use two different gene targets for microorganism identification. Although more isolates were identified to the group rather than species level with the dual-locus module, this observation is a reflection of known and still unresolved phylogenetic ambiguities for particular Mycobacte-riumsp., such as theM. fortuitumandM. aviumcomplexes. In this study, we illustrated the potential of same-tube sequencing of two well-characterized gene regions for the identification of mycobacteria. This tool also has the potential to couple gene regions important in antibiotic resistance, virulence, and/or identification.
REFERENCES
1.Adekambi, T., P. Colson, and M. Drancourt.2003.rpoB-based identification of nonpigmented and late-pigmenting rapidly growing mycobacteria. J. Clin. Microbiol.41:5699–5708.
2.Adekambi, T., and M. Drancourt.2004. Dissection of phylogenetic relation-ships among 19 rapidly growingMycobacteriumspecies by 16S rRNA,hsp65, sodA,recAandrpoBgene sequencing. Int. J. Syst. Evol. Microbiol.54:2095– 2105.
3.Adekambi, T., M. Drancourt, and D. Raoult.2009. TherpoBgene as a tool for clinical microbiologists. Trends Microbiol.17:37–45.
4.Ben Salah, I., T. Adekambi, D. Raoult, and M. Drancourt. 2008.rpoB sequence-based identification ofMycobacterium aviumcomplex species. Mi-crobiology154:3715–3723.
5.Cho, C. H., S. H. Han, B. S. Chin, S. H. Choi, H. S. Lee, C. O. Kim, M. S. Kim, J. Y. Choi, Y. G. Song, and J. M. Kim.2009. Diagnosis and species identification of mycobacterial infections by polymerase chain reaction-restriction fragment length polymorphism analysis of sterile body fluids. Korean J. Intern. Med.24:135–138.
6.Clinical and Laboratory Standards Institute.2008. Interpretive criteria for identification of bacteria and fungi by DNA target sequencing: guideline, CLSI document MM18-A. Clinical and Laboratory Standards Institute, Wayne, PA.
7.da Costa, A. R., M. L. Lopes, S. C. Leao, M. P. da Cruz Schneider, M. S. de Sousa, P. N. Suffys, T. C. Corvelo, and K. V. Lima.2009. Molecular identi-fication of rapidly growing mycobacteria isolates from pulmonary specimens of patients in the State of Para, Amazon Region, Brazil. Diagn. Microbiol. Infect. Dis.65:358–364.
8.Ewing, B., L. Hillier, M. C. Wendl, and P. Green.1998. Base-calling of automated sequencer traces using phred. I. Accuracy assessment. Genome Res.8:175–185.
9.Hall, L., K. A. Doerr, S. L. Wohlfiel, and G. D. Roberts.2003. Evaluation of the MicroSeq system for identification of mycobacteria by 16S ribosomal DNA sequencing and its integration into a routine clinical mycobacteriology laboratory. J. Clin. Microbiol.41:1447–1453.
10.Kim, B. J., S. H. Lee, M. A. Lyu, S. J. Kim, G. H. Bai, G. T. Chae, E. C. Kim, C. Y. Cha, and Y. H. Kook.1999. Identification of mycobacterial species by comparative sequence analysis of the RNA polymerase gene (rpoB). J. Clin. Microbiol.37:1714–1720.
11.Kim, H., S. H. Kim, T. S. Shim, M. N. Kim, G. H. Bai, Y. G. Park, S. H. Lee, G. T. Chae, C. Y. Cha, Y. H. Kook, and B. J. Kim.2005. Differentiation of Mycobacteriumspecies by analysis of the heat-shock protein 65 gene (hsp65). Int. J. Syst. Evol. Microbiol.55:1649–1656.
12.Kommedal, Ø., B. Karlsen, and Ø. Saebo.2008. Analysis of mixed sequenc-ing chromatograms and its application in direct 16S rRNA gene sequencsequenc-ing of polymicrobial samples. J. Clin. Microbiol.46:3766–3771.
13.Kommedal, Ø., K. Kvello, R. Skjastad, N. Langeland, and H. G. Wiker.2009. Direct 16S rDNA sequencing from clinical specimens with special focus on poly-bacterial samples and interpretation of mixed DNA chromatograms. J. Clin. Microbiol.47:3562–3568.
14.Leao, S. C., E. Tortoli, C. Viana-Niero, S. Y. Ueki, K. V. Lima, M. L. Lopes, J. Yubero, M. C. Menendez, and M. J. Garcia.2009. Characterization of mycobacteria from a major Brazilian outbreak suggests that revision of the taxonomic status of members of theMycobacterium chelonae-M. abscessus group is needed. J. Clin. Microbiol.47:2691–2698.
15.Macheras, E., A. L. Roux, F. Ripoll, V. Sivadon-Tardy, C. Gutierrez, J. L. Gaillard, and B. Heym.2009. Inaccuracy of single-target sequencing for discriminating species of theMycobacterium abscessusgroup. J. Clin. Micro-biol.47:2596–2600.
16.McNabb, A., D. Eisler, K. Adie, M. Amos, M. Rodrigues, G. Stephens, W. A. Black, and J. Isaac-Renton.2004. Assessment of partial sequencing of the 65-kilodalton heat shock protein gene (hsp65) for routine identification of Mycobacteriumspecies isolated from clinical sources. J. Clin. Microbiol.
42:3000–3011.
17.Schinsky, M. F., R. E. Morey, A. G. Steigerwalt, M. P. Douglas, R. W. Wilson, M. M. Floyd, W. R. Butler, M. I. Daneshvar, B. A. Brown-Elliott, R. J. Wallace, Jr., M. M. McNeil, D. J. Brenner, and J. M. Brown.2004. Taxo-nomic variation in theMycobacterium fortuitumthird biovariant complex: description ofMycobacterium boenickeisp. nov.,Mycobacterium houstonense sp. nov.,Mycobacterium neworleansensesp. nov. andMycobacterium bris-banensesp. nov. and recognition ofMycobacterium porcinumfrom human clinical isolates. Int. J. Syst. Evol. Microbiol.54:1653–1667.
18.Simmon, K. E., J. I. Pounder, J. N. Greene, F. Walsh, C. M. Anderson, S. Cohen, and C. A. Petti.2007. Identification of an emerging pathogen, My-cobacterium massiliense, byrpoBsequencing of clinical isolates collected in the United States. J. Clin. Microbiol.45:1978–1980.
19.Tamura, K., J. Dudley, M. Nei, and S. Kumar.2007. MEGA4: molecular evolutionary genetics analysis (MEGA) software version 4.0. Mol. Biol. Evol.
24:1596–1599.
20.Zelazny, A. M., J. M. Root, Y. R. Shea, R. E. Colombo, I. C. Shamputa, F. Stock, S. Conlan, S. McNulty, B. A. Brown-Elliott, R. J. Wallace, Jr., K. N. Olivier, S. M. Holland, and E. P. Sampaio.2009. Cohort study of molecular identification and typing ofMycobacterium abscessus,Mycobacterium massil-iense, andMycobacterium bolletii. J. Clin. Microbiol.47:1985–1995.