A THEORETICAL METHOD FOR ELECTRONIC
STRUCTURE CALCULATIONS ON SYSTEMS OF
BIOLOGICAL IMPORTANCE - THE GROUP FUNCTION
APPROACH
Donatella Paci
A Thesis Submitted for the Degree of PhD
at the
University of St Andrews
1994
Full metadata for this item is available in
St Andrews Research Repository
at:
http://research-repository.st-andrews.ac.uk/
Please use this identifier to cite or link to this item:
http://hdl.handle.net/10023/15439
A T H E O R E T IC A L M E T H O D
F O R E L E C T R O N IC S T R U C T U R E C A L C U L A T IO N S O N S Y S T E M S O F B IO L O G IC A L IM P O R T A N C E
T H E G R O U P F U N C T IO N A P P R O A C H
-A Thesis Presented fo r the Degree o f D o c to r o f P hilosophy
in the F a culty o f Science of the U n ive rsity o f St. Andrew s
b y D o n a te lla Paci
ProQuest Number: 10167023
All rights reserved INFORMATION TO ALL USERS
The quality of this reproduction is dependent upon the quality of the copy submitted. In the unlikely event that the author did not send a com plete manuscript and there are missing pages, these will be noted. Also, if material had to be removed,
a note will indicate the deletion.
uest
ProQuest 10167023
Published by ProQuest LLO (2017). Copyright of the Dissertation is held by the Author.
All rights reserved.
This work is protected against unauthorized copying under Title 17, United States C ode Microform Edition © ProQuest LLO.
ProQuest LLO.
789 East Eisenhower Parkway P.Q. Box 1346
D E C L A R A T IO N S
I, D o n a te lla Paci, hereby ce rtify th a t th is thesis has been composed by m yself, th a t it is a record o f m y ow n w ork, and th a t it has not been accepted in p a rtia l o r com plete fu lfilm e n t, o f any o th e r degree or professional q u a lifica tio n.
D o n a te lla Paci.
I was a d m itte d to the F a culty o f Science at the U n ive rsity o f St. Andrew s under O rdinance General No. 12 on 1st December 1988 and as a candidate fo r the Degree o f P h.D . on 1st Decem ber 1989.
D o n a te lla Paci.
I hereby c e rtify th a t the candidate has fu lfille d the conditions o f the R esolution and R egulations a p p ro p ria te to the Degree o f P h.D .
C O P Y R IG H T
In s u b m ittin g this thesis to the U n iv e rs ity o f S t.A ndrew s, I understand th a t I am g ivin g perm ission for it to be made available fo r use in accordance w ith the regulations o f the U n iv e rs ity L ib ra ry fo r the tim e b eing in force, subject to any copyright vested in the w o rk n o t being affected thereby. I also understand th a t the title and a bstract w ill be published, and th a t a copy o f the w o rk m ay be made and supplied to any bona fide lib ra ry o r research worker.
A C K N O W L E D G M E N T S
I w ould like to th a n k P rof. R. McW eeny fo r his invaluable assistance th ro u g h o u t the w o rk, and D r. C. Thom son fo r all his help d u rin g m y staying in St.Andrew s.
I w ish to acknowledge the Association fo r In te rn a tio n a l Cancer Research fo r p ro v id in g grants.
A B S T R A C T
T h eo re tica l m ethods fo r stu d yin g molecules o f biological im p o rta n ce are re viewed, b o th ab initio and sem i-em pirical. T he G roup F u nctio n A pproach is devel oped in d e ta il in its strong o rtho g on a l fo rm and corrections to the energy are added fo r ta k in g in to account n o n -o rth o g o n a lity effects, depending on the overlaps o f the group functions. A p p ro xim a tio n s are in tro d u ce d and tested so th a t this m e tho d can be applied to large molecules.
In p a rtic u la r, a system (o r a relevant fragm ent o f it ) is b u ilt up fro m localized tw o-electron groups, each one described by a two-electron group fu n c tio n (gem inal). Each group fu n ctio n is o p tim iz e d by using an SCF m ethod w ith an effective h a m ilto - n ian consisting o f the tw o-electron h a m ilto n ia n o f the group tog e the r w ith the effective p o te n tia l due to the presence o f the o ther electron groups (and to the e xternal e n vi ronm ent, eventually). T h e w avefunction fo r the whole molecule is an a ntisym m e trized p ro d u ct o f gem inals. T he energy is com puted as a sum of group co n trib u tio n s. C o r rections, depending on up to the second power o f the overlaps o f tw o groups at a tim e , are p a rtic u la rly im p o rta n t in co nfo rm a tio na l studies. The a pp ro xim a tio ns in tro d u ce d are based on the consideration th a t d ista n t groups consisting o f tw o p ositive and tw o negative charges see each o th e r cis n e u tra l e ntities and thus do n o t c o n trib u te appre cia b ly in the d e fin itio n o f the effective ham ilton ia n : the co m p u tin g e ffort is g re atly reduced in this way, the e rro r in tro d u ce d is sm all and can be estim a te d easily.
The theoretical m ethod presented in th is thesis offers a p o w e rfu l to o l fo r m a kin g q u a lita tiv e predictions o f th e changes re su ltin g from localized effects, such as tw is tin g around a bond, and it can be usefully applied to conform ational studies and geom etry o p tim iza tio n s. T he o th e r p ro pe rtie s w hich can be calculated are fo r the m ost p a rt d ire c tly related to the electron density; th is determines, fo r exam ple, the electrostatic p o te n tia l outside a m olecule and hence the position o f a tta ck by approaching ions o r p o la r species. C hem ical reactions, w hich involve breaking o r re-arrangem ent o f bonds, provide another vast fie ld o f a p p lica tio n. Such processes u sua lly involve o n ly localized regions in a m olecule and the adm ission of in tra g ro u p C l ensures th a t the stu d y o f bond breaking rem ains va lid th ro u g h o u t the whole process.
A T H E O R E T IC A L M E T H O D
F O R E L E C T R O N IC S T R U C T U R E C A L C U L A T IO N S O N S Y S T E M S O F B IO L O G IC A L IM P O R T A N C E
T H E G R O U P F U N C T IO N A P P R O A C H
-C O N T E N T S
P A R T I. T H E T H E O R E T IC A L T R E A T M E N T O F B IO M O L E C U L A R S Y S T E M S
5 L I In tro d u c tio n .
12 1.2 E m p iric a l E nergy C alculations.
15 1.3 Q u a n tu m C hem ical M ethods A p p lie d to the S tudy o f Large Molecules
o f B io log ica l Im portance.
1.3.1 A B IN IT IO M ethods.
1.3.2 S em iem pirical M ethods.
48 1.4 T h e Relevant Fragm ent and its E nviro nm e n t.
P A R T II. T H E G R O U P F U N C T IO N A P P R O A C H
64 , II. 1 T he G roup F u n ctio n A pproach: H isto rica l B ackground.
70 I I . 2 T he G ro up F u n ctio n A pproach: Present A p p lica tio n s.
75 11,3 P o ssib ility o f In c lu d in g N o n -o rth o g o n a lity Effects.
82 I I . 4 A p p ro xim a tio n s.
83 II.5 T he ’E ffective F ie ld ’.
87 I I . 6 Results and Discussion.
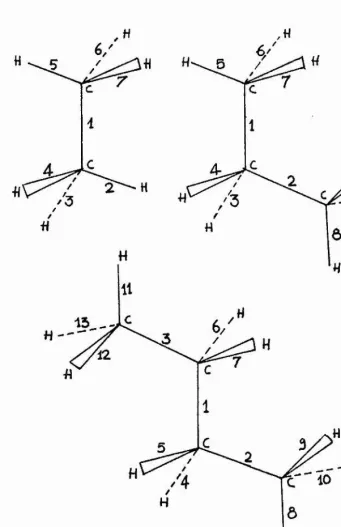
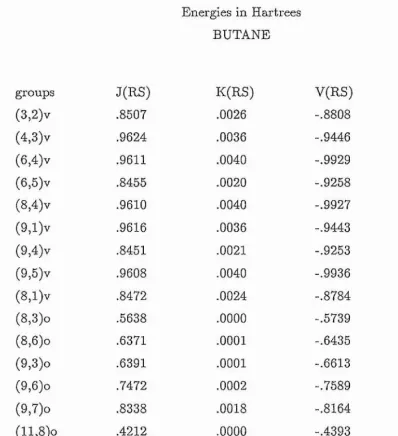
11.6.1 H ydrocarbons.
11.6.2 In tro d u c in g D ouble Bonds and Lone Pairs
11.6.3 A n A m in o A cid : Glycine.
11.6.4 Conclusions.
101 I I . 7 Perspectives.
105 A P P E N D IX A . H ydrogen-like O rb ita ls.
I l l A P P E N D IX B. O p tim iz a tio n o f A to m ic O rb ita l Basis Sets.
PART I
THE THEORETICAL TREATMENT
L I In tro d u c tio n .
T h e th e o re tica l tre a tm e n t o f b io m olecu la r systems is becom ing in cre asing ly im p o rta n t in m odern science [1,2]. Such th e o re tica l studies a llo w in fo rm a tio n to be available th a t cannot be o b ta in e d b y e xpe rim en ta l m ethods. M oreover, w o rkin g h y potheses can be fo rm u la te d fo r th e e xpe rim e n ta list to test. T h e a im o f th is section is to review a num ber o f m ethods in use fo r s tu d yin g large system s, and to in tro d u ce the G ro u p F u n ctio n A p p ro a ch (developed in d etails in P a rt I I o f th is thesis) on w hich o u r a pp lica tio ns are based.
T h e s tu d y o f a chem ical system consists in lo o kin g fo r in fo rm a tio n about its s tru c tu re and its b eh a vio ur in presence o f o th e r chem ical system s (re a c tiv ity ). The m ost useful conceptual fra m e w o rk fo r the d escrip tio n o f chem ical s tru c tu re and reac tiv it y is represented b y P o te n tia l E nergy Surfaces (P E S ) [2a,3]. A P o te n tia l E nergy Surface is the m a th e m a tica l re p re sen tatio n o f the dependence o f th e to ta l energy Etot on the nuclear coordinates needed to define th e nuclear p osition s, assumed fixe d in space. F or a system o f M nuclei and N electrons, the to ta l energy is th e sum o f the k in e tic energies o f the nuclei AT(xn) plus th e k in e tic energies o f the electrons AT(xe) plus the p o te n tia l energy due to th e e le ctro sta tic in te ra c tio n between a ll th e particles (nuclei and electrons), U (x „ ,X e ),
E t o i = AT(Xn) 4- A'(X e) + C/(Xn,Xe) ( I . l . l )
I f we assume th a t the positions o f the nuclei are fixed in space*, the nuclear k in e tic energy is negligible and can be dropped fro m E q .( I .l.l) ; the system o f N electrons m oving in the fie ld o f M nuclei has a to ta l energy given b y
= A T ( X e ) + t / ( x „ , X e ) (1.1.2) w here Z7(xn,Xe) depends p a ra m e tric a lly on the nuclear coordinates { x ^ } . T h e value o f Etot in (1.1.2) is u ne q uivoca lly determ ined b y specifying the nuclear configuration. T h e nuclear co nfig u ra tio n can be specified in term s o f eithe r cartesian o r in te rn a l coordinates. In order to define the la tte r one has to m ention the m any experim ental evidences fo r the p o s s ib ility o f e x tra c tin g th ro u g h the stu d y o f a m o le cular system some lo ca l feature characterizing a sm aller system w h ich thus appears as a m ore or less d is to rte d co n stitu tive u n it. In th is way the concept o f bond was in tro du ce d and defined as a c o n s titu tive u n it localized between tw o neighbouring atom s. In te rn a l coordinates are given by b o n d lengths, bond angles and dihedral (o r to rsio n a l) an gles. A bon d length is the distance between tw o neighbouring atom s fo rm in g a bond, a bon d angle is the angle between tw o bonds w ith an atom in com m on and a d ih e d ra l angle is the angle between the planes form ed b y tw o pairs o f bonds having a bond in com m on. I f M nuclei are present, 3M -6 coordinates are needed in a p o lya to m ic m olecule to define the nuclear co n fig u ra tio n (3M -5 in a lin e a r m olecule). Therefore fo r a system o f M nuclei the P o te n tia l E nergy Surface has 3M -5 dim ensions (3 M -4 i f th e m o le cular system is lin e a r), since to the dimensions fo r nuclear coordinates
m ust be added the dim ension th a t measures the energy. T h e PES co nta in a large a m ount o f in fo rm a tio n a bo u t chem ical s tru ctu re and re a ctivity. Its m in im a represent (m eta )stab le nuclear co nfig u ra tion s (conform ations); its m a xim a and in fle c tio n p oints should be associated w ith unstable states and it is possible to stu d y the response to p e rtu rb a tio n (re a c tiv ity ) m o vin g away over the surface fro m a m in im u m . In fact a chem ical reaction can be th o u g h t o f as the changes a m olecule is subjected to when a p e rtu rb a tio n occurs. Such p e rtu rb a tio n can be due to the presence o f another m o lecule o r another p a rt o f the same m olecule or to an external fie ld . T h e s tru c tu re o f the m olecule in question, o rig in a lly in one o f its stable configurations, reactant (re presented by a m in im u m in a P E S ), changes continuously; it firs t assumes increasing values o f energy, u p to a m a x im u m o f energy corresponding to th e so called tra n s itio n state (represented by a saddle p o in t on the P E S ) and the n decreasing values o f energy dow n to a new stable co n fig u ra tio n (rea ctio n p ro d u ct). In p a rtic u la r, it is possible to evaluate a ro ta tio n a l b a rrie r in a m olecule. One speaks about in te rn a l ro ta tio n if some o f the d ihedral angles are changed w h ile o th e r in te rn a l coordinates rem a in unaffected. W h e n a (p a rt o f a) PES is determ ined as fu n c tio n o f certain selected d ih ed ra l angles, the energy difference betw een a neighbouring m in im u m and m a xim u m is called the b a rrie r to the corresponding ro ta tio n . T h e whole PES is u sually n o t know n; o n ly a p a rt o f it is studied. M a n y the o re tical m ethods are com m only a pp lie d to calculate the energy values. In any case, a series o f energy values are calculated correspon d in g to a p a rtic u la rly in te re stin g s tru c tu re and change; it is com m on p ra ctice to use m in im iz a tio n m ethods fo r fin d in g e q u ilib riu m structures.
T h e to ta l energy o f a m olecule is expressible in term s o f b o n d c o n trib u tio n s e^-, bon d -b on d in te ra ctio n s e*y, and so on. T h is conclusion follow s fro m the analysis o f a great num ber o f e xpe rim en ta l results and leads to a so-called e m p irica l bond system atic [4],
b o nd s bonds bonds bonds bonds bonds
E ^ o + E E E % ' ^ +
w here th e quantities 6^^,... do n ot depend on the environm ent. The very old em p iric a l system atics fo r the heat o f fo rm a tio n o f a molecule are examples o f num erical system atics. G ood results are o btained also if the czilculation is re stricte d to tw o-body in te ra ctio n s €,j between adjacent and vic in a l bonds. A n a ly tic a l system atics are also possible. They co n stitu te the basis fo r em pirical energy calculations which w ill be discussed later. We w ill see th a t o th e r m olecular properties are expressible in term s o f b o n d contributions. A n enorm ous body o f experim ental data can be interpreted by b o n d a d d itiv ity rules and sometimes by re la tive ly sm all deviations which in tu rn can be referred to special effects. Indeed one can id e n tify characteristic bond distances, b o n d angles, force constants, stre tchin g frequencies, in d ire ct spin-spin coupling con stan ts, etc.
For a given system m any in terestin g properties can be calculated by theoretical m ethods. T hey all are related to the changes in the electronic situ a tio n of the system , som etim es not affecting the nuclear configuration.
E le ctric properties are re la ted to the charge d is trib u tio n p{r) (charge density),
p (r) = - p e ( r ) + ^ ZAS{r - r ^ ) (1.1.3) A
w here P c(r) is the electronic d is trib u tio n ; Za is the atom ic num ber, is the p osition o f nucleus A , and
is the D irac delta fu n ctio n .
In te g ra tio n o f />c(r) over the whole space obviously gives the tota l num ber o f electrons (N ) in the m olecule,
T h e in fo rm a tio n contained in Pe(r) can be ve ry useful, fo r instance in m aking predictions on charge-controlled chemical reactions. One is often interested in the influence o f d iffe re n t atom s in the molecule and thus it is useful to approxim ate the charge d is trib u tio n in term s o f p o in t charges associated w ith the atom s fro m w hich the m olecule is b u ilt up. In this m anner the concept o f atom ic charge can be introduced. The a to m ic charge is not a physical quantity. I t can have many different theoretical d efinitions, re fle ctin g the a m b ig u ity of the concept. A conceptually sim ple d efin itio n is based on th e d ivisio n o f the whole space in to dom ains, one for each atom [5]. The a tom ic charge ça is then defined as
ÇA ^ Za - [ p e { r ) d V (1.1.4)
Jv^
where Va is the dom ain containing atom A. It is n ot always easy to fin d an appropriate d ivisio n o f space and the integrals in (1.1.4) are ra th e r d iffic u lt to evaluate.
Charge d is trib u tio n s can also be described in term s o f m ultipoles. The p ote ntia l energy E o f the charge d is trib u tio n Pe(r) in an e xternal field is
E = I Pc{r)V,r,{r)dV.
B y expanding V'ext(r) in a T a ylo r series, we have
B - e v . . . , ™ +
-where Q is th e to ta l charge o f the system, and r,- is x, y or z according to i = l , 2 or 3. T h e dipole m om ent has components pi given by
Pi —
J
/?e (r)r,d UQ i k = 5 y P e (r)[n rfc
-S im ila rly, hig he r m u ltip o le m om ents can be defined. T o ta l d ip o le m om ents can also be e stim a te d as v e c to ria l sums o f tra nsferable bond co n trib u tio n s to a s u rp risin g ly good accuracy.
A ve ry in fo rm a tiv e p ic tu re o f m olecular electrostatics emerges fro m an in te rn a l tra n s fo rm o f the m olecule charge d is trib u tio n , the M o le cular E le c tro s ta tic P o te n tia l (M E P ). T h e M E P U ( r ) th a t is created in the space around a m olecule b y its electrons and nuclei is given by
where is the e le ctron ic charge d en sity and Zm and Bure th e charge and p o s itio n ve cto r o f the m -th nucleus. F ( r ) m ay be regarded as th e p o te n tia l o f the m olecule fo r in te ra c tio n w ith an electric charge located at the p o in t r. T h u s a p o sitive charge is a ttra c te d to those regions in w h ich U ( r ) is negative since th is leads to a negative sta b iliz in g in te ra c tio n energy, and it is repelled fro m regions o f p o sitive p o te n tia l in w h ich the in te ra c tio n energy is destabilizing.
T h e accuracy o f in fo rm a tio n obtained fro m a ca lcu latio n is s tric tly dependent on the d e fin itio n o f a m odel fo r the system . T h is m odel is q uite sim ple fo r sm a ll systems in the gas phase and it contains o nly those n uclei and electrons w h ich d ire c tly p a rtic ip a te in th e process. In the m o d e llin g o f a b iom olecular s tru c tu re o r process the solvent m ust be included and a d is tin c tio n m ust be m ade between tw o b ro a d classes. In the firs t, p ra c tic a lly a ll atom s o f th e system c o n trib u te to the p ro p e rty o r take p a rt in the change, and therefore an o verall tre a tm e n t is necessary; e m p irica l energy ca lculations can be applied w ith some success. In o th e r cases, changes are locaHzed to a re la tiv e ly sm a ll p a rt, the relevant fragm ent. A num ber o f q ua n tu m m echanical m ethods has been used fo r s tu d yin g the la tte r. T h e p ro to ty p e approach is u s u a lly used, where
broken bonds at the fro n tie rs o f the fragm ent are sa turated by some a p p ro p ria te ly |
I
1.2 E m p irica l Energy C a lcu la tion s.
E m p iric a l m ethods are based on the observation th a t bond p ro pe rtie s are more o r less transferable w ith in ce rta in classes o f molecules. Regions o f p o te n tia l energ>* surfaces around th e ir local m in im a can be approxim ated by a sum o f sim ple analytical expressions w ith constants th a t are fitte d to reproduce a great num ber o f experim ental q u a n titie s as closely as possible [1,2b].
In the em pirical approach, the to ta l energy o f a m olecular system is approxim ated as
Etot = Efc + E t -f* Enh (1.2.1)
w here Eb, Et,Enh stand fo r b o n d in g, torsional and non bonding in te ra ctio n s respec tiv e ly .
B o n d in g interactions are a pp ro xim a te d by a sum of stretching, bending and cross te rm s
Eb = ^ 2 ■^‘■(^o) + ^ X / - ro )^ 4- ^ X ] - ^o)^ 4 cross terms (1.2.2)
j r 9
bending degrees o f freedom . Cross terms^ if considered, in clu de stretch-stretch, bend- bend o r bend-stretch c o n trib u tio n s. In general, these sim ple quadratic potentials are used, though m ore re a lis tic M orse potentials can be applied w ith little increase in com plexity.
The general fo rm o f th e torsional potential which accounts fo r the hindered ro ta tio n s around bonds is
= (1.2.3)
where is the m -fo ld ro ta tio n a l b a rrie r and 6 is the phase. The b a rrie r height is derived fro m the rates o f isom erization of molecules w h ile m and 6 are determ ined fro m sym m etry considerations.
The non b o n d in g te rm is usually expressed in the fo llo w in g general form
= e - ' + E C .,l(r ô ) " - (1.2.4)
The firs t te rm corresponds to electrostatic interactions between atom i and jf, bearing net charges o f qi and qj; e is the dielectric constant. T h e second te rm is the Hydrogen b o n d in g p o te n tia l. V an der Waals interactions are accounted fo r in the last te rm , where r*j = C ij and rjy are adjustable param eters, differing for various types of a to m ic pairs. In m ost cases n is equal to 12.
Several approaches are proposed which differ m a in ly in the param etrization.
The atom ic n et charges Çi are calculated by qua n tu m m echanical methods or derived together w ith th e Van der Waals parameters fro m the observed geom etry o f m olecular crystals o r fitte d to quantum -m echanically calculated electrostatic poten tia ls.
w here are adjustable param eters.
For exam ple, fo r am ide-carbonyl hydrogen bonds:
e
w here are fo r H ydrogen bon d in g in te ra ctio n s, A\j^B[j fo r V an der W aals in te ra c tio n s , is th e O — H separation, 9 is the H — N — O angle and a is an a d ju sta b le param eter.
1.3 Q u a n tu m C hem ical M ethods A pp lie d to the S tudy o f Large Molecules o f B io lo g ica l Im portance.
T he proper d e scrip tio n o f the electronic structure o f extended systems by state- o f-th e -a rt m ethods re m a in a challenging task o f quantum chem istry. These large system s are always b u ilt up fro m relatively sim ple b u ild in g blocks. The existence of such b u ild in g blocks o r fragm ents is a fundam ental p rin cip le in experim ental physical ch em istry [6]. The G ro u p F u nctio n T heory provides a the o re tical fram ew ork fo r the q u a n tu m m echanical tre a tm e n t o f this situation. It w ill be extensively discussed in the second p a rt o f th is thesis.
G enerally, those atom s d ire c tly involved in the chem ical process are treated quan tu m m echanically (rele va nt fra gm en t) and the rem ainder (fo r exam ple the solvent, or p a rts o f the m olecule fa r fro m the relevant fragm ent) are tre ate d as classical systems [2b]. M ore precisely, th e system can be fo rm a lly divided in to subsystems, one o f such subsystems being th e relevant fragm ent; the accuracy and the level o f description o f each subsystem decreases w ith the distance fro m the chem ically interesting fragm ent. The to ta l energy o f the system is the sum of the energies o f the subsystems plus the ir in teractio n s.
d e scrip tio n o f the system . I f a djusta ble param eters and e m p irica l q u a n titie s are in tro d u ce d in the q u a n tu m m echanical ca lcu latio n , the so-called se m i-e m p irical m ethods are o bta in ed .
1.3.1 A B / m r / O m ethods.
1.3.1.1 T h e Schodinger E qu a tion .
T h e ele ctron ic s tru c tu re and pro pe rtie s o f any m olecule m a y be d eterm ined in p rin c ip le fro m the com plex fu n c tio n w h ich is a s o lu tio n o f S chodinger’s (tim e - in d e p e n d e n t) e quation [7]. For a system o f N electrons th is equ a tion takes the fo rm ,
Ê ^ ( X i , X 2, . . . , X a t ) = E ^ ( x i , X 2, . . . , X ; v ) (1.3.1.1)
w here x,- sym bolizes a ll the variables needed in re fe rrin g to e le ctron %,
x,‘ = (r,', y,‘, 0,’, S{),
Xi,yijZi sp a tia l variables, s,- spin variable (see A P P E N D IX A );
H is the H a m ilto n ia n o p e ra tor, associated w ith the observable energy, fo llo w in g fro m q u a n tu m m echanical p rin cip le , in a.u., *
Eh-fc(i) = - i v ^ ( 0 + V ( 0 (1.3.1.16)
— is the kin e tic energy o pe ra tor o f electron i;
V { i ) — — 7^ is the p o te n tia l energy o f electron i in the field of the nuclei n
w h ich are assumed fixed in space (B o rn-O ppenheim er app ro xim a tio n), r „ , being the m a g n itu d e o f the position vector o f electron i re la tive to nucleus n, and the charge o f nucleus n;
g { i , j ) = ^ is the electrostatic repulsion of electrons i and j , being the separation between electrons i and j ;
T h e fu n c tio n called the w avefunction, describes the state o f the N -electron system in the sense th a t can be in terprete d as being the p ro b a b ility o f fin d in g electrons 1 ,2 ,..., TV in (space-spin) volum e elements d x i,d x2, r e s p e c t i v e l y (d r — d x id x2--.dxN)i provided ^ is norm alized, th a t is to say, f = 1.
G ive n this physical m eaning o f the w avefunction it follows th a t it must be co ntin uous, single-valued and q u a d ra tica lly integrable; th is im plies th a t not every solution o f the Schrodinger equation is acceptable. Besides, P a u li’s P rin cip le states th a t the w avefunction m ust be antisym m etric.
< i4 > =
J
À p i { x iy x \ ) d r (1.3.1.2)X'. = X l
Here  is the q u a n tu m m echanical (h e rm itia n ) o pe ra tor * associated w ith the observable A ; it w orks on fun ctio ns o f X i only. We w ill p u t x[ = X i afte r operating w ith À b u t before com pleting the in te g ra tio n
p i { x i ] x [ ) = N
J
^ (x i,X 2 ,...,X A /)^ f* (x i,X 2 ,...,X iv )d x 2 ...d x /v (I.3.1.2a)is a generalized one-particle density fu n ctio n (the one-particle density m a trix ). In p a rtic u la r the diagonal elem ent p i( x i) = p i ( x i ; x i ) is the p ro b a b ility o f finding any o f the N electrons in d x i, independently o f the positions o f o th e r electrons. The ordinEir}' electron density fu n ctio n measured by X -ra y crystallographers, defining the electronic d is trib u tio n in space (pe in (1.1.3)), is obtained by in te g ra tin g p j( x j) over spin, P i( r i ) = J p i{x i)d s I. A tw o-particle density m a trix can be defined in the same way,
/3 2 (X i,X 2 ;x ',,X ;) = N ( N - 1) / ^ ( X l,X 2 , —, Xn) 9 ' ( Xj,X2, ...,XN)dX3...dXN-(1.3.1.26)
The energy E, in B orn-O ppenheim er app ro xim a tio n, is the energy o f the N elec tro n s m oving in the field provided by the nuclei. E assumes a set of values, cor responding to various electronic states. The to ta l energy o f the molecule, Etot, is o b ta in e d by adding the nuclear-nuclear repulsion energy to the electronic energy":
* A n h e rm itia n o p e ra to r À is defined fro m J <})*{À<j)j)dT = j [À(f>iY(j>jdr. It has re a l eigenvalues and orthogonal eigenfunctions, i.e. f 4>*<f>jdT = 0 if i ^ j .
E,ot = E + y ] ^ ^ (I.3 .:.3 )
Ba b being th e distance between nuclei A and B. E depends p a ra m e tric a lly on the nuclear coordinates. I t represents an effective P o te n tia l E nergy fo r th e nuclei, in the B orn-O p p en h eim e r a p p ro xim a tio n . In th is case the w avefunction fo r the w hole system (nuclei and electrons) can be w ritte n as a p ro d u ct = C ^ 2 (x n )^ (x i, X2, x / v r , P q), w here ^ represents the ele ctron ic p a rt o f the system in one o f its s ta tio n a ry states; it depends on the e q u ilib riu m nuclear co nfig u ra tion Rq. ^ J ( x „ ) is a m em ber o f a set o f nuclear w avefunctions o b ta in e d by solving the equation
one such equation fo r any electronic configuration. In o th e r w ords, Etot is used fo r d e fin in g p oints o f the P o te n tia l E nergy Surfaces fo r the given nuclear configuration, one fo r each electronic state.
1.3.1.2 T h e Independent P a rticle M odel. T he H artree-Fock Theory.
U p to now the essential principles o f qua n tu m ch em istry have been sum m arized. T h e y are the fo u n d a tio n o f th e o re tica l m ethods and a pp ro xim a tio ns used fo r descri b in g atom s and m olecules and the processes they are subjected to and co n stitu te the rem ote background o f the a pplications o f the G roup F u n ctio n approach we w ill develop in the second p a rt o f th is thesis, b u t they cannot be em ployed in calcula tio ns on systems o f great co m p le xity w ith o u t in tro d u c in g app ro xim a tio ns. In fact the S chrodinger equation is e xa ctly solvable o nly in a few cases. For the H ydrogen atom , fo r exam ple, one finds one-electron wavefunctions, called a to m ic sp in -o rb ita ls (S O ’s) (see A P P E N D IX A ).
one-electron h am ilto n ia n s H = - h(^i) and the corresponding w a ve fu n ction could be take n as a p ro d u ct o f one-electron eigenfunction, (a to m ic o r m olecular sp in o rb ita ls)
!
one fa c to r fo r each electron, I
I ' ï = i/’i(Xi)i/>2(x2)...i/'n (x n)- i I I In th is case we could easily o b ta in an exact so lu tion b y using the m e tho d o f se- j p a ra tio n o f variables and by solving a one-electron eigenvalue equation; th is is the j so-called Independent P a rticle M odel (IP M ). P roducts o f th is k in d are eigenfunctions | o f the h e rm itia n ope ra tor h and therefore the y fo rm a com plete set according to the | postulates o f q ua n tu m mechanics. Thus it is possible to describe the w avefunction ! ^ o f a m any-electron a to m o r m olecule as a lin e a r co m b ina tio n o f a ntisym m e trized {
pro du cts. I
A n a n tisym m e trize d p ro d u c t o f one-electron fun ctio ns can be constructed as a ! d e te rm in a n t b u ilt up fro m one-electron S O ’s {ipi}^ each one ’o ccup ie d ’ b y an electron, |
i.e., describing one electron, i
det
/ ^ i ( x i ) ^ i( x z ) . . . ^ i ( x ^ ) \ V '2 (x i) 1/'2(X2) . . . V'2(XN)
\V 'N (X i) ^2V(X2) . . . V ^ N (X ^ )/
(1.3.1.4a)
T h e d e te rm in a n ta l w avefunction (S later d e te rm in a n t) is often w ritte n in the sh ort hand n o ta tio n , \'ipixp2...i^N\ [S] and it is equivalent to the a n tisym m e trize d p ro d u ct
i[i/> i(x i)î/)2 (x 2 ). . . ^iv(xjv)] (1.3.1.46)
w here
 = cpP, (1.3.1.4c)
p
T h e Hartree-Fock m e th o d [9] uses these principles. T h e to ta l m any-electron w a ve fu n ctio n is w ritte n in the fo rm o f a single determ inant o f norm alized and orthog o n a l S O ’s.
1.3.1.3 The V a ria tio n M e th o d .
N o w we consider the p ro b le m o f o p tim izin g the SO ’s in the o ne-determ inant wave fu n c tio n . A fundam ental theorem states th a t the energy, calculated by m in im izin g th e fu n ctio n a l
o b ta in e d from the energy e xp e cta tio n value form ula E [^ ] = on replacing th e exact w avefunction 'I' w ith an approxim ate one containing variable param eters, p ro vid e s an upper bound to the tru e energj' (vn ria tio n a l p rin cip le ).
A system in its g round state is usually well described by a single determ inant of d o u b ly occupied o rb ita ls (closed-shell ground state); this means th a t the set o f SO’s used fo r the determ inant (occupied SO ’s) consists o f pairs o f S O ’s, the tw o o rb itals o f th e p a ir differing o n ly in the spin factor. We em ploy S later rules [8] to derive the e x p lic it fo rm of (1.3.1.4) in the case o f a norm alized one-determ inant wavefunction. T h e re s u lt is
E = ^ < 4>i\h\<i)i > ^ > (1.3.1.6)
» iJ
w h ere the sum m ations ru n over the occupied spin-orbitals and we have introduced th e ’’ antisym m etrized 2-electron in te g ra l”
< > = < 4>i<l>jWi(l>j > - < >
<
(f>i4>k\9\<l>j<l>i > =J
( f > * { x i ) 4 > l { x 2 ) ( f > j { x i ) à i { x 2 ) d x i d x :To o bta in a s ta tio n a ry value, we let -b Spk and set 6E = 0 (to first order) fo r all S(f>kj rem em bering th a t (1.3.1.6) remains valid only when the spin-orbiteds rem ain norm alized and orthogonal. W ith the change we obtain a corresponding firs t order va ria tio n o f the energy
= < 6(f)k\h\4>k > + C . C . + ^ ( ^ < Hk<f>)\\4>k<Pj > > ) + c.c.
j I
where each c.c. is the com plex conjugate o f the preceding term . The two sums in the parentheses, however, are identical, because the integrals are in va ria n t against exchange o f the indices on b o th sides of the ||, sim ultaneously, while the name o f the sum m ation in d ex (i o r j ) is im m ate ria l. Thus
6E^*‘^ = ( < S(f)k\h\<l)k > + ^ < S(j}k<l>i\\Ok4>i > ) + c.c. i
T he re su ltan t firs t order change 6E is SE =
I t is usual to in tro d u ce tw o new operators J, and A ',, defined as follows,
A ( l ) ( ^ ( X i ) = [
J g { l , 2 ) ( l ) i { X 2 ) ( l ) * { X 2 ) d X 2 ] < f > i X i )
I<i{l)(f){xi) = [
j
g { l ,2)(f)i{xi)(l)*(x2)o{x2)dx2]energy o f an electron at X i in th e fie ld due to a charge d is trib u tio n |<^,(x2)|^ (in
electrons per u n it volume). K i is the exchange operator.
I t is also convenient to in tro d u c e the to ta l coulomb and exchange operators, J = Ji and K = w here the sum m a tio n is over a ll occupied SO’s.
From these definitions o f J and K , it follows th a t the tw o-electron te rm in the energy expectation value (1.3.1.6) can be expressed in terms o f expectation values of a one-electron operator, thus
».J 3 3
= < 4> i\J - K \ ( f ) i > = < <f>i\G\(f>i >
where G = J — K.
T h e energy expression can be w ritte n as occ.spinorba
£ = E <<f:i\h + iG\<f>i>
I
F o r a closed-shell system, a fte r sp in in teg ra tio n occ.apac.orbs
E = ^ 2 < (pi\h 2 J — A )y% > = Yli 2 (^ ii + — A,-,) t
where y* is now the o rb ita l fa c to r o f the sp in -o rb ita l and
y /( r ) o = <^2/ - i( x )
y /(r)/? = (j>2iix)
ha = < <Pi\h\(pi > Jii = < y»jt7|yi > K ii = < y,-|A'|y,- >
D efining the Fock h a m ilto n ia n operator F = /i - f O , it results
= < 6y t | F | y t > + < y t|F |< ^ y t > = 0
Since this e v id e n tly gives the first order v a ria tio n o f a 1-electron energy expe ctatio n value €& = < y t | F | y t > , the Fock o p e ra to r m ay be regarded as the effective h am ilto n ia n fo r a single electron m o \in g in the fie ld o f the nuclei (contained in h) together w ith an effective " coulomb-exchange'' fie ld representing the presence o f a ll o th e r electrons.
T h e requirem ent th a t 6E^^^ be stationary stro n g ly suggests th a t the o rb ita l en ergies €k m ight be eigenv’alues o f F, b u t to ve rify th is conjecture we must take proper account o f the o rth o g o n a lity constraints,
< > = ^jk
where
^ L = I ^
^ 1 0 otherwise.
T h e new SO ’s y& - f S<pk m ust satisfy these co n d itio n , i.e. must be o rthog onal to ipk fo r p reserving norm alization and to every w ith j 0 for ensuring o rth o n o rm a lity in the new set o f orbitals,
< Sipkl'pk > + < ^k\Sipk > ~ 0
< <5y*|yj > = 0; < y ; |6y * > = 0; j k
< <^k\F\8ipk > - ^kj < y y l^ y t > = 0
w here ejk^^kj are a rb itra ry constants.
Since F is h e rm itia n ,
> = 0 V ;, A;
T h u s
^jk =
and
F ^ k - X ! = 0 VA; J
o r, in m a trix n o ta tio n ,
F ^ —# 6
w here $ = ( y i , y2, •••) and e is a square m a trix w ith elements h a vin g a rb itra ry values. A o n e -d e te rm in a n t w a ve fu n ction is in va ria n t u nder u n ita ry m ix in g o f th e o rb ita ls; new o rb ita ls # = # U m a y be in tro du ce d; and then
T hus, w ith o u t changing the m any electron w avefunction itse lf, we m ay always change the o rb ita ls so th a t a diagonal ë is obtained. D ro p p in g the bars,
In o th e r words, every o rb ita l satisfies = ey, and it is the n an eigenfunction o f an one-electron eigenvalue equation. Since F depends on $ th ro u g h the electron in te ra c tio n term s, the eigenvalue equation m ust be solved ite ra tive ly. T h is is the Self C onsistent F ie ld (S C F) m e tho d fo r solving an H artree Fock equation.
E ve ry tim e an eigenvalue equation has to be solved where the d e fin itio n o f the o p e ra to r is dependent on the fo rm o f the w avefunctions to be o p tim ize d a SCF m ethod can be em ployed. T h is is the case, fo r exam ple, o f the G roup F u n c tio n M e th o d ; fo l lo w in g fro m the d ire ct a p p lica tio n o f the v a ria tio n a l theorem to a v a ria tio n a l wave fu n c tio n defined as an a ntisym m e trized p ro d u ct o f (a n tisym m e tric) g roup wavefunc tio n s, an eigenvalue equation rem ains to be solved fo r each electron g roup, having an effective h a m ilto n ia n defined as a fu n c tio n o f the w avefunctions o f the o th e r groups and describing the effective fie ld the y generate around the electrons o f th e group in question. A great n um ber o f the o re tical procedures are in fa ct a pp lica tio ns of the SC F m ethod.
Reasonable firs t a pp ro xim a tio ns to the a to m ic o rb ita ls fo r heavier atom s are hydrogen-like o rb itals, o b ta in ed fro m the H ydrogen o rb ita ls b y s u b s titu tin g th e ir exponents w ith variable param eters. L in e a r com binations o f S later o rb ita ls o r gaus- sian o rb ita ls are often used to reproduce a to m ic o rb ita ls (see A P P E N D IX A ). T he SC F m e th o d has been extensively applied to calculations on m olecules too after the in tro d u c tio n o f the R oothaan L C A O (L in e a r C o m b in atio n o f A to m ic O rb ita ls) ap p ro x im a tio n fo r describing one-electron m olecular w avefunctions (M o le cu la r O rb ita ls, M O s) [10].
In general it is always possible to expand each y , as a lin e a r co m b ina tio n o f
y : = = XT 3
— $ € becomes, fo r a closed-shell system,
F T = eST
where
F fil/ — hfn, -f- G fif/
Sfit/ = < XmIx*' ^
hfii/ = < ^
= E > ,„ ^ a K /ji^ IA c t) - K/JC'lAy)]; (/ii/|A<r) = < ^A1^ |i/<7 >
occ
F<ta — 2 ^ TffjT^j 3
I
1.3.1.4 The E le ctro n Density. |
A to m ic and m olecular states can be described generally by using lin e ar com bi n a tio n s o f determ inants o f spin-orbitals, the Hartree-Fock w avefunction being sim ply a o ne-determ inant a pp ro xim a tio n. Since each term is defined by sta tin g a ’’ configu ra tio n ” o f occupied SO ’s, this expansion procedure is often referred to as the m ethod o f ’’ C o n fig u ra tio n In te ra ctio n ” (C l). For a state described by a 'F w hich is a linear co m b in a tio n o f S later determ inants the density fu n ctio n can be expanded in the form
p ( x i; x i) = X ^ p R S 0 /t(x i)V ’J ( x i) (1.3.1.7a) R,s
T h e one-electron energy becomes,
< X ] ^ (0 > = X / ^ > .
i R,S
T h e re q uired pRs is thus s im p ly the coefficient o f < > in the usual o rb ita l expression fo r th e one-electron energy. T h e d ensity m a trix w ith o u t reference to spin is s im ila rly
= X I PR S ^^R ^ri ) y g ( r i ) (1 .3 .1 .7 6 ) R,S
w here Pr s is a n um erical coefficient obtained as the result o f spin in te g ra tio n . I f we
express each one-electron fu n c tio n as a lin e ar co m b in a tio n o f a rb itra ry basis fun ctio ns X = ( x i) X 2, - ” jX m ), th e n y i t ( r i ) = and the e le ctron d e n sity takes the
general appearance
-P (n ) = P ( r i ; r i ) =
= P R sTm T-s R,S
F o r a closed-shell system Pr s = 26r s and PX becomes P ^x as defined in the
pre viou s section.
W h e n the basis consists o f o rb ita ls (e.g. A O ’s) w ith a w e ll-de fin ed lo ca liza tio n in space th e coefficients p e rm it a q u a n tita tiv e analysis o f the fo rm o f th e electron d is tri b u tio n . W e define n orm a lize d o rb ita ls and overlap densities (assum ing fo r convenience real o rb ita ls )
d%;(r) = % i( r ) x ;( r ) /5 ':;, overlap density, w ith S ij = < X i ( ^ ) \ X j i ^ ) > , and fin d
^ ( r ) = X Z + X Z W (L3.1.8)
* %<;
w here qi = P a, qij — 2 S i j P i j , {i j ) .
Since in te g ra tio n o f P ( r ) over all space m ust y ie ld N (th e n u m b e r o f electrons)
it follo w s th a t
N i<3
T h e to ta l charge is d ivid e d o ut in such a w ay th a t an a m o u n t qi arises fro m each o rb ita l density d{ and qij fro m each overlap den sity dij. These q u a n titie s, firs t proposed b y M cW eeny, are usually referred to as o rb ita l and overlap populations. T h e idea o f p o p u la tio n analysis has been extensively developed b y M u llik e n [11] b u t th e p o p u la tio n s assigned to the various regions o f space are n o t unique, depending e n tire ly on the choice o f th e basis o rb itals; it is therefore clear th a t the n ature o f chem ical bonds cannot be studied sim p ly fro m an in sp e ctio n o f b o n d p op u la tion s.
1.3.1.5 Valence B on d m ethod.
T h e H P m odel is n ot apt to describe the s p littin g o f a m olecule in to tw o parts when a bond is broken. T h e re su ltin g pieces, in fa c t, are open-shell system s and are therefore b a d ly described by single determ inants o f d o u b ly occupied o rb ita ls.
A b e tte r tre a tm e n t o f this phenom enon is given by the Valence B on d theory where each electron occupies an atom ic o rb ita l and each chem ical b o n d is associated w ith th e p a irin g o f the spins of two-electrons in the valence o rb ita ls o f the atom s they belong to ; fo r a p o ly a to m ic m olecule w ith N sin g ly occupied valence a to m ic o rb ita ls, y i , y 2, the o rb ita l p ro d u ct fu n ctio n y i ( x i ) y 2( x2) . . . y ^ ( x ^ ) , is com bined w ith
single o r p a ra lle l coupled electron. T he P au li p rin cip le is satisfied a p p ly in g the usual a n tisym m e trize r  = Y2p e p P defined in E q.I.3.1.4c.
W ith neglect o f overlap one m ay define
Q — < y i y 2 . . . y N | F | y i y 2 . . . y N >
and
Kij
= <yiy2---yi---yi-.-yiv|F|yiy2-.-yi---yi---yiv
> and o bta in s the energy e xp e cta tio n valueE = Q + /- C .- y - i 5 3 K i j - 5 3 K i j (I.3.1.9)
( i j ) p a i r e d ( i j ) u n c o u p l e d { i j ) p a r a l l e l
T h is fo rm u la gives the "p e rfe ct a irin g ” a pp ro xim a tio n, using one electronic con fig u ra tio n corresponding to the spin coupling scheme associated w ith a h yp o th e tica l a llo ca tio n o f chem ical bonds. W h e n a single co nfig u ra tion does n o t give an adequate p ic tu re o f the bonding, a ll the plausible stru cture s are a d m itte d w ith various p a irin g schemes and the w avefunction is represented as a lin e ar co m b ina tio n. T h e o p tim a l m ix in g coefficients are obta in ed , as usual, by solving a m a trix secular equation. A t large distance, afte r b reaking a bond, a correct w avefunction is o bta in ed , describing isolated pieces o f the m olecule.
T h e m a in d iffic u lty o f th is m e tho d consists in ta kin g in to account o rb ita l over lap, w h ich g re a tly increases the m a th e m a tica l com plexity. T h is is w h y the m ethod has been displaced w ith the developm ent o f ab in itio m ethods o f co m p u ta tio n . Se veral form s o f the th e o ry are now available, able to face th is p ro ble m and in d ic a tin g p ro m isin g developm ents.
1.3.1.6 C o rre la tio n.
due to th e a n tis y m m e try o f the w avefunction, b u t it is n o t considered fo r electrons w ith opposite spins and in tro du ce s an e rror o f ro u g h ly one per cent; u n fo rtu n a te ly , th is corresponds to energy differences com parable, fo r exam ple, w ith m o le cu la r b in d in g energies, b a rrie r heights, m olecular in te ra c tio n energies th a t the th e o ry should be able to p re d ict. T h e co rre la tio n energy is usua lly defined as th e difference between the exact energy and the best o ne-determ inant to ta l energy.
T h e greatest efforts have been em ployed by the o re tical chem ists to stu d y the co rre la tio n problem .
T h re e m a in m ethods have been applied:
(i) one can determ ine th e coefficients o f a C l expansion d ire ctly, along w ith the firs t few energy levels, b y d ire ct solution o f the secular e q u a tion , using the m ost p o w e rfu l tecniques available fo r evaluating m a trix elements and so lvin g the m a trix eigenvalue problem ;
( ii) long C l expansions can also be handled by s ta rtin g fro m a leading te rm 0Q, w h ich w o u ld be an exact eigenfunction o f a m odel h a m ilto n ia n Hq, and re g ardin g
H — H q as a p e rtu rb a tio n u nd e r whose influence the rem a in in g C l coefficients w ill as
sume non-zero values, w h ich can be estim ated b y m a n y-b od y p e rtu rb a tio n th e o ry [12], using d ia g ra m m a tic tecniques, w ith o u t a c tu a lly solving secular equations. R ayleigh- S chrodinger p e rtu rb a tio n th e o ry [7], where the energy is expanded in term s o f sum - over-states form ulae, fo rm s th e basis o f m a n y-b od y p e rtu rb a tio n th e o ry ; ca nce lla tio n o f u nphysical, n o n -lin e a r term s in the sum -over-the states form u la e is a fea tu re o f m a n y -b o d y fo rm a lism w ith in the algebraic a pp ro xim a tio n , i.e., b y using fin ite ana ly tic basis sets. T he M ^lle r-P le sset p e rtu rb a tio n th e o ry [13], is e ssentially one fo rm o f th is p e rtu rb a tio n approach and is now w id e ly used. T a kin g a sin g le -d e te rm in a n t
^ 0 as reference fu n c tio n fo r the C l expansion ^ = #o 4- Y2 th e re m a in in g
x > o
single excitations can be e lim in a te d ; fo r electrons double e xcitations are o f the great est im portance, h igher-order corrections to the w avefunction being well expressed as antisym m etrized p ro d u cts o f th e m . Such an approach leads to a C orrelated E lectron P a ir A p p ro xim a tio n , C E P A [14], o f w hich there are m any V2irietes; if higher-order corrections are neglected, i.e. the electron pairs are considered com pletely indepen dent, an Independent E le c tro n P a ir A pp ro xim a tio n , lE P A [15], results in w hich the co rre la tion energy, at least fo rm a lly , can always be expressed as a sum o f electron p a ir co ntribu tio ns. A separate S chrodinger equation is then solved fo r each electron pair, and in firs t a pp ro xim a tio n th e re su ltin g p a ir correlation energies are sim ply summed to give an estim ate o f the to ta l co rre la tion energy.
( iii) A system can be fo rm a lly d ivided in to weakly in te ra ctin g subsystems, cor re la tio n being calculated b y C l w ith in each subsystem (G ro u p Function Approach). T h is m ethod is conceptually d iffe re n t from the other illu s tra te d in (i) and (ii) be cause it stresses the im p o rta n ce o f local co n trib u tio n s to the properties of the whole m olecule. The group w avefunctions, each one describing a group o f electrons, are u sua lly b u ilt up by m a kin g an a p rio ri assum ption o f lo ca liza tion o f the electrons. For exam ple, we can suppose th a t two-electron groups are able to represent bonds. I f every bond o rb ita l is b u ilt as a lin e ar com bination o f tw o one-electron wavefunc tions, a bonding ( r i) and an a n ti-b o n d in g ( t2) o rb ita l, then the a ntisym m etric group fu n c tio n can be defined as
+ A ^ ||r2f2||)
w here N r is a n o rm a liza tio n fa cto r. The to ta l w avefunction is an antisym m etric p ro d u c t of group fun ctio ns
’F = Â [0 ^ $ g . . .0 A...]
— ^ a ) + A ^ (rg r2( a ^ — ^Of))]
^ = c ° {Â ^ [a ia i6i6i.. . r ir i.. . ] + V ' ^ ,^ Q ia i6i6^ .. 1 1
“ n r i
T T + _ }
R ^ n n s i s i
In th is case the antisym m etrizer is À' ~ Y lp ^ p E where P is any p e rm u ta tio n between the variables and c® = first te rm in is a S later dete rm ina n t
b u ilt up fro m the one electron functions { r i } , the rem aining term s are wavefunc tions o b ta in e d fro m ^ on replacing r i f \ by r2T2, i.e., by double su b stitu tio n s, o r by qua d ru ple su b s titu tio n s and so on. and the y give rise to the correlation energy.
1.3.1.7 Localized O rbitals.
M o le cu la r ca lcu latio n s would stand to gain considerable in tu itiv e appeal if they contained some d ire c t recognition o f localized bonds [4,2c, 16].
L o ca l b o n d fu n ctio n s were n a tu ra lly employed in the valence-bond the o ry of m o le cular e le ctro n ic stru ctu re [6]: each bon d was described as a tw o-electron closed- shell system w ith an antisym m etric spin fun ctio n, — ^ a ) and a sym m etric sp a tia l fu n c tio n assumed as a linear com bination o f aa, bb and ^ ( n 6 -f 6a), a and b being a to m ic w avefunctions for the two atom s o f the bond respectively.
share th e p e rm u ta tio n p ro pe rtie s o f bonds, being inconverted b y the sym m e try ope ra tio n s o f the m olecular p o in t group, and are h ig h ly localized in the bon d regions. Lennard-Jones and P ople suggested th a t, since equivalent o rb ita ls are h ig h ly lo ca li zed, an electron occupying one o f them w ould have m a xim u m C oulom b in te ra ctio n w ith th e electron sharing th a t o rb ita l . E dm iston and Ruedenberg used th is idea and devised an ite ra tiv e n u m erical m ethod fo r im p le m e n tin g th is self-energy lo ca liza tion c rite rio n also fo r cases where sym m e try equivalence could n o t be defined [18]. A steepest-ascents m e tho d has also been suggested [19]. Foster and Boys th o u g h t o f localized o rb ita ls w h ich m in in rize the sum o f separations between o rb ita l centroids o f charge [20]. T h is ty p e o f lo ca liza tio n has been called an ’’in trin s ic lo ca liza tio n ” c rite rio n since it in no w ay it depends on the L C A O basis. A n o th e r in trin s ic c rite rio n is due to von Niessen. T h e vo n Niessen or D ensity c rite rio n makes use o f the electron den sity ope ra tor m a xim izin g th e self-overlaps o f the L M O densities [21].
density m a trix [27]. P ro je c tio n m ethods give a set o f quasi localized o rb ita ls. T he p rin c ip le is as follows: a set o f ad hoc com pletely localized o rb ita ls is p ro je cte d in to the space spanned by the p rim itiv e delocalized o rb ita ls, and the re s u ltin g o rb ita ls are reorthogonalized by the L o w d in tra n sfo rm a tio n , g ivin g the fin a l set o f quasi-localized o rb ita ls [28].
T h e d ire ct ca lcu latio n o f local o rb ita ls has been discussed b y E d m in sto n and Rue denberg [18] and in m ore details b y G ilb e rt [29], A dam s [30], Peters [31], W ilh itte and W h itte n [32], D audey [33], Letch er and D u n n in g [34], A nderson [35], vo n Niessen [36], H uzinaga [37]. There are also the so called ’bond fu n c tio n s ’ w h ich are n o t centered on any o f the atom s, b u t are placed between tw o neighbouring ones to represent bonds [38]. These d ire ct lo ca liza tio n schemes, tho u gh the y are c e rta in ly ve ry im p o rta n t fro m the th e o re tica l p o in t o f view , d id n o t help too m uch in p ra c tic a l applications.
T h e concept o f localized o rb ita ls goes fa r beyond the fra m e w o rk o f the pure H F th e o ry: it can also be used in connection w ith correlated fu n ctio n s [39,33].
transform ed in to o rb ita ls w h ich are directed towards two-centre bonds o r lone pairs.
P a u lin g form ulated the firs t d e fin itio n s o f h yb rid o rb itals (HA O s) [6). His analysis o f th e bond revealed th a t the bond is stronger if the concentration o f o rb ita ls in the in te m u cle a r region is greater and he was the first who discussed the prin cip le o f m a xim u m overlapping. However, instead o f calculating overlap integrals, he defined the b o n d strengths of th e o rb ita l as the m axim um value o f the angular p a rt of the o rb ita l fu n ctio n norm alized to 4 7t over the surface o f a sphere.
Since the 1-q u a n tiza tio n does n o t hold anymore in p e rtu rb e d atom s embedded in th e in tra m o lecu la r fie ld , one can freely m ix s, p, d, f... A O ’s. Assum ing h ybrid o rth o n o rm a lity and em ploying a m a xim u m bond strength c rite rio n , equivalent te tra h ed ra l sp^ hybrids are easily constructed:
XI = § [(n s) -b ( n p i) 4- {npy) + (n p .)l X2 = |[( n s ) 4- ( % ) - {npy) - (n p ^)]
X3 = |[(n s ) - (n p x) -f (npj,) - {np,)] X4 = |[( n s ) - (n p x) - {npy) 4- (n p ^)]
P au lin g observed th a t the degree o f h ybridizatio n , when it is n o t determ ined p u re ly by sym m etry, fo r exam ple in the presence o f lone pairs, is stro n g ly dependent on th e (n s)-(np ) energy gap, being m ost effective fo r the firs t row atom s where the gaps are small.
F o r Ein h yb rid o rb ita l the s-character can be defined as
(.% )
c2(2s) 4- c2 (2 p i) 4- c2(2py) 4- c - ( 2 p j
Interestingly, P au lin g explained double and trip le bonds by tw o tetrah e dra shar in g a common edge and side, respectively [40], i.e., by tw o bent bonds, where the e lectron density is concentrated in tw o regions o f space more d ista n t fro m each o th e r th a n in a (7 — 7T description. T h e stru ctu re w ith bent bonds can be, therefore, the energetically favoured.
D uffey and co-workers used the c rite rio n o f m axim um a m p litu d e in stu d yin g a n um ber o f large molecules [44].
A p a rt from the m a xim u m a m p litu d e crite rio n , there are m any other ways o f con s tru c tin g local hybrids A O ’s. One o f them is the Localized A to m ic O rb ita l (L O A ) m e th o d designed by A ufderheide and C hung-P hillips [45]. They m axim ize the fun c tio n La fo r each atom A , where
A occ occ
-a
and denotes the conventional bond order m a trix. The basis set o rb ita ls w h ich yie ld m axim al value o f L a are local orthogonal h yb rid o rb itals. In a ca lcu latio n on H2O tw o ’ra b b it-e a r’ lone pairs result; and the lone pairs hybrids are found to have a h ig he r s-content.
In the Generalized Valence B on d (G V B ) m ethod, h yb rid iza tio n follows as a conse quence o f the fu ll va ria tio n a l ca lcu la tio n and is not introduced as an a p rio ri postulate. E ssentially the G V B w avefunction can be viewed as generalization o f a closed-shell H F fu n c tio n  ('0 iî^ iî/j2'0 2- - ) where À represents a custom ary antisym m etrizer, in w hich each p a ir of o rb itals is replaced by {^iaK'ib + ^ib'^ia)ct0 where the firs t p osition in p roducts im plies electron 1 and the second electron 2.
A large num ber o f molecules have been treated by the G V B procedure and the corresponding hybrids are available [46]. U sing this m ethod Messmer et al. have show n th a t a bent bond p ictu re describes m u ltip le bonds in C O2, C2P4, and C2H2
tures. Inclusion o f co rre la tion between pairs o f coupled electrons makes the bent bond p ictu re even m ore accurate [47]. We shall use a s im ila r m odel w ith in the G roup Func tio n form alism in the second p a rt o f the present w ork. The localized o rb ita l p icture retains its v a lid ity even in archetypal delocalized molecules like benzene, p yrid in e and the related six-m em bered rings [48]. It appears th a t arom atic s ta b ility arises more fro m the mode o f spin co up lin g th a n fro m o rb ita l delocalization.
A no th er way o f e x tra c tin g h y b rid iz a tio n param eters fro m m olecular wavefunc tio ns is by m aking use o f the firs t o rder density m a trix . L et us first consider the early p re scrip tion o f M cW eeny and D el Re [49].
S ta rtin g w ith A O ’s a fte r L o w d in ortho g on a liza tio n, the localized bond form ed b y coupled h ybrids o rb ita ls X r and x» takes the fo rm cos 6rsXr + sin^raXa, where 6rs is the ionic-character param eter. M cW eeny-D el Re m ethod im plies a u n ita ry tra n sfo rm a tio n o f in ita l pure A O ’s and subsequent SCF calculations. A m a trix P can be defined
(
:
: \1 -f cos 2^, sin 20t
sin 29r3 . . . 1 — cos 2â,
t . . . . . . . . . . . . ... 2 J
where Pit = 2 describes the lone p a ir corresponding to tw o coinciding hybrids. If Qra ~ 45® one has a p u re ly covalent bond where Prr — Psa = Pra — Par = 1. In general, neith e r the h yb rid s are com pletely localized n o r the bonds are 1 0 0% covalent. However H A O ’s can be constrained to satisfy one o r b o th o f these conditions leading to an increase in to ta l energy o f a molecule. In th is way effects o f io n ic ity and delocalization can be e xtim a te d .
h y b rid o rb ita ls [50]. S im ilarly, if one diagonalize extended su b m a trix associated w ith b on d ed atom s, one obtains eigenfunctions describing bond o rb itals.
Inste a d o f diagonalizing the density m a trix P , a stra ig h tfo rw a rd calculation of h y b rid s- and p-character is possible [51]. T he idea was based on an early observation o f W ib e rg th a t the square o f the bond order in CH bonds is related to th e ir s-character. T h e b o n d charge can be expressed as:
T h e b o n d in g charge o f all fo u r 2s and 2p A O ’s o f the atom A ta kin g p art in the A — B bond is D efining Wg(^AB) ^ the p a rt o f the 2sa o rb ita l involved in the A — B covalent bond W^(^ab) = P2sa„^2sau^ the s-character o f the x a b h yb rid is given by:
{ l / 2 ) W ^ ^
T h e firs t general m ethod fo r co nstru ctin g h yb rid o rb itals used the m axim um o verlap co n d itio n . M a xim u m overlap m olecular o rb ita ls (M O M O ) received a detailed d e scrip tio n a nd this m ethod is easily im plem ented even in largest systems.
D e l Re discussed a way o f o b ta in in g h y b rid o rb ita ls A O ’s w ith in the fram ework o f th e effective one-electron ham ilton ia n where the wavefunctions are expressed in a fo rm o f lin e a r com bination o f atom ic o rb ita ls, solving by a u n ita ry transform ation of the p u re A O ’s so th a t the overlap m a trix has the pseudo-diagonal 2 x 2 form as close as possible [52], the equation
H C = S C Ê
w here 5 jj = < ^i|x> > , being a lig a nd o rb ita l in a system o f the type ...rjt) ^ and X j being an o rb ita l om atom M .
o rb ita ls
X» = ai(2s) + (1 - a,)^/^(2p)<
is varied u n til the sura o f bond energies is m axim ized.
A plausible assum ption th a t the bond energies are lin e ar fun ctio ns o f the cor responding overlap in teg ra ls Ea b — ^a bSab 4- Ia b is made, where Ua b and Ia b are e m p iric a lly adjusted param eters. The hybridizatio n indices a,- are subject to o r th o n o rm a lity conditions
aiGj 4- (1 — cosOij = <5,y
fo r z, j = 1...4, w here 6ij is the angle between the a xia l sym m etric h ybrids tpi and x})j sharing the same atom . D u rin g the calculations the H A O ’s are allowed to follow the directions o f the s tra ig h t hnes passing through the d ire c tly bonded nuclei. To be q u ite precise, bond angles are determ ined by the in te rh yb rid angles o f the o p tim a l h yb rid s. O rth o g o n a lity and perfect o rb ita l follow ing o f h yb rid s are not com pletely ju s tifie d suppositions b u t the y w ork very well in most cases. T h e y considerably add to the sim p lic ity o f the m e tho d .
F in a lly, in te ra to m ic overlap integrals are forced to follo w a predeterm ined linear b o n d o ve rla p /b o n d distance correlations.
In the firs t ite ra tiv e step the in itia l bond distances are selected b y an educated guess and the independent h y b rid iz a tio n parameters a, are varied u n til the m axim um o f Eh is achieved. T h e n the n ext set o f bond distances is deduced and the h y b rid iz a tio n indices are reoptim ized. T h e w hole ite ra tive procedure is repeated u n til a consistency between the in p u t and th e o u tp u t bond distances is extablished. Hence, the IM O scheme is a constrained w eighted m axim um overlap m ethod.
1.3.2 S em iem pirical Developm ents.