CHARACTERIZATION OF DEGRADATION PROFILES OF ACETALATED DEXTRAN MICROPARTICLES AND THEIR APPLICATION FOR IMMUNE MODULATION
Naihan Chen
A dissertation submitted to the faculty at the University of North Carolina at Chapel Hill in partial fulfillment of the requirements for the degree of Doctor of Philosophy in Division of Pharmacoengineering and Molecular Pharmaceutics in the Eshelman School of Pharmacy.
Chapel Hill 2018
Approved by: Kristy M. Ainslie Michael Jay
iii ABSTRACT
Naihan Chen: Characterization of Degradation Profiles of Acetalated Dextran Microparticles and their Application for Immune Modulation
(Under the direction of Kristy M. Ainslie)
Immune modulation has been widely explored as a robust tool given the essential role played by the immune system at establishing host defense. Immune responses can be up or down regulated in a specific manner to prevent and/or treat various diseases. Vaccination and
autoimmune disease treatment represent two arms of immunomodulation applications, with vaccination prophylactically boosting host immunity against a specific antigen, while
iv
v
ACKNOWLEDGEMENTS
I would like to first of all thank my advisors Drs. Ainslie and Bachelder, without whom I would not be where I am today. Their constant support, expert guidance, and precious
understanding helped me through many ups and downs of my PhD career. My committee members Drs. Jay, Hingtgen, Tisch, and Matsushima have also been instrumental in inspiring and helping me through my graduate study.
Moreover, I am appreciative of all past and present members of the Ainslie Laboratory. In particular, I would like to thank Dr. Monica Johnson for her contribution to the intracellular microparticle trafficking experiment in Chapter 3, Dr. Matthew Gallovic for helping with experiment planning and conducting in Chapter 4, and Dr. Charles Kroger from Prof. Roland Tisch’s lab for contributing significantly to work demonstrated in Chapter 6. From experiment trouble shooting to delicious birthday cakes, my lab mates witnessed my progression as a scientist and supported me through many happy and frustrating moments as dear friends.
Finally, I am particularly grateful to my parents and family for their support and
vi
TABLE OF CONTENTS
LIST OF TABLES ... xvi
LIST OF FIGURES ... xviii
LIST OF ABBREVIATIONS... xxi
CHAPTER 1. INTRODUCTION ... 1
1.1Overview of the Immune System from the Aspect of Immune Activation ... 1
1.2Use of Immune Activation in Vaccination ... 2
1.2.1Traditional Vaccines and their Complications ... 3
1.2.2Subunit Vaccines as Promising Alternatives ... 3
1.2.3Importance of Tunable Immune Regulation for Successful Vaccine Development .. 4
1.3Overview of the Immune System from the Aspect of Immune Suppression ... 5
1.4Use of Suppressive Immunotherapies for Autoimmune Diseases ... 5
vii
1.4.2Type 1 Diabetes ... 7
1.4.3Tolerance Induction for Antigen-Specific Therapy ... 7
1.5Drug Delivery for Better Immunotherapies ... 8
1.5.1Use of Polymeric Particles for Immune Activation and Suppression ... 8
1.5.2Example Formulation Methods for Polymeric Particles ... 11
1.6Objective and Research Contribution ... 12
CHAPTER 2. DEGRADATION CHARACTERIZATION OF ACETALATED DEXTRAN MICROPARTICLES ... 14
2.1Introduction ... 14
2.2Materials and Methods ... 17
2.2.1Materials ... 17
2.2.2Cell culture ... 18
2.2.3Synthesis and CAC analysis of Ace-DEX polymer ... 18
2.2.4Wettability testing using water contact angle assay ... 19
2.2.5Preparation of blank or resiquimod-loaded Ace-DEX MPs ... 19
viii
2.2.7Size determination of Ace-DEX MPs... 20
2.2.8Scanning electron microscopy (SEM) ... 20
2.2.9Endotoxin analysis ... 20
2.2.10 Degradation analysis of Ace-DEX MPs by optical density measurement ... 21
2.2.11 Degradation analysis of Ace-DEX MPs by bicinchoninic acid based assay ... 21
2.2.12 Release kinetics of resiquimod-loaded Ace-DEX MPs ... 22
2.2.13 Cell viability analysis ... 22
2.2.14 Cytotoxicity analysis ... 23
2.2.15 Nitrite production analysis ... 23
2.3Results and Discussion ... 24
2.3.1Synthesis and characterization of Ace-DEX polymer and MPs ... 24
2.3.2In vitro bioactivity of resiquimod-loaded Ace-DEX MPs ... 30
2.4Conclusion ... 33
CHAPTER 3. TUNABLE DEGRADATION OF ACETALATED DEXTRAN MICROPARTICLES ENABLES CONTROLLED VACCINE DELIVERY ... 47
ix
3.2Materials and Methods ... 52
3.2.1Materials ... 52
3.2.2Cell culture ... 52
3.2.3Synthesis of acetalated dextran and analysis of relative cyclic acetal coverage ... 52
3.2.4Microparticle fabrication via electrospray ... 53
3.2.5Encapsulation efficiency of murabutide and OVA ... 53
3.2.6Encapsulation efficiency of FITC-BSA ... 54
3.2.7Physical characterization of Ace-DEX MPs ... 54
3.2.8Degradation analysis of Ace-DEX MPs ... 54
3.2.9Release kinetics of Ace-DEX MPs ... 55
3.2.10 In vitro cell viability analysis ... 55
3.2.11 MP intracellular trafficking via confocal laser scanning microscopy analyses .. 56
3.2.12 In vitro bioactivity of murabutide-loaded Ace-DEX MPs ... 57
3.2.13 In vivo vaccination and antibody titer analysis ... 58
3.2.14 Ex vivo antigen recall cytokine analysis ... 59
x
3.3.1Characterization of electrospray Ace-DEX MPs ... 59
3.3.2In vitro bioactivity of murabutide-loaded Ace-DEX MPs ... 63
3.3.3In vivo vaccination and antibody titer analysis ... 64
3.3.4Ex vivo antigen recall analysis ... 69
3.4Conclusions ... 71
CHAPTER 4. DEVELOPMENT OF UNIVERSAL INFLUENZA VACCINE WITH CONTROLLED IMMUNE ACTIVATION ... 86
4.1Introduction ... 86
4.2Materials and Methods ... 89
4.2.1Materials ... 89
4.2.2Synthesis and characterization of acetalated dextran ... 90
4.2.3Formulation of blank, M2e-, or cGAMP-loaded microparticles ... 90
4.2.4Physical characterization of Ace-DEX MPs ... 90
4.2.5Quantification of Encapsulated M2e and cGAMP ... 91
4.2.6Release profiles of cGAMP-loaded MPs of varying CACs ... 91
xi
4.2.8Antibody titer analysis ... 93
4.2.9Ex vivo antigen recall analysis ... 93
4.2.10 Virus titration, influenza viral challenge, and mouse survival analysis ... 94
4.2.11 Evaluation for cross reactivity against different peptide sequences ... 95
4.2.12 Statistical analysis ... 95
4.3Results and Discussion ... 95
4.3.1Ace-DEX MPs enables tunable release of encapsulated cargos ... 95
4.3.2Ace-DEX MPs induce controlled humoral responses via tunable vaccine delivery 96 4.3.3Ace-DEX MPs of different CACs promote tailored cellular immunity ... 100
4.3.4Ace-DEX MPs protect mice against lethal influenza challenge ... 101
4.3.5Sera antibodies generate cross reactivity against variant M2e sequences ... 102
4.4Conclusions ... 104
CHAPTER 5. TREATMENT OF MULTIPLE SCLEROSIS USING ANTIGEN- SPECIFIC TOLERANCE VIA ACETALATED DEXTRAN MICROPARTICLES ... 125
5.1Introduction ... 125
xii
5.2.1Materials ... 130
5.2.2Animals ... 130
5.2.3Cell Culture ... 130
5.2.4Synthesis and CAC Analysis of Ace-DEX polymer ... 131
5.2.5Preparation of Empty Ace-DEX MPs... 131
5.2.6Preparation of Rapamycin Encapsulated Ace-DEX MPs ... 131
5.2.7Preparation of Protein or Peptide Encapsulated Ace-DEX MPs... 132
5.2.8Preparation of Co-encapsulated Ace-DEX MPs ... 132
5.2.9Physical Characterization of Ace-DEX MPs ... 132
5.2.10 Quantification of Encapsulated Rapamycin ... 132
5.2.11 Quantification of Encapsulated P31 ... 133
5.2.12 Quantification of Encapsulated FITC-BSA ... 133
5.2.13 Quantification of Encapsulated OVA or PLP ... 133
5.2.14 Quantification of Foxp3 Expression ... 134
5.2.15 In Vitro Cytokine Measurements... 134
xiii
5.2.17 In Vivo MP Trafficking ... 135
5.2.18 Delayed-Type Hypersensitivity (DTH) ... 135
5.2.19 Immunization and Treatment of EAE in SJL/J Mice ... 136
5.2.20 Antigen Recall Measurements of IFN-γ, IL-17A, and GM-CSF ... 136
5.3Results ... 137
5.3.1Particle Formulation and Characterization ... 137
5.3.2 Foxp3 Expression in CD4+ T Cells Co-cultured with BMDCs and MPs ... 137
5.3.3Cytokine Production by CD4+ T Cells Co-cultured with BMDCs and MPs ... 138
5.3.4T Cell Proliferation after Co-cultured with BMDCs and MPs ... 138
5.3.5Trafficking of MPs post Subcutaneous Injection ... 138
5.3.6 In Vivo Immunosuppression by Co-encapsulated Rapamycin and OVA MPs ... 139
5.3.7EAE Treatment in SJL Mice ... 140
5.4Discussion ... 141
5.5Conclusion ... 146
xiv
6.1Introduction ... 156
6.2Materials and Methods ... 160
6.2.1Materials ... 160
6.2.2Mice ... 160
6.2.3Cell Culture ... 160
6.2.4Ace-DEX Synthesis and CAC Analysis ... 161
6.2.5Preparation of Empty or Drug-loaded Ace-DEX MPs ... 161
6.2.6Ace-DEX MP Characterization ... 161
6.2.7In Vivo Live-animal Imaging ... 162
6.2.8In Vivo MP Trafficking ... 162
6.2.9Induction and Treatment of Diabetes ... 162
6.2.10 Insulitis Assessment ... 163
6.2.11 Flow Cytometry Analysis ... 163
6.2.12 Ex Vivo Cellular Analysis... 163
6.2.13 DC Phenotype Analysis ... 164
xv
6.2.15 Statistics ... 165
6.3Results and Discussion ... 165
6.3.1Ace-DEX MP characterization ... 165
6.3.2MP trafficking in vivo... 165
6.3.3T1D prevention using Rapa/P31/MPs ... 166
6.3.4Rapa/P31/MPs treatment modulates CD4+ T cells in vivo ... 169
6.3.5Rapa/P31/MP treatment modifies DC and CD4+ T cell in vitro ... 171
6.4Conclusion ... 175
CHAPTER 7. CONCLUSIONS AND FUTURE DIRECTIONS ... 186
7.1Summary ... 186
7.2Evaluation of the Acetalated Dextran Platform for Controlled Delivery ... 187
7.3Tunable Delivery of Model Vaccines... 188
7.4Tunable Delivery of Universal Influenza Vaccine ... 189
7.5Treatment of Multiple Sclerosis via Immune Tolerance ... 191
7.6Prevention of Type 1 Diabetes via Immune Tolerance ... 193
xvi LIST OF TABLES
Table 2.1 Relative cyclic acetal coverage analysis of acetalated dextran. ... 35
Table 2.2 Mean diameter of acetalated dextran microparticles. ... 37
Table 2.3 Degradation half-lives of acetalated dextran microparticles... 39
Table 2.4 Mean diameter of resiquimod-loaded acetalated dextran microparticles. ... 41
Table 2.5 Drug loading of of resiquimod-loaded acetalated dextran microparticles. ... 43
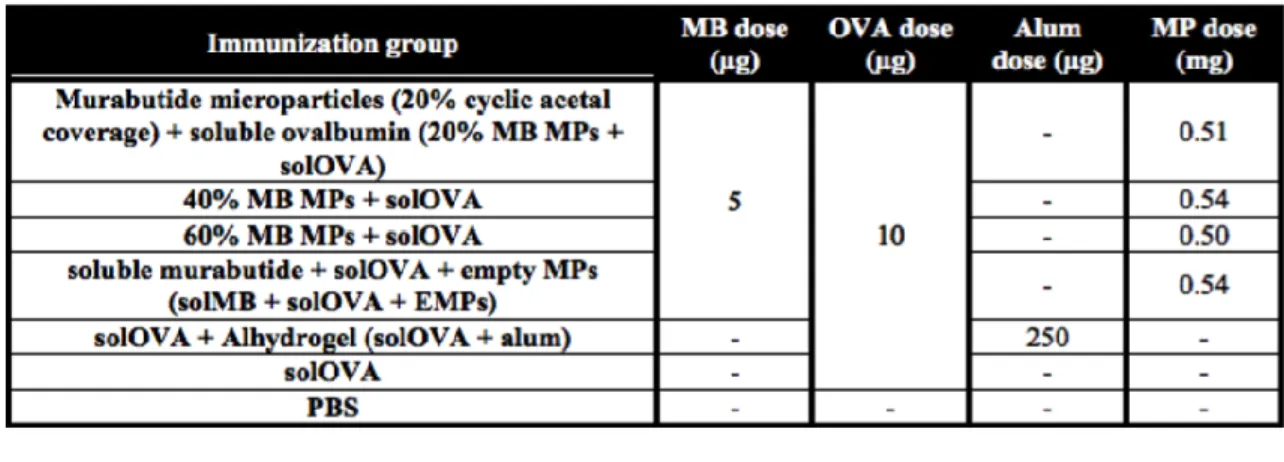
Table 3.1 Summary of immunization groups testing controlled murabutide delivery. ... 73
Table 3.2 Summary of immunization groups testing controlled ovalbumin delivery. ... 74
Table 3.3 Physical characteristics of acetalated dextran microparticles. ... 75
Table 4.1 Summary of immunization groups investigating the effect of microparticle CAC on humoral immunity. ... 105
Table 4.2 Summary of immunization groups investigating the effect of microparticle CAC on cellular immune response and protective efficacy. ... 106
Table 4.3 Statistical significance between Day 28 anti-M2e total IgG antibody titers... 113
Table 4.4 Statistical significance between Day 28 anti-M2e IgG1 antibody titers. ... 114
Table 4.5 Statistical significance between Day 28 anti-M2e IgG2a antibody titers. ... 115
xvii
Table 4.7 Statistical significance between Day 28 anti-M2e IgG2a antibody titers. ... 119
Table 4.8 Statistical significance for cross-reactivity of serum antibodies. ... 124
Table 5.1 Physical characteristics and SEM images of acetalated dextran microparticles. ... 148
xviii LIST OF FIGURES
Figure 2.1 Water contact angles of acetalated dextran polymers. ... 36
Figure 2.2 Scanning electron micrographs of acetalated dextran microparticles. ... 38
Figure 2.3 Cytotoxicity of blank acetalated dextran microparticles. ... 40
Figure 2.4 SEM images of resiquimod-loaded acetalated dextran microparticles. ... 42
Figure 2.5 Release profiles of resiquimod-loaded microparticle. ... 44
Figure 2.6 Cytotoxicity of resiquimod-loaded microparticle. ... 45
Figure 2.7 Tunable innate immunity activation using acetalated dextran microparticles. ... 46
Figure 3.1 Scanning electron micrographs of acetalated dextran microparticles. ... 76
Figure 3.2 Degradation profiles of blank acetalated dextran microparticles. ... 77
Figure 3.3 Intracellular trafficking of acetalated dextran microparticles. ... 78
Figure 3.4 Innate signaling of murabutide at different concentrations. ... 79
Figure 3.5 Humoral responses of tunable adjuvant delivery. ... 81
Figure 3.6 Humoral responses of tunable antigen delivery. ... 83
xix
Figure 3.8 Number of cytokine-expressing cells following tunable vaccine delivery. ... 85
Figure 4.1 Physical characteristics and SEM images of acetalated dextran microparticles. ... 107
Figure 4.2 Release profiles of encapsulated cGAMP. ... 108
Figure 4.3 Antibody titer heat map following tunable M2e and cGAMP vaccination. ... 110
Figure 4.4 Antibody titer levels following tunable M2e and cGAMP vaccination. ... 112
Figure 4.5 Humoral responses of M2e and cGAMP vaccination. ... 116
Figure 4.6 Cellular responses of M2e and cGAMP vaccination. ... 120
Figure 4.7 Mouse survival following influenza virus challenge. ... 122
Figure 4.8 Cross reactivity of serum antibody against different virus sequences. ... 123
Figure 5.1 Foxp3 expression post microparticle treatment. ... 149
Figure 5.2 Cytokine expression post microparticle treatment. ... 150
Figure 5.3 CD4+ T cell proliferation post microparticle treatment. ... 151
Figure 5.4 In vivo trafficking of acetalated dextran microparticles. ... 152
Figure 5.5 Inhibition of local inflammation assessed via delayed-type hypersensitivity. ... 153
Figure 5.6 Slowed disease progression by acetalated dextran microparticles. ... 154
xx
Figure 6.1 In vivo trafficking of acetalated dextran microparticles. ... 178
Figure 6.2 Delayed onset of type 1 diabetes using acetalated dextran microparticles. ... 180
Figure 6.3 Rapa/P31/MP treatment reduces T cell expansion at early time points. ... 182
Figure 6.4 Rapa/P31/MP treated mice exhibit an elevated tolerogenic T cell ratio ex vivo. ... 183
Figure 6.5 Rapa/P31/MPs inhibit dendritic cell maturation and activation. ... 184
xxi
LIST OF ABBREVIATIONS
Ac-DEX Methoxy Acetal Derivatized Acetalated Dextran Ace-DEX Ethoxy Acetal Derivatized Acetalated Dextran Ace-IN Acetalated Inulin
ACN Acetonitrile
Alum Aluminum-Based Salts APC Antigen-Presenting Cell
BCA Bicinchoninic Acid Based Assay
BDC2.5 NOD.Cg-Tg(TcraBDC2.5, TcrbBDC2.5)1Doi/DoiJ 3 NOD.129P2(C)- Tcratm1Mjo/DoiJ
BMDC Bone Marrow Derived Dendritic Cell BSA Bovine Serum Albumin
CAC Relative Cyclic Acetal Coverage CDN Cyclic Dinucleotide
CD4+ T cell Cluster of Differentiation 4 Helper T Cell CD8+ T cell Cluster of Differentiation 8 Cytotoxic T Cell CFA Complete Freund’s Adjuvant
cGAMP 3'3'-cGAMP
CNS Central Nervous System
CpG CpG ODN1826
DC Dendritic Cell
xxii DMSO Dimethylsulfoxide
DTH Delayed-Type Hypersensitivity DXM Dexamethasone
EAE Experimental Autoimmune Encephalomyelitis EE Encapsulation Efficiency
ELISA Enzyme-Linked Immunosorbent Assay ELISpot Enzyme-Linked Immunosorbent Spot Assay Electrospray Electrohydrodynamic Spraying
EMP Empty Ace-DEX MP
FDA Food and Drug Administration FFU/mL Focus Forming Units per mL
FITC-BSA Fluorescein Isothiocyanate Conjugated to Bovine Serum Albumin GA Glatiramer Acetate
GM-CSF Granulocyte-Macrophage Colony-Stimulating Factor HPLC High Performance Liquid Chromatography
IACUC Institutional Animal Care and Use Committee ICG Indocyanine Green
IFN Interferon
LDH Lactate Dehydrogenase
IGRP Islet-Specific Glucose-6-Phosphatase Catalytic Subunit-Related Protein
IL Interleukin
xxiii
LN Lymph Node
LPS Lipopolysaccharide
MB Murabutide
MDCK Mandin-Darby Canine Kidney MHC Major Histocompatibility Complex MOG Myelin Oligodendrocyte Glycoprotein
MP Microparticle
MPL 3-O-Desacyl-4’-Monophosphoryl Lipid A MS Multiple Sclerosis
MTT 3-(4,5-Dimethylthiazol-2-Yl)-2,5-Diphenyltetrazolium Bromide
MW Molecular Weight
M2e Ectodomain of Matrix Protein 2 NMR Nuclear Magnetic Resonance
Nod Nucleotide Oligomerization Domain NOD Nonobese Diabetic Mouse
NOD.scid NOD.CB17-Prkdcscid/J Nod2 Nod-Like Receptor 2 OVA Ovalbumin
PA Anthrax Antigen Protective Antigen PAA Poly(Acrylic Acid)
PAMP Pathogen-Associated Molecular Pattern PBAE Poly(ß-Amino) Esters
xxiv PD-1 Programmed Cell Death Protein-1 PGA Poly-Glycolic Acid
pI Isoelectric Point PLA Poly-L-Lactic Acid
PLGA Poly(Lactic-Co-Glycolic Acid) PLN Pancreatic Lymph Node PLP Proteolipid Protein POE Poly(Ortho Esters)
poly I:C Polyinosinic:Polycytidylic Acid PRR Pattern Recognition Receptor
PR8 Influenza Virus A/Puerto Rico/8/1934 PVA Poly(Vinyl Alcohol)
P31 BDC2.5 Mimitope
Rapa Rapamycin
Resi Resiquimod
R848 Resiquimod
SEM Scanning Electron Microscopy SJL Swiss Jim Lambert
STING Stimulator of Interferon Genes TCR T Cell Receptor
xxv TGF Transforming Growth Factor Beta Th1 T Helper 1 Cell
Th2 T Helper 2 Cell
TMB 3,3′,5,5′-Tetramethylbenzidine TNF Tumor Necrosis Factor
Treg Regulatory T Cell T1D Type 1 Diabetes
1
CHAPTER 1. INTRODUCTION
Considering the intrinsic importance of the immune system at establishing protective immunity, immune modulation acts as a promising therapy for many diseases [1-4]. Immune responses can be elicited or amplified as in vaccination, or reduced for treatment of autoimmune diseases. My thesis focuses on both aspects of immunomodulation.
1.1 Overview of the Immune System from the Aspect of Immune Activation
The immune system can be divided into innate and adaptive immunity. Innate immunity offers short-term, non-specific responses to foreign entities as a first line of defense. It is often triggered when specialized immune cells (e.g., macrophages and dendritic cells) recognize pathogen-associated molecular patterns (PAMPs) that are a diverse family of small molecular motifs usually conserved among microorganisms [5, 6]. After PAMP detection via pattern recognition receptors (PRRs), antigen-presenting cells (APCs), such as dendritic cells (DCs), are activated and present antigens (i.e., molecules capable of eliciting an immune response) to other immune cells, leading to the induction of adaptive immunity [7, 8]. Unlike innate immunity, adaptive responses are long-term and antigen-specific, which are enabled by both humoral and cell-mediated pathways.
2
co-stimulators (Signal 2), CD8+ T cells become activated and programmed to destroy the APC, which can be cancerous, pathogen-infected, or otherwise damaged, contributing to part of the cell-mediated immunity. On the other hand, CD4+ helper T cells (CD4+ T cells) are activated by binding to MHC II with concurrent Signal 2 stimulation. CD4+ T cells can first promote the proliferation and differentiation of B cells into antibody-producing plasma cells. Antibody molecules can bind and inhibit pathogen infectivity (i.e., neutralization), induce phagocytosis of antibody-coated pathogens (i.e., opsonization), and promote complement activation, in turn enhancing opsonization and enabling direct pathogen killing, which together contribute to the majority of the humoral immune response [10]. While type 2 (Th2)-biased CD4+ T cells primarily focus on antibody-based responses, Th1-biased CD4+ T cells also enhance pathogen clearance via CD8+ T cells through cytokine production, which is advantageous when fighting against intracellular pathogens. Therefore, a balanced Th1:Th2 response is essential for
successful pathogen clearance [11]. Since Th1- and Th2-biased cells induce different cytokines (IFN-γ and IL-2 for Th1-biased, and IL-4 and IL-5 for Th2-biased) and distinct subtypes of antibody production (IgG2a, IgG2b, and IgG2c for Th1-biased, and IgG1 for Th2-biased in mouse), the balance of Th1:Th2 response can be gauged by determining the level of various cytokine and antibody production [12-14].
1.2 Use of Immune Activation in Vaccination
3
1.2.1 Traditional Vaccines and their Complications
There are several types of vaccines depending on their components and manufacturing process. Live-attenuated or inactivated vaccines are two types of traditional vaccines where pathogens are weakened or inactivated, respectively. Despite strong immunogenicity of live-attenuated vaccines, potential concerns of pathogen replication urge the need for safer
alternatives. Inactivated vaccines alleviate this safety concern by inactivating pathogens through heat or chemical treatment. However, the problem, in turn, becomes weaker immunogenicity, resulting in the need for multiple boosters. Applicability to immunocompromised patients (e.g., patients on immunosuppressive therapies or with allergies) is also limited for live-attenuated or inactivated vaccines, which further calls for safer and more effective alternatives [16, 17].
1.2.2 Subunit Vaccines as Promising Alternatives
4
problem of imbalanced immune response by incorporating PAMP analogs [22-24]. MPL, derived from a lipopolysaccharide from gram-negative Salmonella enterica, binds to toll-like receptor (TLR) 4 on cell plasma [25], and CpG, a bacterial DNA analog, activates TLR 9 on phagosomal membranes [26]. Although incorporation of these Th1-skewing molecules improves the status of balanced immune response for subunit vaccines, other drawbacks remain from traditional vaccines. One disadvantage that is worth noting is the vaccine’s limited control in delivery kinetics. Antigens or adjuvants are released either rapidly (traditional antigens and those delivered with MF59, AS03, CpG 1018, or AS01) or over a prolonged period of time (antigens delivered with alum or AS04). However, the release kinetics is uncontrolled under either scenario, and little tunability can be placed over the amount of cargo released over time.
1.2.3 Importance of Tunable Immune Regulation for Successful Vaccine Development
Controlled immune activation via tunable delivery of vaccine antigen and adjuvant is important to develop safer and more effective vaccines. Many processes and signaling pathways are involved between the initial vaccine administration and the ultimate development of
protective immunity. Steps include trafficking and distribution of vaccine antigen and adjuvant, cellular uptake, antigen processing, cell-cell interaction, cell activation and proliferation, and antibody production. These processes follow precise, distinct timelines, which likely also vary for different diseases. Moreover, development of protective immunity for various diseases also relies on distinct cell populations. Activation of effector T cells, which is important in
5
understanding of immune activation kinetics coupled with fine tunability in vaccine delivery can provide optimally timed peak and duration of protective immunity to generate robust responses for diseases of interest. Conventional subunit vaccines that have little control over antigen and adjuvant delivery profiles can greatly benefit from controlled cargo delivery.
1.3 Overview of the Immune System from the Aspect of Immune Suppression
While vaccination establishes protective immunity by amplifying immune responses, down regulation of the immune system (tolerance) can sometimes be beneficial. Tolerance is defined as a lack of inflammatory response towards specific antigens. It is part of the natural immune regulation that acts towards food antigens, commensal bacteria, and apoptotic debris from healthy tissues. In the same way a vaccine induces an antigen-specific response towards the vaccinated pathogen, immune tolerance creates tolerance specific towards an antigen. Most tolerance mechanisms are mediated through dendritic cells (DCs) that can be tolerized towards distinct self-antigens [31]. Tolerogenic DCs are characterized by self-antigen presentation in the absence of co-stimulatory signals (e.g., CD80/86), which results in deletion, anergy (lacking reactivity), or differentiation of autoreactive T cells into alternative T cell types like regulatory T cells (Tregs), which can further inhibit autoreactive effector cells.
1.4 Use of Suppressive Immunotherapies for Autoimmune Diseases
Suppressive immunotherapies are beneficial for autoimmune diseases or organ transplant, where immune responses are dampened to alleviate disease symptoms or to prevent tissue
6
conditions such as systemic lupus erythematosus, or of a specific organ or site, causing diseases including multiple sclerosis (MS) and type 1 diabetes (T1D).
1.4.1 Multiple Sclerosis
MS is a chronic demyelinating inflammatory disease of the central nervous system (CNS) that affects nearly 2.5 million people worldwide [32]. T cell targeting auto-antigens in the CNS leads to immune cell invasion across the blood-brain barrier, activation of macrophages and microglia in the brain parenchyma, and ultimately demyelination, neuroaxonal dysfunction, and neurodegeneration. Current therapies, including immunomodulatory, antibody, or small molecule strategies, can successfully alleviate disease symptoms via immunosuppression. Interferon beta (IFN-β) and glatiramer acetate (GA) are the most common immunomodulatory therapies. While IFN-β suffers from low patient compliance due to flu-like symptoms [33, 34], GA (four synthetic polypeptide resembling that outcompetes endogenous myelin protein) is seen as a viable
alternative with less adverse side effects (e.g., chest tightness and injection site reactions) [35, 36]. Antibody therapies are the second treatment class for MS. They block key immune
7
1.4.2 Type 1 Diabetes
Patients receiving treatment for T1D face the same problem that the underlying causes of disease is not getting treated. T1D is characterized as a T cell mediated autoimmune disease that results in destruction of insulin producing pancreatic β-cells, leading to improper glucose
regulation. Approximately 80 people are diagnosed daily in the United States with T1D, equating to $14.9 billion in annual healthcare costs [41]. T1D is treated with everyday exogenous insulin injections or through continuous administration with a pump. Despite early effort at the
development of CD3 antibody or CTLA-4 fusion protein for T1D treatment via immune suppression, little efficacy was shown in clinic [42, 43]. Therefore, alternative therapies are in urgent need to address the underlying disease cause.
1.4.3 Tolerance Induction for Antigen-Specific Therapy
One alternative for autoimmune disease treatment is to exploit the immune system and achieve antigen-specific therapy that addresses the core of pathogenesis via immune tolerance. A majority of the previous tolerance work has focused on either introduction of antigens to mucosal surfaces or ex vivo cell manipulation. As much peripheral tolerance naturally occurs at mucosal surfaces (e.g., lung and gastrointestinal tract), administration of soluble antigens to mucosal surfaces was first explored. This approach was later proven largely ineffective due to antigen degradation or denaturation [44-46]. For ex vivo cell manipulation, T and dendritic cells are isolated from patients, cultured ex vivo with cytokines (transforming growth factor beta and interleukin-2) and Flt3 ligand, respectively, and administered back into patients to inhibit autoimmune responses [47, 48]. T cells incubated with tolerogenic DCs have also shown inhibitory abilities [49]. Other methods to generate antigen-specific tolerance are targeted
8
leukocytes, researchers have developed auto-antigen-bound cells, which eventually become apoptotic and present antigens to induce tolerance [50-52]. However, these processes require cell isolation, ex vivo manipulation, and re-administration back into patients, which are lengthy, expensive, and challenging for regulatory approval. Cell viability, engraftment efficiency, and lymph node homing after transplantation have also shown to be sub-optimal for significant clinical impact [53, 54]. Therefore, new therapies need to be explored to protect patients from long-term disease progression without sacrificing immune competency.
1.5 Drug Delivery for Better Immunotherapies
1.5.1 Use of Polymeric Particles for Immune Activation and Suppression
Delivery systems using polymeric particles could address many concerns associated with current immune modulatory therapies for vaccine or immune tolerance. Drug-loaded polymeric particles can improve cargo solubility and reduce degradation and denaturation, improve pharmacokinetic profile, achieve sustained release and dose sparing, limit systemic exposure to lower the risk of adverse side effects, enhance patient compliance, and potentially avoid cold-chain storage to improve accessibility in underprivileged areas [55-60]. Vehicles of appropriate sizes (0.5-2 μm) can also passively target APCs to boost therapy efficacy [61].
When used for vaccines, polymeric particles can enable sustained antigen release and robust antibody production, which is beneficial for subunit vaccines with poor immunogenicity. Certain delivery platforms can also facilitate antigen cross-presentation on MHC I and II [62, 63], contributing to more balanced Th1/Th2 cellular responses that are important for protection against viral and intracellular infections. Moreover, polymeric platform also allows the
9
flexible, precise control over vaccine delivery provides us the tool needed to examine immune activation kinetics, optimize peak and duration of protective immunity, and establish proper vaccination schedule for diseases of interest for safer and more effective vaccines.
When applied for tolerance-inducing therapies, polymeric platforms can reduce antigen degradation and denaturation and prolong its circulation half-life, mitigating the major drawback for in vivo antigen delivery [55, 56]. Antigen-loaded microparticles (MPs) can also be
phagocytized by APCs, carried to local lymph nodes, and modulate DC and subsequent T cell activities in vivo [61, 66, 67]. This approach bypasses multiple steps required for the lengthy and costly ex vivo expansion of tolerogenic cells, which can likely improve treatment outcome and reduce time and resources required. Overall, using polymeric MP delivery, immune tolerance can be induced for effective therapies of autoimmune diseases.
1.5.1.1Poly(lactic-co-glycolic acid) and its Drawbacks
Poly(lactic-co-glycolic acid) (PLGA) represents one of the most commonly exploited polymers for drug delivery. It is used in pre-clinical and clinical research due to its low cost, limited toxicity, biocompatibility, and biodegradability, and most importantly is used in several FDA-approved products [68]. Degradation half-lives of PLGA formulations can range from weeks to months due to bulk erosion [69, 70]. This extended degradation kinetics allows for slow release of loaded cargos, thereby improving their pharmacokinetic profiles, boosting subsequent immune modulatory capacities, achieving dose sparing, and enhancing patient compliance [71]. Given these beneficial properties, the PLGA delivery platform has been studied extensively in pre-clinical research for many disease indications, including vaccines and autoimmune
10
Despite efforts to mitigate the pH shift by incorporating antacids (e.g., basic salts) or stabilizing excipients (e.g., polyethylene glycol, PEG) or to counteract undesirable modifications of cargo proteins by PEGylation or the use of positively charged ionic salts, there are other concerns such as slow degradation and limited tunability due to bulk erosion [80-85]. While vehicle-assisted delivery prolongs cargo release profiles, acute release is hard to achieve due to minimal pH sensitivity, which is important for diseases requiring rapid intervention. There is also limited tunability within the PLGA platform. Although researchers have shown some flexibility in PLGA degradation rates from weeks to months, it requires chemical modification or a mixture of multiple polymer types [86, 87], leading to complicated formulation procedures. Therefore, alternative polymers are needed for safer and more effective delivery for immune modulatory therapies.
1.5.1.2Acetalated Dextran as an Advantageous Alternative
Acetalated dextran (Ace-DEX) is a more newly developed biodegradable polymer, represents a promising platform for drug delivery, and has been exploited for various
11
other hand are the thermodynamic product, which forms as the reaction proceeds and are more resistant to degradation. The relative ratio between cyclic and acyclic acetals is quantified as Cyclic Acetal Coverage or CAC. By changing the reaction time, and in turn, CAC, Ace-DEX degradation kinetics vary with half-lives ranging from minutes to weeks [91, 92]. This wide time range allows for both quick and sustained delivery and flexible control over cargo delivery kinetics, which is advantageous over other polymeric delivery platforms.
1.5.2 Example Formulation Methods for Polymeric Particles
1.5.2.1Emulsion
A variety of methods are available to formulate polymeric particulate vehicles. One of the most commonly used approaches is the solvent evaporation technique. Both hydrophobic and hydrophilic molecules can be encapsulated via single and double emulsion, respectively, and many antigens, adjuvants, or other immunomodulatory molecules have been encapsulated using this approach [93]. Extra attention is needed when encapsulating sensitive, hydrophilic
protein/peptide compounds as organic solvents and high-energy sources associated with particle formation can affect cargo state, and lengthy exposure to the aqueous buffer may result in low encapsulation efficiency. Despite these pitfalls, emulsion particles take up a significant number of pre-clinical and clinical formulations, with several emulsion-based formulations already FDA-approved [94].
1.5.2.2Electrohydrodynamic Spraying
12
solvent, with hydrophilic cargos (e.g., protein and peptide) in the aqueous phase. The two phases meet at the tip of a conductive needle where a Taylor cone forms due to a high electric voltage. The liquid droplet is driven to the collection plate that is charged with the opposite polarity. As the organic solution evaporates in the aerosol phase, the non-volatile polymer forms particles with cargos encapsulated and are collected on the collection plate. Limited exposure to harsh organic solvent and high-energy sources lowers the risk of cargo denaturation [100]. The lack of an aqueous outer phase enables boosted encapsulation efficiencies [101-103]. Electrospray has also shown to produce more monodispersed particle populations and have the potential for large-scale production using a multiplexed system [104-106]. As electrospray is not as well studied compared to emulsions, both methods were explored in my thesis.
1.6 Objective and Research Contribution
The immune system plays an immensely important role in maintaining a dynamic balance of the host defense system. This thesis aims to exploit innate and adaptive immune responses to induce and control immune activation and suppression for vaccination and autoimmune disease treatment, respectively. Vaccination saves millions of lives and lowers their associated cost as one of the most effective public health measures, while successful treatment of autoimmune diseases can improve the life quality of 23.5 million autoimmune patients in the U.S. Although subunit vaccines circumvent safety concerns of traditional live-attenuated antigens, poor
immunogenicity and the need for fine-tuning of delivery and immune activation profiles call for better delivery systems. Similarly, for autoimmune treatment, despite recent efforts to avoid blanket immune suppression using cell-based antigen-specific therapy, alternative approaches are needed to circumvent the lengthy and costly ex vivo process. Both aspects of immune
13
14
CHAPTER 2. DEGRADATION CHARACTERIZATION OF ACETALATED
DEXTRAN MICROPARTICLES1
2.1 Introduction
Acid-sensitive polymers are widely studied and of increasing interest for drug delivery applications [107]. Because these polymers are stable at neutral environments and degrade rapidly at lower pH levels, they can provide triggered release in biological regions of interest where there is an acidic local environment. Several areas of the body can contain lower pH regions, such as at tumor tissues [108, 109], within intracellular compartments [110, 111], and at sites of inflammation [112]. Utilizing the natural pH gradient within biological systems,
microparticles (MPs) composed of acid-labile polymers can enhance biological responses as it allows for targeted delivery of chemotherapeutic agents to the acidic tumor microenvironment [108], increased transfection efficiency of DNA vaccine antigens via the pH-gradient in the endocytic pathway [113], and localized delivery of immunosuppressants to inflammatory sites of the gastrointestinal tract [114]. Moreover, acid-sensitive polymeric carriers can also reduce the frequency of adjuvant administration (i.e., dose-sparing) [103, 115] and promote antigen cross presentation on both major histocompatibility complex (MHC) I and MHC II complexes following phagosomal escape of encapsulated antigens [90], which are both advantageous for vaccine applications.
1
15
There are currently a few acid-sensitive polymers under investigation for drug delivery, such as poly(ß-amino) esters (PBAEs), poly(ortho esters) (POEs), and polyketals [116, 117]. PBAEs have been developed primarily for DNA vaccine applications [113, 118]. Due to their acid-sensitivity, PBAEs particles dissolve rapidly after being phagocytosed by antigen presenting cells (APCs), leading to an increased transfection efficiency and higher levels of dendritic cell activation compared to poly(lactic-co-glycolic acid) (PLGA) particles. Despite this promising outcome, PBAEs are associated with potential toxicity and may cause severe side effects for long-term treatment [113, 118]. POEs, on the other hand, have good biocompatibility and degrade rapidly under acidic conditions. However, the ortho ester group hydrolyzes quickly in neutral environments and is stable only under basic conditions (pH 8.4) [119, 120]. Polyketals display reduced cytotoxicity and have high acid sensitivity as suggested by studies performed using a library of polyurethanes and polyureas containing the same dimethyl ketal moiety [121]. However, all these polymers have complex synthesis reactions, which makes them challenging to be broadly accessible. Therefore, due to the limitations of existing acid-labile polymeric systems, further work must be performed to design an easily synthesized polymer with acid-sensitivity, and minimal toxicity that allows for vaccine and drug delivery.
One alternative acid-sensitive polymer is acetalated dextran. It is synthesized from the dextran polysaccharide with the hydroxyl groups on the glucose backbone replaced by
16
PLGA and some polyanhydrides that degrade into acidic byproducts, all degradation metabolites of acetalated dextran (dextran, acetone, and an alcohol) are pH-neutral. Thus, hydrolytic
degradation of Ac-DEX or Ace-DEX should not disrupt the local pH environment or harm the encapsulated payload. This is important because acidification of the local microenvironment, as seen with PLGA hydrolysis, may result in tissue toxicity and damage to sensitive cargos [78, 122, 123].
Besides neutral degradation products, acetalated dextran is advantageous also due to its tunable degradation profiles. Depending on the length of the synthesis reaction, varying degrees of cyclic or acyclic acetal groups will form. Cyclic acetal groups, which are the thermodynamic product, form with longer reaction times and exhibit more hydrolytic stability. On the contrary, acyclic acetal groups, which are the kinetic product, form with shorter reaction times and hydrolyze faster. Therefore, the degradation rate of acetalated dextran can be tuned by varying the length of the synthesis reaction. Half-lives of Ac-DEX range from minutes to months [90], with its hydrolysis rate relying on the reaction time and dextran MW [124]. Precise control of the polymer's degradation profile allows for both burst and sustained cargo release, which makes it an ideal carrier for desired therapies. Its stability outside cold chain conditions serves as another advantage of acetalated dextran. Kanthamneni et al. showed that Ac-DEX MPs containing horseradish peroxidase remained stable when stored at -20 °C, 4 °C, 25 °C, or 45 °C for 3 months [60]. This property makes acetalated dextran easy to handle and desirable for use in developing nations.
17
been shown to protect cargos outside the cold chain [60], and can be sized more aptly for passive targeting of relevant immune cells [128]. In order to demonstrate an acid-responsive platform that has tunable degradation profiles, Ace-DEX MPs composed of polymers of different CAC and MWs were characterized for degradation kinetics. First, we analyzed the hydrophobicity of Ace-DEX polymers of varying CAC and MW. The polymers were then formulated into MPs, which were evaluated for size and morphology. The degradation properties of these MPs were investigated for their acid-sensitive degradation characteristics and the ability to tune degradation rate with CAC and MWs. Lastly, we examined the effect of tunable degradation profiles on the release kinetics and bioactivity of encapsulated cargos. Using resiquimod (an intraphagosomal toll-like receptor 7/8 agonist) as a model drug, we characterized the release kinetics of different MP sets and studied the dependence of degradation profiles on drug bioactivity using RAW 264.7 macrophages. Cyto-compatibility of blank and resiquimod-loaded MPs was also assessed. The aim of this study is to characterize the tunable degradation kinetics of the acid-sensitive Ace-DEX polymer based on CAC and MW, and demonstrate how variability in degradation impacts drug bioactivity and cyto-compatability by changing the release kinetics of encapsulated cargos.
2.2 Materials and Methods
2.2.1 Materials
All chemicals were purchased from Sigma-Aldrich (St. Louis, MO) and used unmodified unless otherwise noted. Water used in these experiments was purified using a Millipore Milli-Q Integral Water Purification System (Billerica, MA). In the presence of acetal-containing
18
Fluorescence and absorbance readings were generated using a Molecular Devices SpectraMax M2 (Sunnyvale, CA).
2.2.2 Cell culture
RAW 264.7 macrophages (ATCC, Manassas, VA) were grown and cultured according to the manufacturer's protocol. Cells were maintained at 37 °C with 5% CO2 and 100% relative humidity. Culture media consisted of Dulbecco’s Modified Eagle’s Medium (Fischer Scientific, Pittsburgh, PA), 10% fetal bovine serum (Hyclone, Pittsburgh, PA), and 1%
penicillin-streptomycin (Fischer Scientific, Pittsburgh, PA).
2.2.3 Synthesis and CAC analysis of Ace-DEX polymer
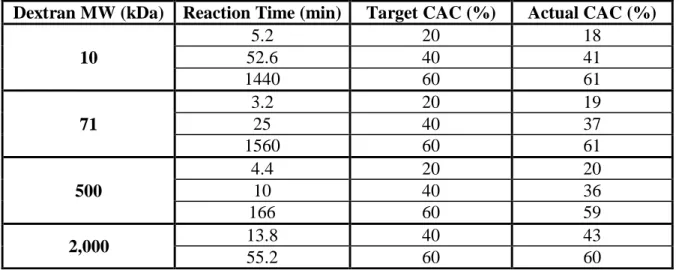
Ace-DEX polymer was synthesized from 10, 71, 500, or 2,000 kDa dextran with some modifications to the previously described procedure [89]. To synthesize 10 or 71 kDa Ace-DEX, lyophilized dextran (10 or 71 kDa) and pyridinium p-toluenesulfonate (0.0617 mmol) were dissolved in anhydrous dimethyl sulfoxide (DMSO, 10% vol/wt poly). The mixture was reacted with 2-ethoxypropene (37 mmol; Waterstone, Carmel, IN) under nitrogen gas at room
temperature. In order to obtain 20%, 40%, or 60% CAC, the 10 kDa reaction went for 5.2, 52.6, or 1440 min, while the 71 kDa reaction went for 3.2, 25, or 1560 min. To synthesize 500 or 2,000 kDa Ace-DEX, lyophilized dextran (500 or 2,000 kDa) and pyridinium p-toluenesulfonate (0.0308 mmol) were dissolved in DMSO (6.25% vol/wt poly). The mixture was reacted with 2-ethoxypropene (18.5 mmol). To obtain a 20%, 40%, or 60% CAC, the 500 kDa reaction
continued for 4.4, 10, or 166 min. The 2,000 kDa reaction went for 13.8 or 55.2 min to achieve a 40% or 60% CAC. 2,000 kDa 20% CAC polymer was reacted, but it could not form particles. At desired time points, the reactions were quenched with TEA. The quenched reactions were
19
Sorvall Legend XTR, Waltham, MA) to remove the supernatant. The resulting pellet was then frozen and lyophilized. To further purify the polymer, the product was dissolved in ethanol (200 proof) and centrifuged at 14,400 rpm for 10 min at 4 °C. The supernatant was precipitated in basic water and lyophilized for another 2 days to yield Ace-DEX polymer. CAC was determined using 1H nuclear magnetic resonance (NMR) based on a previously developed method [88, 89].
2.2.4 Wettability testing using water contact angle assay
Ace-DEX of various dextran MWs and CAC was dissolved in hexafluoroisopropanol (2% vol/wt poly). The solution was dropped onto a silicon wafer (test grade, University Wafer). The wafer was placed on the spin coater (Chemat Technology KW-4A Spin Coater, Northridge, CA) and rotated at 500 rpm for 5 seconds and at 2,500 rpm for another 60 seconds. The wafer was air dried for 30 minutes at room temperature before water (15 µL) was added using the sessile drop method. Images were obtained immediately using a ThorLabs camera (DCC1645C; Newton, NJ) and analyzed using ImageJ to determine the contact angle.
2.2.5 Preparation of blank or resiquimod-loaded Ace-DEX MPs
20
were fabricated following the same procedure, except no resiquimod was added. Both blank and resiquimod-load MPs were made using polymers of different CAC and MW.
2.2.6 Encapsulation efficiency of resiquimod
Resiquimod encapsulation efficiency was determined as previously described [103]. Briefly, samples of each resiquimod-loaded Ace-DEX MP set were prepared in triplicates and dissolved in dimethyl sulfoxide (DMSO). The solution was loaded onto a solvent-resistant 96-well plate together with a calibration curve containing free resiquimod. The amount of
resiquimod was quantified based on its autofluorescence (260-360 nm) using a SpectraMax M2 microplate reader (Molecular Devices, Sunnyvale, CA). Blank MPs with corresponding MW and CAC were used to measure the background signal.
2.2.7 Size determination of Ace-DEX MPs
The average diameter of Ace-DEX MPs was measured by Mean Diameter by Number using dynamic light scattering (DLS) following a previously described protocol [103]. Ace-DEX MPs were resuspended in basic water (33.3 µg/mL) and sized using a Brookhaven NanoBrook 90Plus Zeta Particle Size Analyzer (Holtsville, NY).
2.2.8 Scanning electron microscopy (SEM)
Ace-DEX MPs were observed and characterized by; Hitachi s-4300 Cold Field
Emission). SEM samples were prepared by mounting MPs directly onto the SEM pin stub using carbon tape. The samples were sputter-coated with a 7 nm layer of palladium/gold alloy before imaging [89, 124].
2.2.9 Endotoxin analysis
21
on the day of particle fabrication. An endotoxin assay was conducted to confirm the absence of endotoxin of all Ace-DEX MPs. To perform the assay, MPs were resuspended in nanopure water (1 mg/mL) and incubated at 4 °C overnight. They were centrifuged at 14,800 rpm for 10 min, and supernatant was used for the assay using the Pierce LAL Chromogenic Endotoxin
Quantitation Kit following the manufacturer’s directions (Thermo Scientific, Waltham, MA).
2.2.10 Degradation analysis of Ace-DEX MPs by optical density measurement
Blank Ace-DEX MPs with different CAC and MWs were resuspended in either PBS (pH=7.4) or 0.3 M sodium acetate buffer (pH 5.0) in triplicates (1.5 mg/mL). Samples were agitated on a shaker plate (150 rpm) at 37 °C. At 0, 0.5, 2, 4, 8, 24, 48, 72, 168, and 336 hours, the solution was vortexed, and its absorbance at 600 nm was read using a SpectraMax M2 microplate reader (Molecular Devices, Sunnyvale, CA). The degradation profile of MPs was determined by comparing their absorbance levels at different time points to that at 0 hours. The percent degradation of each sample is equal to the difference between its absorbance reading and that at 0 hours divided by the absorbance reading at 0 hours. The degradation half-life was defined as the time when 50% of the MPs had degraded.
2.2.11 Degradation analysis of Ace-DEX MPs by bicinchoninic acid based assay
22
Protein Assay Kit; Thermo Scientific, Waltham, MA). This assay measures the amount of degradation product dextran in the supernatant over time. The percent degradation of each sample was obtained by normalizing its reading at a certain time point to that of the respective MP set at pH 5 at the last time point when it was fully degraded.
2.2.12 Release kinetics of resiquimod-loaded Ace-DEX MPs
Resiquimod-loaded Ace-DEX MPs with different CAC and MW were resuspended in either PBS (pH=7.4) or 0.3 M sodium acetate buffer (pH 5.0) in triplicates (1.5 mg/mL).
Samples were agitated on a shaker plate (150 rpm) at 37 °C. At 0.5, 2, 4, 8, 24, 48, 72, 168, and 336 hours, the solution was vortexed, and an aliquot (170 μL) was collected. The aliquots were centrifuged at 14,800 rpm for 15 min. Supernatant (150 µL) was transferred to a 96-well polystyrene plate, combined with 30 µL ethanol, and stored at -20 °C. At the last time point (hour 336), a sample (150 µL) was removed from the homogenous suspension and treated with 30 µL ethanol to degrade any remaining MPs. This sample contained the total amount of
resiquimod (100% released). The amount of resiquimod in the supernatant was quantified based on its autofluorescence (260-360 nm) using a SpectraMax M2 microplate reader (Molecular Devices, Sunnyvale, CA). The percentage of resiquimod released was calculated relative to the fluorescence of the respective MP set after complete degradation at the last time point.
2.2.13 Cell viability analysis
23
incubation, MTT assay was performed to examine cell viability. Briefly, culture media in each well was replaced with a solution of MTT reagent in fresh media (0.6 mg/mL, 170 µL). The plate was incubated at 37 °C for around 1 hour until purple formazan crystals formed. The supernatant was replaced with isopropanol (200 µL), and the solution was pipetted up and down to dissolve the crystals. The plate was read at 560 and 670 nm using a plate reader. Background absorbance at 670 nm was subtracted from all 560 nm readings. Cell viability was determined by comparing the readings of the treated groups to those of the negative (media-only) and positive (10% v/v Tween 80) controls. Signals of positive controls were subtracted from all readings to obtain background-adjusted values. Percent viability was calculated to be the result of the adjusted-reading for MP-treated cells divided by the adjusted-reading for the negative control.
2.2.14 Cytotoxicity analysis
Cytotoxicity of various Ace-DEX MPs on RAW 264.7 macrophages was evaluated using a lactate dehydrogenase (LDH) assay (Pierce LDH Cytotoxicity Assay Kit; Thermo Scientific, Waltham, MA). Cells were seeded overnight in a 96-well plate at a density of 40,000 cells per well. Ace-DEX MPs (10, 71, 500, or 2,000 kDa with 20%, 40%, or 60% CAC) were
resuspended in culture media and added on the next day (0.0625, 0.125, 0.25, 0.5, and 1 mg/mL). After 24 or 48 hour incubation, 100 µL media was transferred to a 96-well V-bottom plate, which was then centrifuged at 4,200 rpm for 10 min at room temperature. Supernatant (50 µL) was transferred to a solvent-resistant 96-well microplate and analyzed in accordance with the manufacturer’s protocol.
2.2.15 Nitrite production analysis
24
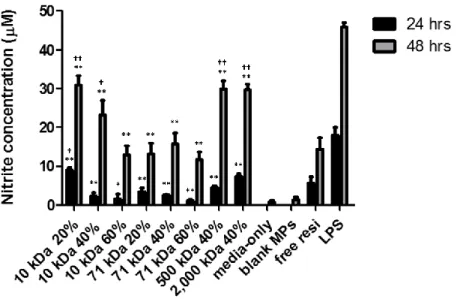
WI). Cells were seeded overnight in a 96-well plate at a density of 40,000 cells per well. Resiquimod-loaded Ace-DEX MPs (10, 71, 500, or 2,000 kDa with 20%, 40%, or 60% CAC) were resuspended in culture media and added on the next day. Particle concentrations (around 0.21 mg/mL) were adjusted based on their resiquimod encapsulation efficiency so that the final resiquimod concentration was the same across all groups (0.15 µg/mL). Media-only, free resiquimod (0.15 µg/mL), and lipopolysaccharide (LPS, 100 ng/mL) were included as controls. Blank Ace-DEX (10 kDa, 60%) MPs were included as a negative control. This MP set was selected because its correspondent resiquimod-loaded MP set required the highest particle concentration (0.23 mg/mL) in order to achieve the same resiquimod dose due to its low encapsulation efficiency. After 24 or 48 hour incubation, 100 µL media was transferred to a 96-well V-bottom plate, which was then centrifuged at 4,200 rpm for 10 min at room temperature. The supernatant (50 µL) was transferred to a solvent-resistant 96-well microplate and analyzed following the manufacturer’s instructions.
2.3 Results and Discussion
2.3.1 Synthesis and characterization of Ace-DEX polymer and MPs
25
Water contact angle measurements of Ace-DEX polymers with varying CAC and MW are shown in Figure 2.1. There was no strong correlation between the water contact angle and either polymer CAC or dextran MW. Ace-DEX polymers with varying CAC and dextran MW had comparable water contact angles, which suggested similar levels of hydrophobicity. The contact angles of Ace-DEX polymers are similar to those of PBAEs. Metter et al. showed that PBAEs synthesized from isobutylamine and 1,4-butanediol diacrylate had a contact angle of 66.7°, while PBAEs synthesized from isobutylamine and poly(ethylene glycol)-200 diacrylate or diethylene glycol diacrylate showed a contact angle of 16.2° or 35.5°, respectively [129]. Both Ace-DEX and PBAEs are less hydrophobic than PLGA, whose water contact angle is 98° [130].
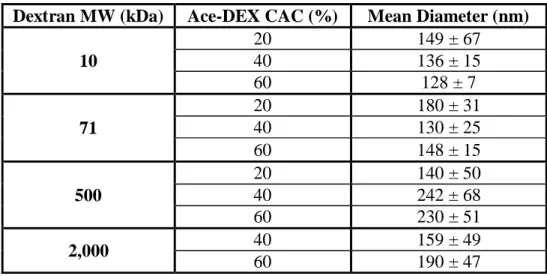
Ace-DEX polymers of varying CAC and MW were than formulated into MPs, which were characterized for size and morphology using DLS (Table 2.2) and SEM (Figure 2.2). The SEM micrographs highlight that the MPs’ spherical morphology was not affected by CAC or dextran MW.
26
the greater hydrolytic stability of the thermodynamically-favored cyclic acetals compared to kinetically-favored acyclic acetals.
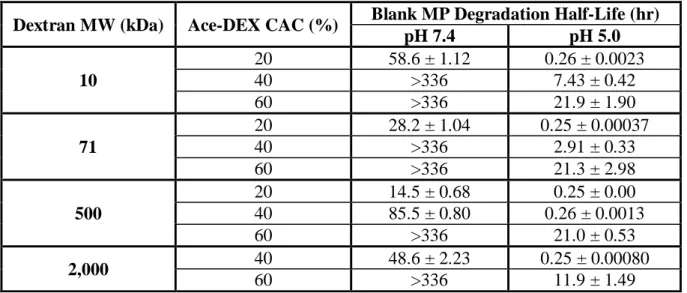
When CAC was held constant, degradation half-life of Ace-DEX MPs was inversely related to dextran MW (Table 2.3). Higher MW MPs underwent faster hydrolysis and had shorter half-lives. For example, the half-life of Ace-DEX (10 kDa, 20%) MPs was 58.6 hours, much longer than 28.2 hours of Ace-DEX (71 kDa, 20%) or 14.5 hours of Ace-DEX (500 kDa, 20%) MPs. This trend may be due to different packing densities of polymers with various MWs. Because dextran with smaller MWs has shorter chains, MPs made from Ace-DEX of lower dextran MWs were presumably more densely packed. This makes it harder for water molecules to diffuse into the polymer matrix, which results in a smaller area being exposed to water and slower MP hydrolysis. The same trend was observed for PLGA polymers. Wu et al. suggested that MPs made from PLGA polymers of higher MW degrade faster because their longer polymer chains are more likely to get hydrolyzed compared to their lower MW counterparts. The
hydrolysis process was further accelerated due to autocatalysis of the newly-generated carboxylic end groups [71].
27
irregularly linked chains to pack densely. We hypothesized that both processes might accelerate the hydrolysis process. Because polymers synthesized from higher MW dextran have longer chains, they are affected by the folding to a greater extent, which leads to faster degradation. Future experiments are needed in order to confirm this hypothesis. This correlation between Ace-DEX degradation rate and dextran MW was different than expected based on previous findings. Instead of a positive relationship between the two, Kauffman et al. previously showed that MPs composed of 10 kDa Ac-DEX degraded faster than those composed of 71 kDa Ac-DEX [124]. This could be explained by different properties of Ac-DEX and Ace-DEX. Although Ace-DEX shares many characteristics with its analog Ac-DEX, Ace-DEX has an additional carbon on its acyclic acetal groups, which introduces additional hydrophobicity and steric hindrance, possibly leading to different degradation profiles.
28
To confirm the accuracy of the optical density measurement, degradation characteristics of Ace-DEX (10 kDa 20%, 40%, and 60%) MPs were also examined using the BCA assay. Degradation profiles generated by the BCA assay qualitatively and quantitatively overlapped with those generated by optical density measurements (data not shown), which indicated consistency of the two methods. Optical density assay was used for all degradation
measurements in this study because it provided more reliable readings across different pH levels and could be used for polymers of different MWs. In the BCA assay, a copper ion reacts with only the reducing end group of the dextran molecule [133]. Because polymers of higher dextran MW have larger units and fewer terminal groups, BCA was less reactive to Ace-DEX polymers with a MW larger than 10 kDa. For these reasons, the optical density measurement was used because it can be applied for varying dextran MWs across different pH levels. By studying the degradation profiles of Ace-DEX MPs, we concluded one advantage of this system to be the broad tunability as suggested by the wide range of degradation rates at both neutral and acidic pH. Degradation profiles can be precisely controlled and easily tuned for the desired therapeutic purpose by simply changing the dextran MW and/or CAC (i.e., reaction time) of the polymer. Flexibility of this system gives it a major advantage over other acid-sensitive polymers, such as PBAEs, POEs, and polyketals, of which degradation profiles are less variable.
29
and 0.25 mg/mL) showed high levels of cellular metabolic activities (>81%) across all treatment groups (Figure 2.3A). Macrophages treated with Ace-DEX (10 and 71 kDa, 60%) MPs at 1 mg/mL showed lower levels of metabolic activity. This dampened activity was observed previously by Reddy et al. where phagocytosis of a large number of apoptotic cells by
30
2.3.2 In vitro bioactivity of resiquimod-loaded Ace-DEX MPs
After evaluating degradation profiles and cytotoxicity of blank Ace-DEX MPs of varying CAC and MW, we encapsulated resiquimod as a model drug to further characterize the system. Resiquimod is an immunomodulatory molecule used in vaccine formulations as a TLR 7/8 agonist. It has also been used for treatment of type 2 herpes simplex virus (HSV-2) and human papilloma [136]. Unlike TLR 9 expression that is only limited to plasmacytoid dendritic cells in humans, TLR 7/8 expression is similarly expressed in both mouse and humans, as well as other species [137]. Furthermore, because TLR 7/8 reside in the phagosome [103, 138-140],
resiquimod is an ideal drug to study the effects of Ace-DEX MPs’ degradation kinetics on drug bioactivity after Ace-DEX MPs are phagocytosed by macrophages [141].
Resiquimod-loaded Ace-DEX MPs were characterized first for size and morphology using DLS (Table 2.4) and SEM (Figure 2.4). Their average diameters were comparable to those of blank MPs (Table 2.2). The SEM micrographs of resiquimod-loaded MPs highlight that the MPs’ spherical morphology was not affected by CAC, dextran MW, or resiquimod
encapsulation.
Endotoxin levels and resiquimod encapsulation efficiency were then examined for these resiquimod-loaded Ace-DEX MPs. All MP sets had endotoxin levels below 0.25 EU/mL (data not shown), which is the FDA's guideline for sterile water (U.S. Food and Drug Administration, 2015). Resiquimod encapsulation efficiency and final weight loading of Ace-DEX MPs are listed in Table 2.5. MPs with similar drug loading (0.65 - 1.0 µg resiquimod per mg MP) were
31
concentrations due to close drug loadings, a consistent particle to cell ratio across groups was ensured. This was important because higher particle to cell ratios has been associated with enhanced drug bioactivity [103, 142].
Release kinetics of encapsulated resiquimod was characterized for Ace-DEX MPs of different CAC and MW. At pH 7.4, for MPs of the same dextran MW (10 and 71 kDa),
resiquimod was released faster from MPs of lower CAC (20% compared to 40%) (Figure 2.5A and 2.5B). This inverse relationship between cargo release and polymer CAC can be explained by the shorter degradation half-lives of MPs of lower CAC (Table 2.3). When polymer CAC was held constant (40%), a higher level of resiquimod was released into the supernatant from higher MW MPs (Figure 2.5C). This correlation between cargo release and polymer MW resulted from the faster hydrolysis of higher MW MPs (Table 2.3). Similar trends were observed under the acidic environment (pH 5.0), where resiquimod was released faster from MPs of lower CAC or higher MW due to their faster degradation (Figure 2.5). All MP sets released the drug faster at pH 5.0 compared to their counterpart under the pH neutral condition, suggesting acid-sensitivity of Ace-DEX polymer. The difference between resiquimod release profiles among various MP sets was smaller under acidic conditions compared to that at a pH neutral environment. This may be due to the acid-sensitive nature of Ace-DEX polymer. Because all Ace-DEX MP sets
32
sustained release profile was observed with the compound released faster at pH 7.4 [103]. This discrepancy may be due to differences in fabrication methods and varying drug weight loading.
After evaluating the tunable and acid-sensitive release kinetics of resiquimod-loaded MPs, their cytotoxicity was analyzed on RAW macrophages using a MTT and LDH assay. For the 24 hour incubation, all treatment groups showed comparable viability and proliferation levels to cells treated with blank MPs (Figure 2.6A). Except for the LPS-treated positive control, all groups showed larger than 100% metabolic activity, which was due to increased proliferation. The higher proliferation level was previously observed by Peine et al., where RAW macrophages were treated with resiquimod-loaded liposomes [143]. For the 48 hour incubation, treatment groups showed lower metabolic activity than that observed for blank MPs, which might result from limited proliferation. Little to no cytotoxicity was observed for either treatment period as measured by the LDH assay (Figure 2.6B). Thus, resiquimod-loaded Ace-DEX MPs exhibited similar levels of cyto-compatibility to blank MPs, which were both better than previously reported with PBAE MPs, causing a 60% drop in the viability of P388D1 macrophages at 0.1 mg/mL [118].
33
amount of resiquimod after phagocytosis, resulting in less robust nitrite production. This trend was less obvious for 71 kDa MPs because 71 kDa Ace-DEX degraded faster than 10 kDa. By the end of the incubation period, most of the 71 kDa MPs that had been phagocytosed by
macrophages were nearly completely degraded, minimizing the effect of varying CAC. When polymer CAC was held constant (40%), nitrite production was in general more potent for higher MW MPs. MPs fashioned from 500 or 2,000 kDa dextran induced greater nitrite production than those formulated using polymer of lower MW. This trend may be due to different release profiles of resiquimod from MPs composed of varying dextran MW. A larger amount of resiquimod may be released after phagocytosis for higher MW MPs due to their faster hydrolysis (Table 2.3 and Figure 2.5), resulting in more robust nitrite production. Therefore, macrophage activation and nitrite production can be precisely controlled by CAC and MW of Ace-DEX MPs, which makes Ace-DEX an ideal carrier for therapeutic delivery.
2.4 Conclusion
34
the tunability of this platform. Scalable production, in vivo efficacy, and in vivo effect of tunable degradation of Ace-DEX MPs will also be studied to further demonstrate the novelty and
35
Table 2.1 Relative cyclic acetal coverage analysis of acetalated dextran.
Relative cyclic acetal coverage (CAC) analysis of acetalated dextran polymer (Ace-DEX) synthesized from dextran with various molecular weights (MW) as determined by 1H NMR.
Dextran MW (kDa) Reaction Time (min) Target CAC (%) Actual CAC (%)
10
5.2 20 18
52.6 40 41
1440 60 61
71
3.2 20 19
25 40 37
1560 60 61
500
4.4 20 20
10 40 36
166 60 59
2,000 13.8 40 43
36
Figure 2.1 Water contact angles of acetalated dextran polymers.
37
Table 2.2 Mean diameter of acetalated dextran microparticles.
Number-weighted mean diameter of blank acetalated dextran (Ace-DEX) microparticles (MPs) as determined by dynamic light scattering. MPs were composed of Ace-DEX polymers of different relative cyclic acetal coverage (CAC) and synthesized from dextrans of different molecular weights (MW). Data are presented as mean ± standard deviation (n = 5).
Dextran MW (kDa) Ace-DEX CAC (%) Mean Diameter (nm)
10
20 149 ± 67
40 136 ± 15
60 128 ± 7
71
20 180 ± 31
40 130 ± 25
60 148 ± 15
500
20 140 ± 50
40 242 ± 68
60 230 ± 51
2,000 40 159 ± 49
38
Figure 2.2 Scanning electron micrographs of acetalated dextran microparticles.
Representative scanning electron micrographs of blank acetalated dextran (Ace-DEX)
39
Table 2.3 Degradation half-lives of acetalated dextran microparticles.
Degradation half-lives of blank microparticles (MPs) composed of acetalated dextran (Ace-DEX) with different dextran starting material molecular weights (MW) and relative cyclic acetal coverage (CAC) incubated at pH 7.4 and 5.0. Data are presented as mean ± standard deviation (n = 3). Half-lives of some MP sets are reported as greater than 336 hours, because less than 50% MPs were degraded at this final time point.
Dextran MW (kDa) Ace-DEX CAC (%) Blank MP Degradation Half-Life (hr)
pH 7.4 pH 5.0
10
20 58.6 ± 1.12 0.26 ± 0.0023
40 >336 7.43 ± 0.42
60 >336 21.9 ± 1.90
71
20 28.2 ± 1.04 0.25 ± 0.00037
40 >336 2.91 ± 0.33
60 >336 21.3 ± 2.98
500
20 14.5 ± 0.68 0.25 ± 0.00
40 85.5 ± 0.80 0.26 ± 0.0013
60 >336 21.0 ± 0.53
2,000 40 48.6 ± 2.23 0.25 ± 0.00080
40
Figure 2.3 Cytotoxicity of blank acetalated dextran microparticles.
(A) Cellular metabolic activity and (B) cytotoxicity of RAW macrophages incubated with blank acetalated dextran microparticles for 48 hours. Data are presented as mean ± standard deviation (n = 3). Groups are indicated by dextran molecular weight and relative cyclic acetal coverage of the acetalated dextran polymer.
41
Table 2.4 Mean diameter of resiquimod-loaded acetalated dextran microparticles.
Number-weighted mean diameter of resiquimod-loaded acetalated dextran (Ace-DEX) microparticles (MPs) as determined by dynamic light scattering. MPs were composed of Ace-DEX polymers of different relative cyclic acetal coverage (CAC) and synthesized from dextrans of different molecular weights (MW). Data are presented as mean ± standard deviation (n = 5).
Dextran MW (kDa) Ace-DEX CAC (%) Mean Diameter (nm)
10
20 161 ± 92
40 129 ± 38
60 174 ± 126
71
20 119 ± 12
40 99.6 ± 19
60 135 ± 51
500
20 N/A
40 157 ± 45
60 N/A
2,000 40 156 ± 87
42
Figure 2.4 SEM images of resiquimod-loaded acetalated dextran microparticles.
43
Table 2.5 Drug loading of of resiquimod-loaded acetalated dextran microparticles.
Encapsulation efficiency (EE) and final weight loading of resiquimod-loaded acetalated dextran microparticles (Ace-DEX MPs). MPs were composed of Ace-DEX polymers of different relative cyclic acetal coverage (CAC) and dextran molecular weights (MW). Data are presented as mean ± standard deviation (n = 3).
Dextran MW
(kDa) Ace-DEX CAC (%) Resiquimod EE (%)
Resiquimod Loading (µg drug/mg MP)
10
20 6.70 ± 0.77 0.67 ± 0.08
40 6.78 ± 0.36 0.68 ± 0.04
60 6.45 ± 1.91 0.65 ± 0.19
71
20 6.54 ± 1.03 0.65 ± 0.10
40 7.38 ± 1.24 0.74 ± 0.12
60 7.64 ± 1.06 0.76 ± 0.11
500 40 9.78 ± 1.85 0.98 ± 0.19
44
Figure 2.5 Release profiles of resiquimod-loaded microparticle.