Copyright © 2000, American Society for Microbiology. All Rights Reserved.
Genomic Variations in Echovirus 30 Persistent Isolates
Recovered from a Chronically Infected Immunodeficient Child
and Comparison with the Reference Strain
JEAN-LUC BAILLY,1* MARTINE CHAMBON,1CE´CILE HENQUELL,1JOSETTE ICART,2 ANDHE´LE`NE PEIGUE-LAFEUILLE1
UFR Me´decine, Laboratoire de Virologie, BP38 F-63002, Clermont-Ferrand, cedex 1,1and
Service de Bacte´riologie-Virologie-Hygie`ne, CHU Rangueil, F-31403 Toulouse, cedex 4,2France
Received 7 June 1999/Returned for modification 25 August 1999/Accepted 2 November 1999
Seven sequential isolates of echovirus type 30 (EV30) were recovered over 22 months from a child with severe combined immune deficiency syndrome. The nucleotide sequences of the 5ⴕ halves of the genomes (4,400 nucleotides) of the first (S1) and last (S7) isolates were determined and compared with that of the EV30 Bastianni reference strain, also determined in this study. In genome regions P1 and P2, 101 variations were identified between the two isolates. Synonymous differences far outnumbered nonsynonymous differences. Amino acid changes affected both capsid and nonstructural polypeptides (particularly 2B). The VP1 nucleotide sequences of the seven isolates were determined to analyze genome evolution during the chronic infection. In the phylogenetic tree, the seven isolates were directly related to the prototype strain in an individual mono-phyletic group, strongly suggesting that the chronic infection in the child arose from a single persistent EV30 isolate. Four lineages were observed in the persistent isolates. Isolates S2, S4, S5, and S6 were close relatives of one another, whereas isolates S1 and S3 formed individual lineages. Isolate S7, distantly related to all other isolates, formed the fourth lineage. These findings suggest the quasispecies nature of the genomes of the seven sequential EV30 isolates. Grouping of persistent isolates on the basis of replicative capacities was consistent with phylogenetic relationships. Overall, the results indicate that genetically related EV30 variants with different replicative capacities coexisted in a carrier state, probably in the gastrointestinal tract, during the infection of the child.
Enteroviruses are common human pathogens responsible for asymptomatic infections and several clinical manifestations including aseptic meningitis (7, 33), encephalitis (5, 33), and severe diseases in the newborn (6, 9, 29) and immunocompro-mised hosts (18, 27).
Enterovirus infections in humans are normally self-limited by humoral immunity. In persons with immune deficiencies (for instance, agammaglobulinemia), enteroviruses have been responsible for chronic (persistent), sometimes fatal, infections (16, 18, 27). Prolonged excretion of an enterovirus over a period of several months has been described in immunodefi-cient patients. Echovirus type 11 was the most common cause of chronic infection, but other enteroviruses, like echovirus type 30 (EV30), were also involved (10). EV30 is one of the most prevalent enteroviruses (13, 28) and is usually associated with acute aseptic meningitis (1), a common illness that occurs both as sporadic cases of infection and as seasonal outbreaks (2, 12, 15, 24, 26), which contribute to the active circulation of the serotype in the general population.
In this study, we report on the genetic analysis of seven EV30 isolates collected over a period of 22 months from stool specimens of a young girl with combined immune deficiency syndrome associated with cartilage-hair hypoplasia (34). The patient had congenital short-limbed dwarfism, and her immune deficiency was unusually severe, affecting both cell-mediated immunity and antibody-mediated immunity. She developed au-toimmune manifestations and chronic EV30 infection. The
first isolate was recovered in September 1989 when the child, then age 6, presented with gastrointestinal manifestations. Six further, consecutive isolates were collected in the period up to July 1991.
These isolates were studied retrospectively to determine whether the patient had been reinfected or whether the infec-tion was persistent. To analyze the genetic characteristics of the seven consecutive isolates, the study was performed in two stages. First, the nucleotide sequences of the 5⬘halves of the genomes (about 4,400 nucleotides) of the first and seventh isolates, which were recovered 22 months apart, were deter-mined. In addition, because only partial sequences were known for the EV30/1958/USA/Bastianni prototype strain (12, 14), the nucleotide sequence of the 5⬘ half of the genome of this strain was also determined and was compared with those of sequential isolates. In the second part of the study, we used the molecular data for the amplification and sequencing of the complete VP1-coding sequences of the other five isolates. Phy-logenetic relationships, inferred from the nucleotide se-quences, showed four lineages in the seven isolates. Finally, we analyzed the replicative capacities of the consecutive isolates and showed phenotypic variations that were consistent with the phylogenetic differences observed.
MATERIALS AND METHODS
Virus strains and identification.Seven enterovirus isolates (Table 1) were sequentially recovered from a child with severe combined immune deficiency (34). Each isolate was recovered from one stool specimen after inoculation of the specimen onto standard cultures of MRC5 cells (human lung embryonic fibro-blasts) at the Laboratory of Virology of the Centre Hospitalier Universitaire Rangueil (Toulouse, France). Virus identification by neutralization tests with the antiserum pools of Lim and Benyesh-Melnick (25) was performed and showed that all isolates were EV30. Propagation of the isolates was performed in MRC5 cells by previously described methods (3) and was limited to a maximum of two
* Corresponding author. Mailing address: UFR de Me´decine, Labo-ratoire de Virologie, 28 Place Henri-Dunant, BP38, F-63002 Cler-mont-Ferrand cedex 1, France. Phone: 33 4 73 60 80 17. Fax: 33 4 73 44 90 29. E-mail: [email protected].
552
on May 15, 2020 by guest
http://jcm.asm.org/
passages. The EV30 prototype strain, EV30/1958/USA/Bastianni (32), and two recent isolates (91CF670 and 98CF746) collected from patients with aseptic meningitis were included in this study as controls for the phylogenetic analyses.
Single-cycle infection and virus titration assays.Confluent MRC5 cell mono-layers in 35-mm tissue culture dishes were washed once with phosphate-buffered saline (PBS) and were infected with one of the viruses examined at a multiplicity of infection of one to five cytopathic units (most probable number of cytopathic units [MPNCU]) per cell (3). After 1 h of incubation at 37°C, the inoculum was discarded, and the cells were washed twice with PBS and fed Eagle’s minimum essential medium containing 2% fetal calf serum. At the postinfection times indicated below, the infected cells were recovered in the medium and the virus was released by three cycles of freezing and thawing. Each virus was studied in duplicate (two independent single-cycle infections). Virus production was deter-mined in each sample, by end-point dilution in microtitration assays with MRC5 cells, as described elsewhere (3).
Preparation of cytoplasmic RNAs from infected MRC5 cells and reverse transcription.Cytoplasmic RNAs were isolated from infected MRC5 cells grown in 60-mm tissue culture dishes at 5 to 7 h postinfection (4). Infected MRC5 cell monolayers were washed once with cold PBS and were lysed in 200l of cold lysis buffer (10 mM Tris-HCl [pH 7.4], 150 mM NaCl, 2 mM EDTA, 0.5% [vol/vol] Nonidet P-40). Cytoplasmic RNAs were separated from cellular pro-teins by three consecutive extractions, one with acid phenol (pH 4.3), one with phenol-chloroform-isoamyl alcohol (25/24/1), and one with chloroform. The synthesis of cDNA was performed in a volume of 50l from 5g of cytoplasmic RNA and with oligo(dT)18as a primer. First-strand cDNA synthesis kit (Strat-agene, Montigny-le-Bretonneux, France) was used according to the manufactur-er’s recommendations.
Oligonucleotides used in the study. Oligonucleotides 5NCNOT, VP4b, 5NC622, and 2C were designed from the available sequences of prototype strains, and oligonucleotides EV30P1 and EV30P2 were constructed from se-quences determined in this study for strain Bastianni and isolates S1 and S7. Oligonucleotides 5NCNOT (5⬘-TCA GCG GCC GCT TAA AAC AGC CTG TGG-3⬘) and VP4b (5⬘-GTT GAC ACT TGA GCT CCC-3⬘) were constructed from the first 16 nucleotides of the enterovirus genome and from 18 nucleotides at the 5⬘end of the coding region (sites 744 to 761 in the genome of coxsackie B virus type 3 [CBV3]), respectively. Oligonucleotides 5NC622 (5⬘-TAT TGG ATT GGC CAT CCG G-3⬘) and 2C (5⬘-CGG CAT TTG GAC TTG AAC TGT AT-3⬘) were constructed from two motifs conserved in the 5⬘noncoding region (5⬘NCR; sites 624 to 642 in CBV3) and in the 2C-coding sequence (sites 4376 to 4398). EV30P1 (5⬘-TCC GCG TGC AAC GAT TTC TC-3⬘) and EV30P2 (5⬘ -CTC CCA CAC GCA GTT CTG CC-3⬘) were designed from conserved motifs in sequences encoding VP3 and 2A, respectively.
PCR amplification of cDNAs.The 5⬘NCR of the EV30 RNA was amplified by a previously described method (4) with primers 5NCNOT and VP4b. In addition, a large fragment of the viral RNA (approximately 3,800 bases long) was ampli-fied with the Expand Long Template PCR System (Roche Molecular Biochemi-cals, Meylan, France) and primers 5NC622 and 2C. The amplification reactions were performed from 2 to 5l of the cDNA in a mixture containing each of the primers at a concentration of 300 nM, each of the four deoxynucleotides at a concentration of 350M, and 1.75 U of the enzyme mix (thermostableTaqand PwoDNA polymerases) in 35 cycles. The first cycle consisted of denaturation for 2 min at 94°C, hybridization for 30 s at 52°C, and elongation for 2 min and 10 s at 68°C and was followed by 34 cycles each of 20 s at 94°C, 30 s at 52°C, and 2 min and 10 s at 68°C, Samples were run on the Omnigene thermocycler (Hybaid, Paris, France). The VP1-coding sequences of the EV30 isolates were specifically amplified with the two primers EV30P1 and EV30P2 by using the conditions described above but with a shorter elongation time (1 min). For each isolate, six to eight PCRs were performed, and the amplification products were brought together and were purified from low-melting-point agarose by standard phenol-chloroform extractions.
Nucleotide sequencing of PCR products.The nucleotide sequences of the purified PCR products were determined at Euro Se´quences Ge`nes Service (Montigny-Le-Bretonneux, France). The nucleotide sequences were determined with the ABI PRISM Dye Terminator Cycle Sequencing Ready Reaction Kit (Perkin-Elmer Corporation), resolved with the 373 DNA sequencer, and ana-lyzed with the ABI PRISM 373 Genetic Analyzer (Applied Biosystems).
Phylogenetic analysis.The phylogenetic relationships of reference strain Bas-tianni with previously characterized prototype enteroviruses and with the se-quential EV30 isolates characterized in this study were estimated from compar-isons of their nucleotide and amino acid sequences. The nucleotide and amino acid sequences used in this study were aligned with the computer programs ESEE3 (8) and CLUSTALW (39). For optimal alignment, a core alignment was produced with VP1 amino acid sequences of poliovirus type 1 Mahoney (PV1M) and CBV3 to provide information about the three-dimensional structure of the viruses (17, 30). A master alignment was then produced by aligning the amino acid sequences of 30 other enteroviruses with the core alignment. The nucleotide sequences were aligned by using amino acid alignment as a guide to obtain the final data set. Pairwise sequence comparisons from the VP1 data set were performed with the MEGA program (23). A phylogenetic tree was constructed from the alignment by the neighbor-joining method (35) as implemented in CLUSTALW. Sites at which there was a gap in any of the aligned sequences were excluded from all comparisons, and distances were corrected by using Kimura’s two-parameter method (20). The reliability of the branching orders was esti-mated by bootstrapping (1,000 samples). A phylogenetic analysis of the VP1 sequences was also performed with the Puzzle computer program (37). The phylogenetic tree was reconstructed by the quartet puzzling method from mo-lecular distances estimated by maximum likelihood. Distances were calculated with the model of nucleotide substitutions of Tamura and Nei (38). The transi-tion/transversion parameter, nucleotide frequencies, and the parameter␣for a gamma distribution of substitution rates were estimated directly from the data set.
Nucleotide sequence accession numbers.The nucleotide sequences of EV30/ 1958/USA/Bastianni and of the seven isolates studied have been deposited with the EMBL data Library under the accession numbers AJ131523 and AJ133656 to AJ133662, respectively.
RESULTS
[image:2.612.58.551.83.195.2]Sequencing of the 5ⴕhalf of the genome for the EV30 Bas-tianni prototype strain and for the first (S1) and seventh (S7) EV30 sequential isolates. Two independent PCR products spanning 4,400 nucleotides were produced from the genome of the EV30/1958/USA/Bastianni prototype strain and of sequen-tial isolates S1 and S7. The nucleotide sequences of the over-lapping cDNAs were determined. The genome organization of the three viruses was determined by comparison of the ge-nomes with those of other reference enteroviruses. As for all other previously characterized enteroviruses, the 3⬘end of the 5⬘NCR was defined by the first AUG triplet that was not closely followed by a stop codon. Translation of the viral RNA probably started at the 7th AUG triplet (nucleotides 757 to 759) in strain Bastianni and at the 10th AUG triplet in the sequential isolates. Alignment of the amino acid sequence de-rived from the open reading frame in EV30 allowed confident prediction of the boundaries for each functional polypeptide. TABLE 1. Echovirus type 30 isolates used in this studya
Virus isolate Isolation (yr, month, city, country) Characteristic Designation
89T2090 1989, September, Toulouse, FR First sequential isolate S1
90T248 1990, January, Toulouse, FR Second sequential isolate S2
90T423 1990, February, Toulouse, FR Third sequential isolate S3
90T2328 1990, August, Toulouse, FR Fourth sequential isolate S4
90T2657 1990, September, Toulouse, FR Fifth sequential isolate S5
90T2917 1990, October, Toulouse, FR Sixth sequential isolate S6
91TLC 1991, July, Toulouse, FR Seventh sequential isolate S7
91CF670 1991, July, Clermont-Ferrand, FR Control isolate 91CF670
98CF746 1998, May, Clermont-Ferrand, FR Control isolate 98CF746
Bastiannib 1958, NK, New-York, USA Reference isolate EV30
aAbbreviations: FR, France; USA, United States; NK, not known.
bThe reference strain was obtained from the World Health Collaborating Center, National Reference Center for Enteroviruses and Hepatitis A.
on May 15, 2020 by guest
http://jcm.asm.org/
Nucleotide sequences of 5ⴕNCRs of EV30 sequential iso-lates.In the 5⬘NCR, isolates S1 and S7 shared 84% nucleotide similarity with the Bastianni strain and 98% nucleotide simi-larity with each other. Half of the 18 nucleotide differences observed between the two isolates were clustered in the last 90 nucleotides located before the initiation codon. Four differ-ences were observed in spacers connecting the secondary-structure domains of the 5⬘NCR, and five were scattered in three domains. The nucleotide sequence of the 5⬘NCR was also determined for isolates S3 and S5. Interstrain variations observed in sequential isolates ranged from 1.2 to 4.2% and were consistent with the intratypic variations observed in EV25 and CBV5 isolates collected independently from different in-dividuals (4, 22). The proportion of observed differences in sequential isolates was also consistent with that determined in poliovirus type 3 (PV3) isolates collected during an outbreak in Finland in 1984 (21). There were more differences between isolates S1 and S7 than between the most distant PV3 isolates. Hence, the 5⬘NCR was not suitable for determination of whether the EV30 sequential isolates were derived from a persistent virus or whether they were acquired independently. Nucleotide sequences of the 5ⴕhalves of the coding regions in EV30 sequential isolates S1 and S7.In the 5⬘half of the open reading frame that was sequenced (3,650 nucleotides), isolates S1 and S7 shared 83.7% nucleotide sequence similarity with strain Bastianni and 97.7% similarity with each other. Of the 83 nucleotide differences observed between the two iso-lates, 21 were missense differences, and 16 of these resulted in amino acid variations in capsid polypeptides. Nucleotide dif-ferences were also observed in genome region P2. In the se-quence encoding polypeptide 2Apro, of 11 differences only 1 led to an amino acid change. In polypeptide 2B, three amino acid changes were observed. Finally, eight nucleotide differ-ences were observed in the 320 nucleotides examined for polypeptide 2C, and only one resulted in an amino acid differ-ence between isolates S1 and S7.
Phylogenetic clustering of the EV30 sequential isolates on the basis of the complete VP1 nucleotide sequence.The phy-logenetic relationships of the seven EV30 sequential isolates were constructed from the entire VP1-coding sequence (876 nucleotides). The VP1-coding sequence was chosen for general and specific reasons. First, VP1 is the largest capsid polypep-tide in enteroviruses and comprises both stable and variable domains that correspond approximately, in the complete virion structure, to internal-sheets and exposed loops, respectively. Accordingly, the sequences encoding these domains would be expected to give different phylogenetic information. More spe-cifically, the VP1-coding sequence was the most divergent se-quence of the genome in the closely related isolates S1 and S7 and thus was the best suited for analysis of genetic evolution from closely related sequences.
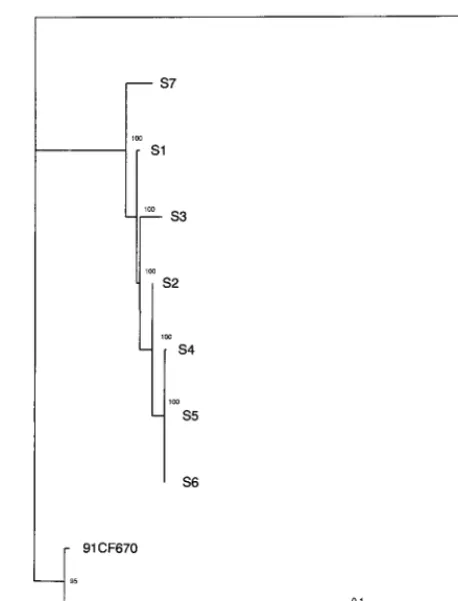
The phylogenetic tree obtained by the neighbor-joining method showed that the seven consecutive isolates were di-rectly related to the Bastianni prototype strain. The branching order of the viral sequences showed that all isolates diverged from a common ancestor and constituted a monophyletic group distinct from the control viruses 91CF670 and 98CF746 isolated from patients with aseptic meningitis. In addition, a major lineage comprising sequences of isolates S4, S5, S6, and S2 was observed. The phylogenetic tree was also reconstructed from the VP1 sequences by the quartet puzzling method with the Puzzle computer program, which estimates pairwise dis-tances by maximum likelihood (37). The Tamura-Nei model (38) of sequence evolution was used with four categories of substitution rates to approximate the gamma distribution. The parameters for the model were estimated directly from the
data set: transition/transversion parameter (10.21⫾2.15), py-rimidine/purine transition parameter (1.13⫾0.25), and nucle-otide frequencies. The parameter␣, which is inversely related to the extent of rate variation at sites, had a low value (0.18⫾
0.04), indicating the existence of a high degree of variation of the substitution rate across nucleotide sites in VP1 sequences. The phylogenetic tree (Fig. 1) reconstructed from maximum likelihood distances was similar to the tree obtained by the neighbor-joining method. The phylogenetic relationships ob-served in Fig. 1 confirmed the existence of a major lineage and showed three additional lineages, one each for isolates S1, S3, and S7. All the internal branches had a very high degree of reliability (⬎95%) and were therefore strongly supported.
[image:3.612.309.538.70.371.2]Evolutionary rate of VP1-coding sequences in sequential isolates.The relationships between the number of nucleotide substitutions and time was examined for the VP1-coding se-quences except for that of the most divergent isolate (the last isolate). The substitution rate was estimated from the nucleo-tide differences observed between isolates S2 to S6 and isolate S1 (Fig. 2). The proportion of the nucleotide differences in each pairwise comparison (differences at all sites, differences at synonymous sites, and differences at the third codon position) were plotted against the divergence time of the two isolates compared. All isolates were distributed along a straight line, suggesting that they evolved at a constant substitution rate from a common ancestor. The substitution rate for the overall observed nucleotide divergence, estimated as the regression
FIG. 1. Phylogenetic relationships of EV30 persistent isolates to one an-other. Pairwise maximum likelihood distances were estimated by the Tamura-Nei model (38) from the VP1 sequences by using all informative nucleotide sites (199 in 876). The tree was reconstructed by the quartet puzzling method (37), and the reliability value of each internal branch indicates (in percent) how often the corresponding cluster was found among the 1,000 intermediate trees. In 210 quartets analyzed, only 7 (3.3%) were unresolved. Branch length was drawn to the indicated scale. EV30 was used as the outgroup to root the trees.
on May 15, 2020 by guest
http://jcm.asm.org/
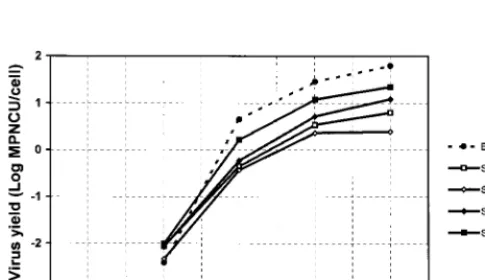
coefficient of the linear relationships, was 8.4⫻10⫺3(0.8%) substitution per year per site. This rate was due to an extremely high rate of synonymous substitutions, 31.2⫻ 10⫺3 (3.12%) per year per synonymous site (Fig. 2). On the assumption that the virus had evolved at a constant rate since the initial infec-tion of the child, the time of the primary infecinfec-tion was esti-mated to be 15 to 20 months before isolation of the first isolate. Phenotypic differences between the EV30 sequential iso-lates.The production of infectious virions in MRC5 cells in-fected with the Bastianni strain or with one of the four sequen-tial isolates studied (isolates S1, S3, S5, and S6) was measured at 37°C and at various postinfection times (Fig. 3). The kinetics of the five strains studied were almost identical in shape; how-ever, major differences in the final virus yields were observed. The virus yield for the four sequential isolates studied was lower than that for the reference strain: when compared with this strain, isolates S3, S1, S5, and S6 produced 25-, 10-, 5-, and 3-fold fewer viruses, respectively. When the virus yields were compared with that of the first isolate, individual differences were noted: isolate S3 produced 2.5-fold fewer viruses, whereas isolates S5 and S6 produced 1.9- and 3.9-fold more viruses, respectively. The third isolate was particularly inter-esting because the maximum virus yield was less than 5 infec-tious units per cell. In a second experiment, performed in
duplicate, all sequential isolates were compared to the refer-ence strain. Virus yield was determined 18 h postinfection under the conditions described above for single-cycle infec-tions. As expected, virus yield was the highest for strain Bas-tianni (1.2 log MPNCU per cell). Isolates grouped in two clusters. The first included isolates S4, S5, and S6 (0.43, 0.69, and 0.96 log MPNCU per cell, respectively), and the second included isolates S7, S3, S2, and S1, which had the lowest virus yield (⫺0.34, 0.05, 0.11, and 0.20 log MPNCU per cell, respec-tively). These results were consistent with the phylogenetic relationships that showed that isolates S4, S5, and S6 were close relatives which evolved significantly from the first isolate and which acquired the highest degree of replicative efficiency in vitro. Isolate S2, which was genetically related but not sim-ilar to isolates S4, S5, and S6, had a lower level of replicative efficiency, and isolates S1, S3, and S7 had significant genetic variations and low levels of replicative efficiency.
DISCUSSION
This is the first report to describe molecular differences and to analyze genetic and phenotypic evolution in isolates of EV30 recovered from a patient with severe combined immune deficiency associated with cartilage-hair hypoplasia, a distinc-tive form of short-limbed dwarfism. Patients with this syn-drome are unusually susceptible to severe infections with vari-cella-zoster virus (40) and are also at risk of enterovirus infection (36). Because enteroviruses are known to cause chronic infection in immunocompromised hosts (27), the main aim of the retrospective study of the EV30 isolates was to determine if the seven isolates were derived from a single persistent virus chronically excreted over at least 22 months.
The phylogenetic analysis of the entire VP1-coding se-quence showed that genetically all isolates were closely related to one another and are directly related to the EV30 Bastianni prototype strain, thereby confirming the serotypes of the vi-ruses determined by the neutralization tests. The sequential isolates grouped in a monophyletic cluster, which shows that the child was persistently infected with one virus strain that diverged into several variants during the 22-month period. Genetic relationships, inferred from the analysis of VP1-cod-ing sequences, showed four lineages for the seven persistent isolates collected from September 1989 to July 1991. In the major lineage, isolates S4, S5, and S6 were identical or differed at only a few nucleotide sites and were closely related to isolate S2. The other lineages comprised isolates S1, S3, and S7, re-spectively. These results show that persistent isolates form a genetically heterogeneous viral population and, thereby, strongly suggest the quasispecies structures of the genomes in the virus population (11).
[image:4.612.53.296.551.691.2]The evolution rates, measured from nucleotide differences in the first six isolates (with isolate S7 being excluded because of its high degree of divergence), showed a constant accumu-lation of changes in VP1 over 13 months. The estimated rate of evolution (3.1% synonymous substitutions per synonymous site per year) is consistent with the rate found for a persistent PV1 strain isolated from an immune-deficient patient with vaccine-associated paralytic poliomyelitis (19) and for a persistent cox-sackie A virus type 15 (CAV15) in an agammaglobulinemic patient (31). The initial infection of the child with EV30 was estimated from the rate of evolution of the VP1-coding se-quences to be 15 to 20 months before the isolation of the first isolate in September 1989. This suggests that infection began long before the isolation of the first EV30 isolate and was not associated with critical clinical symptoms. The interval between the initial infection and the first isolation of an EV30 isolate,
FIG. 2. Duration of chronic EV30 infection estimated from nucleotide sub-stitution rates in VP1-coding sequence. The proportion of nucleotide differences at all sites (diamonds), at synonymous sites (squares), and at the third codon positions (triangles) were calculated for each pairwise comparison with the MEGA computer program (23). Nucleotide substitutions were extrapolated back to zero substitution.
FIG. 3. Time course of virus production in MRC5 cells. MRC5 cells were seeded at 2⫻105per 35-mm culture dish and were cultured for 4 days before inoculation. Data are the means of two independent experiments, and virus titers were determined in duplicate for each experiment.
on May 15, 2020 by guest
http://jcm.asm.org/
which was associated with gastrointestinal manifestations, is in good agreement with clinical data because virus isolation ended a 2-year period during which prophylaxis and adminis-tration of immunoglobulins successfully prevented infectious complications in the child (34).
Despite the divergence, isolate S7 is definitely related to the other persistent isolates by multiple genetic characteristics not only in the VP1-coding sequence but also in other genome sequences. We found 98 nucleotide differences between iso-lates S1 and S7 in the 5⬘halves of their genomes. These dif-ferences correspond to a genetic divergence of 2.3%, equiva-lent to that observed in the VP1 sequence. In genome region P1, we found rates of nucleotide substitutions ranging from 5.3⫻10⫺3 (VP4 sequence) to 14.6⫻ 10⫺3(VP1 sequence) substitutions per nucleotide site per year. These high rates are not consistent with a separate evolution of isolate S7 in com-petition with strains with higher replicative capacities since its replicative efficiency in vitro was lower than those of the other isolates. The isolation of three other virus isolates with low yields indicates instead the coexistence in a carrier state, prob-ably in the gastrointestinal tract, of genetically related viruses with different replicative capacities during the infection of the child. Differences in replication efficiency observed between sequential isolates are consistent with the results of the phylo-genetic analysis. Isolates S4, S5, and S6, which belong to the same genetic lineage and which have identical VP1-coding sequences, have similar replicative capacities. In contrast, iso-lates S1, S3, and S7 are major genetic variants from the former isolates and replicate less efficiently. It is not yet possible to identify exactly the molecular determinants that are responsi-ble for the phenotypic differences observed. However, multiple genetic variations occurred in the 5⬘halves of the genomes of isolates S1 and S7 in regions P1 and P2 and suggest strongly that multiple factors, both in the capsid and in nonstructural polypeptides, are involved in the phenotypic differences ob-served.
In conclusion, our study provides further evidence of the susceptibility of immunodeficient patients to enteroviruses cir-culating in the general population. The genetic characteristics and replicative capacities of EV30 sequential isolates obtained in cell cultures show that viruses with genetic variations in different genome regions and with different biological proper-ties can be isolated from the same individual during the course of a chronic infection. The selection of isolates with different replicative capacities in vitro suggests that new biological fea-tures (cell specificity, tissue tropism, virulence) may be selected in EV30 replicating during a persistent infection in immuno-deficient patients. These isolates are useful tools for under-standing the relationships between nucleotide sequences, pro-tein structure, and biological properties in echoviruses, and the study of such isolates may also enable us to identify virulent and attenuated determinants in virus strains that emerge in different susceptible human populations.
ACKNOWLEDGMENTS
We are grateful to Danielle Thouvenot of the World Health Orga-nization Collaborating Center, National Reference Center for Entero-viruses (Lyon, France), for providing us with the reference strain of EV30. We thank Jeffrey Watts for revision of the English manuscript. This work was supported in part by a grant from Ministe`re de l’Education Nationale de la Recherche et de la Technologie.
REFERENCES
1.Atkinson, P. J., M. Sharland, and H. Maguire.1998. Predominant entero-viral serotypes causing meningitis. Arch. Dis. Child.78:373–374. 2.Aymard, M., J.-J. Chomel, B. Lina, and D. Thouvenot.1997. Annual report
1997. National Reference Center for Enteroviruses and Hepatitis A, Lyon, France.
3.Bailly, J.-L., M. Chambon, H. Peigue-Lafeuille, and F. Charbonne´.1994. Replication of echovirus type 25 JV4 reference strain and wild type strains in MRC5 cells compared with that of poliovirus type 1. Arch. Virol.137:327– 340.
4.Bailly, J.-L., A. M. Borman, H. Peigue-Lafeuille, and K. M. Kean.1996. Natural isolates of echovirus type 25 with extensive variations in IRES sequences and different translational efficiencies. Virology215:83–96. 5.Bello, M.1997. Viral meningoencephalitis caused by enterovirus in Cuba
from 1990–1995. Rev. Argent. Microbiol.29:176–183.
6.Bergman, I., M. J. Painter, E. R. Wald, D. Chiponis, A. L. Holland, and H. G. Taylor.1987. Outcome in children with enteroviral meningitis during the first year of life. J. Pediatr.110:705–709.
7.Berlin, L. E., M. L. Rorabaugh, F. Heldrich, K. Roberts, T. Doran, and J. F. Modlin.1993. Aseptic meningitis in infants⬍2 years of age: diagnosis and etiology. J. Infect. Dis.168:888–892.
8.Cabot, E. L., and A. T. Beckenbach.1989. Simultaneous editing of multiple nucleic acid and protein sequences with ESEE. Comput. Appl. Biosci.5:233– 234.
9.Chambon, M., C. Delage, J.-L. Bailly, J. Gaulme, P. Dechelotte, C. Henquell, C. Jallat, and H. Peigue-Lafeuille.1997. Fatal hepatic necrosis in a neonate with echovirus 20 infection: use of the polymerase chain reaction to detect enterovirus in liver tissue. Clin. Infect. Dis.24:523–524.
10. Cherry, J. D.1992. Enteroviruses: polioviruses (poliomyelitis), coxsackievi-ruses, echovicoxsackievi-ruses, and enterovicoxsackievi-ruses, p. 1705–1753.InR. D. Feigin and J. D. Cherry (ed.), Textbook of pediatric infectious diseases, 3rd ed. The W.B. Saunders Co., Philadelphia, Pa.
11. Domingo, E., and J. J. Holland.1997. RNA virus mutations and fitness for survival. Annu. Rev. Microbiol.51:151–178.
12. Drebot, M. A., C. Y. Nguyan, J. J. Campbell, S. H. S. Lee, and K. R. Forward.
1994. Molecular epidemiology of enterovirus outbreaks in Canada during 1991–1992: identification of echovirus 30 and coxsakievirus B1 strains by amplicon sequencing. J. Med. Virol.44:340–347.
13. Druyts-Voets, E.1997. Epidemiological features of entero non-poliovirus isolations in Belgium 1980–94. Epidemiol. Infect.119:71–77.
14. Gjoen, K., A. L. Bruu, and I. Orstavik.1996. Intratypic variability of echo-virus type 30 in part of the VP4/VP2 coding region. Arch. Virol.141:901–908. 15. Gorgievski-Hrisoho, M., J.-D. Schumacher, N. Vilimonovic, D. Germann, and L. Matter.1998. Detection by PCR of enteroviruses in cerebrospinal fluid during a summer outbreak of aseptic meningitis in Switzerland. J. Clin. Microbiol.36:2408–2412.
16. Hertel, N. T., F. K. Pedersen, and C. Heilmann.1989. Coxsackie B3 virus encephalitis in a patient with agammaglobulinemia. Eur. J. Pediatr.148:642– 643.
17. Hogle, J. M., M. Chow, and D. J. Filman.1985. Three-dimensional structure of poliovirus at 2.9 Å resolution. Science229:1358–1365.
18. Johnson, J. P., R. H. Yolken, D. Goodman, J. A. Winkelstein, and J. E. Nagel.
1982. Prolonged excretion of group A coxsackievirus in an infant with agam-maglobulinemia. J. Infect. Dis.146:712.
19. Kew, O. M., R. W. Sutter, B. K. Nottay, M. J. McDonough, D. R. Prevots, L. Quick, and M. A. Pallansch.1998. Prolonged replication of a type 1 vaccine-derived poliovirus in an immunodeficient patient. J. Clin. Microbiol.36:
2893–2899.
20. Kimura, M.1980. A simple method for estimating evolutionary rate of base substitutions through comparative studies of nucleotide sequences. J. Mol. Evol.16:111–120.
21. Kinnunen, L., T. Po¨yry, and T. Hovi.1991. Generation of genetic lineages during an outbreak of poliomyelitis. J. Gen. Virol.72:2483–2489. 22. Kopecka, H., B. Brown, and M. A. Pallansch.1995. Genotypic variation in
coxsackievirus B5 isolates from three different outbreaks in the United States. Virus Res.38:125–136.
23. Kumar, S., K. Tamura, and M. Nei.1993. MEGA: molecular evolutionary genetics analysis version 1.01. The Pennsylvania State University, University Park, Pa.
24. Leonardi, G. P., A. J. Greenberg, P. Costello P, and K. Szabo.1993. Echo-virus type 30 infection associated with aseptic meningitis in Nassau County, New York, USA. Intervirology36:53–56.
25. Lim, K. A., and M. Benyesh-Melnick.1960. Typing of viruses by combination of antiserum pools. Application to typing of enteroviruses (coxsackie and ECHO). J. Immunol.84:309–317.
26. Lopez-Alcala, M. I., M. Rodriguez Priego, D. de la Cruz Morgado, and J. M. Barcia Ruiz.1997. Outbreak of meningitis caused by type 30. An. Esp. Pediatr.46:237–240.
27. McKinney, R. E., Jr., S. L. Katz, and C. M. Wilfert.1987. Chronic enteroviral meningoencephalitis in agammaglobulinemic patients. Rev. Infect. Dis.
9:334–356.
28. Melnick, J. L.1990. Enteroviruses: polioviruses, coxsackieviruses, echovi-ruses and newer enteroviechovi-ruses, p. 549–605.InB. N. Fields and D. M. Knipe (ed.), Virology, 2nd ed. Raven Press, New York, N.Y.
29. Modlin, J. F.1997. Update on enterovirus infections in infants and children. Adv. Pediatr. Infect. Dis.12:155–180.
30. Muckelbauer, J. K., M. Kremer, I. Minor, G. Diana, F. Dutko, J. Groarke, D. C. Pevear, and M. G. Rossmann.1995. The structure of coxsakievirus B3
on May 15, 2020 by guest
http://jcm.asm.org/
at 3.5 Å resolution. Structure3:653–667.
31.O’Neil, K. M., M. A. Pallansch, J. A. Winkelstein, T. M. Lock, and J. F. Modlin.1988. Chronic group A coxsackievirus infection in agammaglobu-linemia: demonstration of genomic variation of serotypically identical iso-lates persistently excreted by the same patient. J. Infect. Dis.157:183–186. 32. Plager, H., and W. Decher.1963. A newly-recognized enterovirus isolated
from cases of aseptic meningitis. Am. J. Hyg.77:26–28.
33. Rotbart, H. A.1995. Meningitis and encephalitis, p. 271–289. InH. A. Rotbart (ed.), Human enterovirus infections. ASM Press, Washington, D.C. 34. Rubie, H., D. Graber, A. Fischer, M. T. Tauber, P. Maroteaux, A. Robert, F. Le Deist, P. Rochiccioli, C. Griscelli, and C. Reignier.1989. Hypoplasie du cartilage et des cheveux avec de´ficit immunitaire combine´. Ann. Pediatr.
36:390–392.
35. Saitou, N., and M. Nei.1987. The neighbor-joining method: a new method for reconstructing phylogenetic trees. Mol. Biol. Evol.4:406–425. 36. Saulsburry, F. T., J. A. Winkelstein, L. E. Davis, L. H. Hsu, B. J. D’Souza,
G. R. Gutcher, and I. J. Butler.1975. Combined immunodeficiency and vaccine-related poliomyelitis in a child with cartilage-hair hypoplasia. J. Pe-diatr.86:868–872.
37. Strimmer, K., and A. von Haeseler.1996. Quartet puzzling: a quartet max-imum likelihood method for reconstructing tree topologies. Mol. Biol. Evol.
13:964–969.
38. Tamura, K., and M. Nei.1993. Estimation of the number of nucleotide substitutions in the control region of mitochondrial DNA in humans and chimpanzees. Mol. Biol. Evol.10:512–526.
39. Thompson, J. D., D. G. Higgins, and T. J. Gibson.1994. CLUSTALW: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, positions-specific gap penalties and weight matrix choice. Nucleic Acids Res.22:4673–4680.
40. Virolainen, M., E. Savilahti, I. Kaitila, and J. Perheentupa.1978. Cellular and humoral immunity in cartilage-hair hypoplasia. Pediatr. Res.12:961– 966.