Structural Studies of SCR domains in Multidomain
Complement Proteins
Thesis Presented for the Degree of
Doctor of Philosophy
By
Mohammed Aslam
Department of Biochemistry and Molecular Biology
University College London
ProQuest Number: 10016036
All rights reserved
INFORMATION TO ALL USERS
The quality of this reproduction is dependent upon the quality of the copy submitted.
In the unlikely event that the author did not send a complete manuscript and there are missing pages, these will be noted. Also, if material had to be removed,
a note will indicate the deletion.
uest.
ProQuest 10016036
Published by ProQuest LLC(2016). Copyright of the Dissertation is held by the Author.
All rights reserved.
This work is protected against unauthorized copying under Title 17, United States Code. Microform Edition © ProQuest LLC.
ProQuest LLC
789 East Eisenhower Parkway P.O. Box 1346
ABSTRACT
There is a level o f organization in proteins that overlaps the classical definitions
o f tertiary and quaternary structure, i. e. sequentially consecutive residues in polypeptide
chains fold into distinct independently-folded regions called domains. Many
m ultidom ain proteins are flexible at their interdom ain junctions and are therefore not
amenable to X-ray crystallography or are too big for m ultinuclear N M R techniques.
Small-angle X-ray and neutron scattering and analytical centrifugation methods, coupled
with m olecular modelling techniques, are able to locate the relative positions o f these
domains relative to each other within the full protein structure.
This PhD thesis has looked at the short com plem ent/consensus repeat (SCR)
dom ains o f two com plem ent proteins. Factor H (FH) and com plem ent related protein
y (Crry) in order to identify the principles o f their domain arrangem ent. SCR domains
are w idespread in complement proteins and the solution conform ation betw een adj acent
SCR pairs is central to the understanding o f the m olecular m echanism o f com plement
activation. A m ethod o f scattering curve m odelling based on rigid body structures as
constraints provide m edium resolution structures on the solution structure o f these SCR
proteins. U sing an automated constrained fit procedure, w ith com plem ent FH, only
those m odels in which the 20 SCR domains in FH were bent back upon themselves were
able to account for the scattering and sedim entation data. These bent-back FH structures
may perm it the multiple binding sites for C3 and heparin to come into close proximity,
and assist the m ultifunctional role o f FH. It is concluded that, i f the inter SCR linkers
are as long as eight residues, as found in hum an FH, significant flexibility and the
generation o f folded back structures can result. If the inter SCR linkers are o f short
length, this results in only a m odest degree o f bending and this w as observed in rodent
Crry. The averaged solution structure o f Crry w ith five SCR dom ains was found to be
highly extended w ith a slightly bent arrangem ent o f SCR domains. The structural
models for FH and Crry have been useful in the elucidation o f the consequences o f
m utations e.g. FH in haem olytic uraem ic syndrome and the evaluation o f anti
A cknow ledgem ents
I w ish to acknowledge the following people for their assistance and support
during the course o f my studies at the U niversity College London. Foremost, I w ould
like to thank m y supervisor Professor S. J. Perkins for providing m e w ith the
opportunity to study for this thesis and to the mem bers o f the protein structure group for
their help in all aspects o f m y work. In particular, I w ould like to thank Dr M. K.
Boehm for useful discussions and Dr. J. Hinshelw ood for encouragem ent throughout.
I would also like to thank Dr. J. Eaton for useful discussions. I am eternally in debt to
Professor P. Swann for his all round support and thank Professor K.R. Bruckdorfer for
overseeing this studentship.
I thank the Biotechnology and Biological Sciences R esearch Council for a
Special studentship and for access to X-ray and neutron facilities, and the W ellcome
Trust for access to the analytical centrifuge. I would also like to thank M rs S. Slawson,
M r A. Gleeson, Dr. G. Grossman for instrumental support at the SRS Daresbury; Dr P.
A. Tim m ins for instrumental support at the ILL Grenoble and D r R. K. Heenan and Dr
S. M. King for generous instrumental support.
Owing to the lack o f space, I offer a sincere apology to the m any people whom
I have not m entioned but are w orthy o f a mention; no m an is an island and your
influences w ere undoubtedly crucial for the character I now adopt.
Finally I w ould like to thank my family and friends especially, my youngest
brother A jmal for all their support during the past years o f my life. My m other is a key
figure and a huge pillar o f support. I acknowledge patience from m y w ife N aila during
thesis writing up. The birth o f my daughter, Iqra nearly one year ago is a welcome
The work presented in this thesis is my own
Signed M. Aslam
Contents Page Chapter 1
Repetitive Nature o f Extracellular Proteins 1
1.1. Introduction 2
1.2. Evolution and cellular location o f m odules 5
1.3. Extracellular modules and their biological role 13
1.4. Blood Coagulation/Fibrinolysis and the Com plement Systems 14
1.5. Some general observations on modules 18
1.5.1. Introduction 18
1.5.2. Conservation and variability o f disulphide bridges 18
1.5.3. Com m on topological features o f different modules 19
1.5.4. D om ain assembly 20
1.6. Conclusions 21
Chapter 2
Protein Structure Determination M ethods 23
2.1. Introduction 24
2.2. H igh resolution techniques 25
2.2.1. X-ray crystallography 26
2.2.2. N uclear magnetic resonance (NMR) spectroscopy 29
2.3. Small angle solution scattering 32
2.3.1. X-ray scattering theory 3 2
2.3.1.1. The Debye equation 33
2.3.1.2. Two-phase model o f solution scattering 36
2.3.2. X-ray solution scattering 37
2.3.2.1. Sample preparation 37
2.3.2.2. X-ray scattering at SRS Daresbury 37
2.3.2.3. Reduction o f SRS scattering data 41
2.3.3. N eutron scattering theory 44
2.3.3.1. Introduction 44
2.3.3.3. Com parison between X-rays and neutrons 45
2.3.3.4. The hydration shell 46
2.3.3.5. Contrast difference Ap 48
2.3.3.6. M atchpoint determination 48
2.3.3.7. Stuhrmann plot 49
2.3.4. N eutron solution scattering 51
2.3.4.1. Sample preparation 51
2.3.4.2. N eutron scattering on LOQ at the RAL 51
2.3.4.3. Reduction o f LOQ scattering data 53
2.3.4.4. N eutron scattering on D22 and D l l at the ILL 55
2.3.4.4. Reduction o f ILL scattering data 61
2.3.5.1. Guinier analyses 65
2.3.5.2. Cross-sectional radius o f gyration 66
2.3.5.3. Estimations o f macromolecular dim ensions 68
2.3.5.4. Real space distance distribution function 68
2.4. Analytical ultracentrifugation 69
2.4.1. Sedimentation velocity experiments 69
2.4.2. Sedimentation equilibrium experiments 72
2.5. Circular dichroism and Fourier transform infrared spectroscopy 74
Chapter 3
Homology M odelling o f Protein Structures 75
3.1. Introduction 76
3.2. Homology m odelling 76
3.2.1. Sequence analysis 78
3.2.2. Secondary structure predictions 80
3.2.2.1. Accessible surface area 83
3.2.2.2. DSSP (Dictionary o f Secondary Structure o f Proteins) 84
3.2.3. Tertiary structure predictions by analogy m odelling 86
3.2.4. M odel building 88
3.2.4.1. M odel refinement 90
3.2.5. Structure validation o f models 91
3.3. X-ray and neutron solution scattering curve m odelling 92
3.3.1. Analysis o f glycoprotein composition 92
3.3.2. Debye scattering curve calculation 94
3.3.3. X-ray and neutron scattering curves calculation 94
3.3.4. Hydrodynamic analyses 103
Chapter 4
The Short Consensus Repeat: The M ost A bundant Domain Type o f Com plem ent 105
4.1. Introduction 106
4.2. N M R Structures 112
4.2.1. V accinia virus complement control protein 112
4.2.2. Interdomain-orientation in domain pairs o f FH and VCP 113
4.2.3. N M R structure o f two pairs o f SCR domains in active site 2 o f C R l 113
4.3. Crystal Structures 115
4.3.1. Introduction to CD46 115
4.3.2. Interdomain m ovem ent and flexibility o f CD46 116
4.3.3. Oligosaccharide content in CD46 118
4.3.4. H um an p2-glycoprotein I: introduction 118
4.3.5. Interdomain m ovement and flexibility o f p2-glycoprotein I 121
4.3.6. Oligosaccharide content in p2-glycoprotein I 121
4.3.7. Crystal structure o f a SCR domain in C ls 123
4.4. Conclusions 127 Chapter 5
The Solution Structure o f Factor H 128
5.1. Introduction 129
5.2. The FH protein family 129
5.2.1. Introduction 129
5.2.2. The factor H-like protein 1 133
5.2.3. FHR-1 and FHR-2 133
5.2.4. FHR-3 and FHR-4 134
5.2.5. O ther members o f the FH family 135
5.3. Structural studies o f FH 135
5.4. M aterials and M ethods 137
5.4.1. Purification o f FH for scattering and ultracentrifugation experiments 137
5.4.2. Functionality o f FH 137
5.4.2.1. Purification o f serum Factor I 138
5.4.2.2. Purification o f serum C3 139
5.4.2.3. Factor I-dependent cofactor activity o f FH Assay 141
5.4.3. Small angle solution scattering 141
5.4.3.1. X-ray data from Station 2.1 at the Synchrotron Radiation
Source 141
5.4.3.2. Neutron data from Instrument LOQ at ISIS and Instruments
D22 and D l l at the ILL 143
5.4.3.3. Analysis o f reduced X-ray and neutron data 144
5.4.4. Analytical ultracentrifugation 145
5.4.4.1. Sedimentation equilibrium and sedim entation velocity data
for FH 145
5.4.5. Homology modelling o f 17 SCR domains in FH 146
5.4.6. M odelling o f FH by constrained scattering fits 154
5.4.6.1. Random ised domain modelling o f FH by constrained
scattering fits (M ethod 1) 154
5.4.6.2. Rotational search modelling o f FH by constrained scattering
fits (M ethod 2) 156
5.4.6.3. Automated Debye scattering curve modelling o f FH 157
5.4.6.4. Sedimentation coefficient m odelling o f FH 160
5.5. Results and D iscussion 160
5.5.1. Small angle solution scattering 160
5.5.1.1. X-ray scattering data for FH 160
5.5.1.2. N eutron scattering data for FH 163
5.5.2. Analytical ultracentrifugation 165
5.5.2.1. Sedimentation equilibrium and velocity data for FH 165
5.5.3. Homology modelling o f 17 SCR dom ains o f FH 166
5.5.4.1. A randomised search for m odelling the X-ray solution
structure o f FH: method 1 169
5.5.4.2. A rotational search for m odelling the X-ray solution structure
o f FH : m ethod 2 178
5.5.4.3. N eutron scattering curve m odelling 180
5.5.5. Analytical ultracentrifugation 181
5.5.5.1. Sedimentation coefficient m odelling o f FH 181
5.5.6. Electrostatic surfaces o f the FH models 181
5.6. Conclusions 183
Chapter 6
The solution structures o f Crrv and Crrv-Ig 192
6.1. Introduction 192
6.2. M aterials and M ethods 196
6.2.1. Purification o f rCrry and mCrry-Ig 196
6.2.2. Small angle solution scattering 198
6.2.2.1. X-ray scattering data acquisition at the Synchrotron Radiation
Source 198
6.2.2.2. N eutron scattering data acquisition at ISIS and the ILL 199
6.2.2.3. Analysis o f reduced X-ray and neutron data 199
6.2.3. Analytical ultracentrification data acquisition and analysis 200
6.2.4. Homology m odelling o f the SCR domains in rCrry and mCrry-Ig 200
6.2.5. M odelling o f rCrry and mCrry-Ig by constrained scattering fits 201
6.2.5.1 M odelling o f rCrry by constrained scattering fits 201
6.2.5.2. M odelling o f mCrry-Ig by constrained scattering fits 202
6.2.6. Debye scattering curve modelling o f rCrry and mCrry-Ig 204
6.2.7. Sedimentation coefficient modelling o f rCrry and mCrry-Ig 206
6.3. Results and D iscussion 206
6.3.1. Small angle solution scattering 206
6.3.1.1. X-ray scattering analyses for rCrry and mCrry-Ig 206
6.3.1.2. N eutron scattering analyses for rCrry and mCrry-Ig 211
6.3.2. Analytical ultracentrifugation data for rCrry and mCrry-Ig 213
6.3.3. Homology m odelling o f the SCR domains o f rCrry and mCrry-Ig 215
6.3.4. M odelling o f rCrry and mCrry-Ig by constrained scattering fits 221
6.3.4.1. Random ised linker modelling o f rCrry by constrained scattering fits 221 6.3.4.2. Randomised linker modelling o f mCrry-Ig by constrained scattering
fits 228
6.4. Conclusions 236
C hapter 7
Sum m ary and Conclusions 239
7.1. Introduction: Previous knowledge o f SCR domains 240
7.3. Solution structure o f 20 SCR domains in FH 242
7.5. Conclusions 244
References 246
Figures Legend Page Chapter 1
Figure 1.1 Secondary structure o f some common domains found in extracellular
proteins 4
Figure 1.2 N om enclature and captions for Figures 1.1 to 1.11 6
Figure 1.3 Cartoon representations for various m ultidom ain proteins 7
Figure 1.4 Cartoon o f multidom ain proteins with short intracellular dom ains 8
Figure 1.5 Cartoon o f domains in receptors 9
Figure 1.6 Cartoon o f selected enzymes flanked mainly by extracellular domains 10
Figure 1.7 Cartoon o f domains in m atrix molecules 11
Figure 1.8 Cartoon o f domains in vertebrate collagens 12
Figure 1.9 C artoon o f the mosaic proteins involved in blood coagulation and
fibrinolysis. 15
Figure 1.10 Cartoon o f the complement cascade and some o f its regulators 16
Figure 1.11 The activation steps o f the complement system 17
Chapter 2
Figure 2.1 General features o f a solution scattering curve I{Q) m easured over a Q
range 34
Figure 2.2 Diffraction o f electromagnetic radiation in a protein 35
Figure 2.3 X-ray solution scattering at the SRS Daresbury 39
Figure 2.4 Instrum ents o f Station 2.1 SRS Daresbury 40
Figure 2.5 Basic set o f X-ray scattering data 42
Figure 2.6 Vacuum tubing o f Station 2.1 SRS Daresbury and a flow diagram o f the
reduction procedure for SRS Daresbury X-ray scattering data 43
Figure 2.7 Contrast matching 50
Figure 2.8 N eutron solution scattering on LOQ 52
Figure 2.9 V iew from above the LOQ sample pit and flow diagram o f LOQ data
reduction using COLETTE 54
Figure 2.10 N eutron Scattering at ILL, Grenoble 56
Figure 2.11 Beam line arrangement at the high-flux reactor at the ILL 57
Figure 2.12 Schematic diagram and characteristics o f D22 58
Figure 2.13 A D om ier neutron velocity selector 59
Figure 2.14 Picture o f the primary collim ation system on D22 60
Figure 2.15 Instrum ent characteristics on D22 62
Figure 2.16 Schematic diagram and characteristics o f D 11 63
Figure 2.17 Flow diagram o f D22 data reduction procedures 64
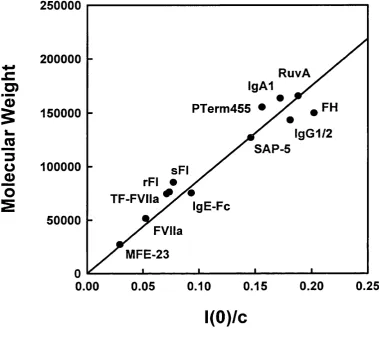
Figure 2.18 Linear relationship betw een the m olecular weight and the neutron 7(0)/c
values for glycoproteins in 100% ^H20 buffer measured on LOQ 67
C hapter 3
for a target sequence based on a know n structure that has the same fold
Figure 3.2 Flow chart o f automated analysis o f m ultidom ain m odels for scattering
curve fits 93
Figure 3.3 Schematic outlines o f six m ultidom ain or oligom er structures to show how
dom ain or subunit translations and rotations were im plem ented during the
curve fit analyses 99
Figure 3.4 The best-fit model from each curve fitting analysis to follow that o f Figure
3.3 100
Figure 3.5 Final X-ray and neutron curve fits based on the best-fit m odels from Figure
3.4 101
Chapter 4
Figure 4.1 Sequence and secondary structure alignm ent o f the PDB structures o f SCR 107
dom ains -108
Figure 4.2 A backbone trace o f the superim position o f individual SCR crystal
structures 110
Figure 4.3 The diverse arrangem ent o f structures o f know n linker conformations. 111
Figure 4.4 O rientations o f SCR domains 114
Figure 4.5 Interdom ain m ovem ent and the flexibility o f the CD46 at the dom ain
interface 117
Figure 4.6 The SC R l and SCR2 interdomain interface o f CD46 119
Figure 4.7 A ribbon representation o f SC R l and SCR5 o f P2GPI showing the 122
structural elements
Chapter 5
Figure 5.1 The factor H protein family 130
Figure 5.2 Structure o f the polysaccharide heparin 131
Figure 5.3 Elution profile for the purification o f C3 140
Figure 5.4 FH cofactor activity 142
Figure 5.5 Time dependence o f successive equilibrium scans o f FH 147
Figure 5.6 Interference scans o f four curves at equilibrium o f FH 148
Figure 5.7 Time dependence o f successive sedim entation velocity scans o f FH 149
Figure 5.8 Fitting o f the tim e derivatives o f sedim entation data o f FH 151
Figure 5.9 Sequence and structure alignm ent o f the FH SCR sequences 152
Figure 5.10 Average carbohydrate structure for FH 155
Figure 5.11 Axes arrangem ent for the rotational search perform ed on FH 158
Figure 5.12 G uinier analyses o f X-ray data on FH 161
Figure 5.13 Concentration dependence o f scattering Rq, R^s.] and Rxs. 2 values for FH 162
Figure 5.14 X-ray and neutron distance distribution functions P(r) for FH 164
Figure 5.15 Sedimentation equilibrium data for FH 167
Figure 5.16 A plot o f sedim entation coefficient against the concentration o f FH 168
Figure 5.17 M odels for FH based on know n linkers 172
Figure 5.19 Structural analysis o f FH models obtained from the Random ised-3 search 176 Figure 5.20 X-ray and neutron curve fits for the four best-fit m odels for FH from the
Random ised-3 search 177
Figure 5.21 A ribbon representation o f the overlay o f SCR-1 o f the four best FH
models from the randomised-3 search 179
Figure 5.22 Plots depicting the structural analysis o f the 2010 FH m odels obtained
from the Random ised-3 search 182
Figure 5.23 Summ ary o f the four best-fit models for FH 184
Figure 5.24 Basic residues responsible for heparin binding 185
Figure 5.25 Sequence alignment o f human, mouse, bovine and barred sand bass
{Parablax neblifer) FH 189
Chapter 6
Figure 6.1 SCR dom ain arrangem ent in rCrry and mCrry-Ig 195
Figure 6.2 Guinier Rq analyses o f rCrry and mCrry-Ig 207
Figure 6.3 Guinier Rxs analyses o f rCrry and mCrry-Ig 209
Figure 6.4 Distance distribution functions P(r) for rCrry and mCrry-Ig 210
Figure 6.5 Sedimentation equilibrium analyses for rCrry 214
Figure 6.6 Sedimentation equilibrium analyses for mCrry-Ig 216
Figure 6.7 Sedimentation velocity fits for rCrry and mCrry-Ig 217
Figure 6.8 A summary o f homology m odelling o f the SCR domains in rCrry and
mCrry-Ig and a schematic o f branched simple carbohydrates 219
Figure 6.9 Sequence and alignment o f the rCrry and mCrry-Ig SCR domains 220
Figure 6.10 Ribbon views o f 20 linker structures seen in crystal and N M R structures 222
Figure 6.11 Structural analysis o f the 2000 rCrry models 226
Figure 6.12 X-ray and neutron curve fits 227
Figure 6.13 Summary o f the conformational search o f best-fit m odels for rCrry 229
Figure 6.14 The rotational search for rCrry 230
Figure 6.15. Structural analysis o f mCrry-Ig models 233
Figure 6.16 X-ray and neutron curve fits for the best-fit m odels for mCrry-Ig 234
List of Tables Description Page Chapter 2
Table 2.1
Table 2.2
Stages in the determination o f a protein structure by X-ray
crystallography and N M R 31
Stages in the determ ination o f a protein structure by SAXS and
SANS and their characteristics 47
Chapter 3
Table 3.1 Scattering curve fit analyses for six m ultidom ain proteins 102
Chapter 5
Table 5.1 Table 5.2 Table 5.3
Summary o f homology modelling o f FH 153
Summary o f modelling searches for FH 171
Linkers residues between SCR domains in R CA protein fam ily 190
Chapter 6
Table 6.1 Summary o f modelling searches for SCR domain arrangements
List o f Abbreviations 2D-NM R APS P2GPI C l/2/3/4/5/6/7/8/9 C lr C ls C3a/4a/5a C3b C3i C3(H20) C3(NH3) C4bp CCP CD CDxx C D lla /b /c /d CD18 CD21 CD55 CD46 C R l/2 CRP Crry DAF DNA DSSP EDTA EOF ELISA Fab FB Fc FH FI FPLC Fn-I/II/III FT-IR Fuc Gal GlcNAc Ig IE M AC M an NeuNAc M ASP-1/-2
two dimensional nuclear m agnetic resonance anti-phospholipid syndrome
p 2 glycoprotein I
complement components 1/2/3/4/5/6/7/8/9 protease com ponent o f complement com ponent 1 protease com ponent o f com plem ent com ponent 1 anaphylatoxins
proteolytically activated form o f C3
hydrolytically activated form o f C3 (synonym o f C3(H20)) hydrolytically activated form o f C3 (synonym o f C3i) amidated C3 (activated form o f C3)
C4 binding protein
complement control protein (synonym o f SCR) circular dichroism
cluster o f differentiation or determ inant (xx is a number) a subunit o f the p2 integrins
p subunit o f the p2 integrins complement receptor 2 complement receptor 1 membrane cofactor protein complement receptor 1/2 C reactive protein
complement receptor-related gene/protein y decay accelerating factor
deoxyribonucleic acid
dictionary o f secondary structure o f proteins ethylenediaminetetraacetic acid
epidermal growth factor
enzyme linked im m unosorbent assay
antigen binding region o f an im m unoglobulin molecule factor B
C-terminal halves o f two heavy chains o f an Ig molecule factor H
factor I
fast perform ance liquid chromatography fibronetin type-I/II/III
Fourier transform infrared spectroscopy fucose
galactose
N-acetyl glucosamine immunoglobulin interleukin
m embrane attack complex mannose
N-acetyl neuraminic acid
MBL M BP M CP m RNA NeuNAc N M R OD P PBS PCR PDB PHD RMS r.p.m. RCA RNA SAS SANS SAXS SCR SDS-PAGE S H l/2 VCP
mannose binding lectin mannose binding protein membrane cofactor protein m essenger RNA
N-acetyl neuraminic acid nuclear magnetic resonance optical density
properdin
phosphate buffered saline polymerase chain reaction protein databank
profile network system from Heidelberg root m ean squared
revolutions per minute
regulation o f complem ent activation ribonucleic acid
small angle scattering
small angle neutron scattering small angle X-ray scattering
short consensus/com plem ent repeat (synonym o f CCP) or the structurally conserved regions (relating to homology m odelling)
sodium dodecyl sulphate polyacrylamide gel electrophoresis Src Homology dom ain 1/2
Vaccinia virus complement protein
Am ino Acid A bbreviations
Am ino acid 3 letter format
1 1(
alanine Ala A
arginine Arg R
aspartate Asp D
asparagine A sn N
cysteine Cys C
glutamate Glu E
glutamine Gin
Q
glycine Gly G
histidine His H
iso leucine He I
leucine Leu L
lysine Lys K
m ethionine Met M
phenylalanine Phe F
proline Pro P
serine Ser S
threonine Thr T
tryptophan Trp W
tyrosine Tyr Y
Chapter 1
1.1. Introduction
The prim ary structure o f a protein is its amino acid sequence i. e. the arrangement
o f am ino acids along a linear polypeptide chain, together with any covalent
m odifications such as the position o f disulphide bridges. Two different proteins that
have significant similarities in their primary structures (generally above 30%) are said
to be homologous, and since their corresponding DNA sequences also are significantly
similar, it is generally assumed that the two proteins are evolutionarily related from a
com m on ancestral gene. Secondary structures in globular proteins occur mainly as a
helices and p strands, connected by loops on the protein surface. The formation o f
secondary structure in a local region o f the polypeptide chain is determined by the
primary structure. Certain amino acid sequences favour either a helices or P strands
whilst others favour the formation o f loop regions. Secondary structure elem ents usually
arrange them selves in one o f several simple motifs. M otifs are form ed by packing side
chains from adjacent a helices or p strands close to each other, and several motifs
usually com bine to form compact globular structures, w hich are called domains. The
proteins tertiary structure describes the sidechain packing w ithin domains. If there is
significant amino acid sequence homology between two domains from different proteins,
these dom ains have sim ilar tertiary structures.
By the analysis o f existing crystal structures, protein folds have been classified
into five classes according to their secondary structure (Richardson, 1981; Kabsch &
Sander 1983). These classes are: (i) all a [22%], w ith all a helices and no P-strands; (ii)
all P[16%], containing only p-strands and no a-helices; (iii) a /p [17%] , in which the
polypeptide chain alternates between a-helices and p-strands; (iv) a + p [30%], where
a-helices and p-strands occur in separate parts o f the structure and (v) coil, in which
there is little or no regular secondary structure [15% allocated to m ulti-dom ain proteins,
membrane and cell surface proteins and small proteins]. The total num ber o f folds in
the database, to date (1 September 2002), is 701 for 17,406 PDB entries according to the
Structural Classification O f Proteins web page (SCOP) ( http://scop, mrc-lmb. cam. ac. uk).
All a class proteins have about 60% o f their residues in a-helices and the helices are
pack against each other. a/(3 class proteins often have one parallel (3-sheet w ith a helix
that occurs betw een pairs o f (3-strands. a+(3 class proteins may have one antiparallel P-
sheet w ith the a helices clustering together at one or both ends o f the p-sheet.
Com puter program s have been developed that analyse the atomic coordinates o f crystal
structures and automatically identify regions o f secondary structure as well as hydrogen
bonding patterns. These programs give a consistent way o f com paring the massive
datasets that result from the determination o f atomic coordinates (for w hich over 15,000
entries are available in the Protein D ata Bank (17 December 2002); Bernstein, et al.,
1977), thus facilitating ready identification o f commonly occurring secondary structure
elem ents (Fasman, 1989; Creighton, 1993).
It has become standard practice to compare new amino acid and nucleotide
sequences with existing ones in the rapidly growing sequence databases. This has led
to the recurring identification o f certain sequence patterns, usually corresponding to less
than 300 am ino acids in length and to a well-defined dom ain structure. Proteins that
contain such domains are widely distributed in biology, but they are particularly
com m on in extracellular proteins. Until a few years ago, structural studies o f intact
proteins w ith extracellular parts had proved difficult, partly because these proteins are
often glycosylated, membrane spanning, large and flexible, and are therefore hard to
crystallise. The 3D structures o f many o f their modular com ponents are now known or
are in the process o f being elucidated. This advance has come about because o f
im provem ents in crystallography, NM R methods and also because recom binant methods
now facilitate the large-scale production o f portions o f the protein that contain
identifiable domains for the use o f structural studies. This ‘dissection’ approach is
leading to a very rapid increase in knowledge o f dom ain 3D structures.
A dom ain is best defined as a spatially distinct structural unit that usually folds
independently o f the rest o f the protein. Domains are repeatedly used as ‘building
blocks’ in functionally diverse proteins. Examples o f domains are depicted in Figure
1.1. They have identifiable amino acid patterns that can be described by a ‘consensus’
sequence. The spread o f a domain type throughout biological systems is likely to have
A n a p h y la to x in
T
C y sta tin -Iik e
C -ty p e le c tin
C y to k in e recep to r N t E G F -lik e
F ib r o n e c tin type-1 F ib ro n ectin t y p e - ll F ib ro n ectin type-111
C -term in a l c y s tin e -k n o t
F o llista tin -lik e
I m m u n o g lo b u lin ‘s u p e r f a m ily ’
K r in g le B PT I in h ib itor uPA recep to r T N F recep to r fa m ily
P -ty p e
Figure 1.1. A figure depicting the secondary structure depicting a helices, p strands and loop regions o f some common domains found in extracellular proteins. The secondary structure o f each domain, derived from its observed pattern o f backbone hydrogen bonding, is shown schematically using MOLSCRIPT (Kraulis, 1991). Taken from the
gene fusion. The presence o f an identified sequence region in another otherwise
unrelated protein and its location between other known dom ain are strong indicators for
proteins o f a m ultidom ain nature. W hile the existence o f exons with compatible phases
at the exon/intron boundaries is good evidence that a dom ain has been spread by exon
shuffling (Patthy, 1991), other means o f domain spreading could exist; thus the term
‘dom ain’ may not only apply to sequence regions w ith com patible phases at exon
boundaries. In the case o f extracellular domains com patible boundary phases are in fact
very often observed. D om ain frequently occur in tandem arrays. M osaic proteins are
com posed o f several different types o f domains. The biological role o f domains may
vary in different settings. For more than 50% o f all described extracellular domain
families, there is at least one member with a known three-dim ensional structure at the
present time. Furthermore, an estimation o f about 40% o f m am m alian protein sequences
are either completely extracellular or have an extracellular part. M any o f them contain
domains (Bork et al., 1996). The intra /extra-cellular division for protein domains is not
always clear since a few domains occur both outside and inside the cell. Other sequence
repeats can occur as units that do not form domains but form super-structures e.g. the
leucine rich repeat (Kobe & Deisenhofer, 1994, 1995).
1.2. Evolution and cellular location o f domains
A m ajor advantage o f building proteins from domains is to facilitate the creation
o f new proteins during evolution. Phylogenetically, ‘o ld ’ proteins such as metabolic
enzymes are usually only composed o f one or two domains, and the creation o f a new
enzyme during evolution required a gene duplication and num erous subsequent point
mutations to acquire a new function for a given fold. A lthough phylogenetically old
enzymes tend not to be m ultidom ain, dom ains can be observed in prokaryotes (Bork et
al., 1996). However, some o f the best-known bacterial m ultidom ain proteins, such as
the extracellular glycohydrolases may have appeared relatively late in evolution as they
are only found in rather specialised bacteria. In eukaryotes, intracellular domains mostly
occur in the cytoskeleton and signal transduction pathways that do not seem to have
equivalent counterparts in bacteria. The best knovm intracellular domains in nuclear
proteins include several DNA binding domains, and the SH2 (Src homology 2), SH3
Figure 1.2. Review o f extracellular domains and their abbreviations. The nomenclature used for dom ains is indicated. It also summarises the dom ain size, the num ber o f di sulphide bonds and the num ber o f tim es the dom ain is found in the sequence database (Bork & Bairoch, 1995). The abbreviations used: in the first column; a 3-5 letter variant for unique identification in databases; second column, a two letter variant. The two letter code and the defined colour codes will be used in subsequent cartoons o f mosaic proteins and other symbols used are explained in figures. The topology o f domains with know n three-dim ensional structure is indicated by the approxim ate arrangement o f their secondary structure elem ents in colum n four: a/p (a/p class), p (all p), a (all a) a+p (a+p) p?a indicates a predom inantly p class domain but an a helix has been observed in some mem bers o f the family. The size is a rough estim ate o f the domain length given in amino acids (rounded up to the nearest 10) depicted in colum n five. The num ber o f cysteines depicted in colum n six, may vary w ithin a dom ain family. 4/6 means 4 or 6 cysteines have been observed, 4-6 m eans that 4 to 6 cysteines may occur. The num ber o f domain occurrences in databases (excluding species redundancies) is mainly based on queries from 1993. The actual num ber m ight now be m uch higher for some o f the
domains. (Diagram adapted from Bork et a l , 1996 and from the EM BL Heidelberg
Abbreviation Full name 3D Size No OCC
5C 2C (AA) Cys
ANATO AT Anaphylatoxin a 70 6 10 APPLE AP Apple - 90 4 20 CIQ CQ Con^lement Clq C-terminal - 140 0-3 10 C345C C3 Conç>lement C3/4/5 C-terminal - 180 4-8 10 CADHE CA Cadherin (3?a 110 0 110 CCP CP CCP (Sushi) (SCR) P 70 4 200 CLECT CL C-type lectin (CTL) P 130 4/6 150 COL4C C4 Collagen IV C-terminal - 110 6 30 COLFI CF Fibrillar collagens C-terminal - 240 8 20 CTCK CK C-terminal cystine knot p?a 90 6/11 90
CUB CU CUB - 110 2/4 30
CYST A CY Cystatin-like a+p 100 0-4 50 CYTR CR Cytokine receptors N-terminal P 90 4/6 40 EGF EG EGF-like p?a 40 6 600 FA58A FA Coagulation factors 5/8 type A - 330 2-4 20 FA58C FC Coagulation factors 5/8 type C - 150 0-2 20 FBG FG Fibrinogen beta/gamma C-terminal - 250 4 20 FIMAC FM Factor I/MAC proteins C6/7 - 70 8/12 20 FNl FI Fibronectin type-I P 40 4 20 FN2 F2 Fibronectin type-II P 60 4 30 FN3 F3 Fibronectin type-III P 90 0 400 FOLLI FS Follistatin-like a+p 50 10 40 FURIN FU Furin-like Cys-rich - 170 26 40 GLA GA Gamma-carboxy-glutamate domain P 60 2 20 HEMOP HX Hemopexin-like - 60 0-2 30 IBPNT IB IGFBP/CTGF N-terminal - 70 12 20 IGSF IG Immunoglobulin "superfamily" P 100 0-6 >999 IGCl 11 Immunoglobulin Cl - 100 0-6 IGC2 12 Immunoglobulin C2 - 100 0-6 IGV IV Immunoglobulin V - 100 0-6 KRING KR Kringle P 80 6 80 KUNIT KU Kunitz/BPTI inhibitor a+p 60 4/6 90 LAMD4 L4 Laminin domain IV (B-type) - 190 8 30 LAMEG LE Laminin EGF-like - 50 8 130 LAMG LG Laminin G-like (A-type) - 190 0-4 60 LAMNT LN Laminin N-terminal (domain VI) - 250 6-10 20 LDLRA LA LDL-receptor class A P 40 6 100 LDLRY LY LDL-receptor YWTD domain - 50 0 140 LINK LK Link (Hyaluronane-binding) - 100 4 10 LRR LR Leucine-rich repeat a/p 25 0 400 LRRN LP LRR preceeding domain (N-flank) - 40 4 20 LRRC LC LRR C-flank - 60 4 30 LY6UP LU Ly6 antigen/uPA receptor P 70 8/10 10 MACPF MA MAC proteins/perforin - 250 8 20
MAM MM MAM - 170 4 10
NOTLI NL Notch/Lin-12 - 30 6 30 PDCM PD P-type (Trefoil) P?a 60 6 30 PKD PK PKDl-like - 80 0 30 SAPOA SA Saposins-like type A - 30 4 10 SAPOB SB Saposins-like type B - 80 6 10
SEA SE SEA - 80 0 20
SOMAB SO Somatomedin B - 40 8 10 SRCR SR Scavenger receptor Cys-rich - 110 6 30 TGFBP TB TGF-beta binding protein - 70 8 20 THYGl TY Thyroglobulin type-I - 50 6/8 30 TNFRC TR TNF family receptors Cys-rich P 40 6/8 40 TSPN TN TSP N-terminal - 210 2/4 20 TSPl T1 TSP type-I - 60 4/6 50 VWFA VA von Willebrand factor type A a/p 200 0-2 60 VWFB VB von Willebrand factor type B - 30 8 10 VWFC VC von Willebrand factor type C - 110 10 30 VWFD VD von Willebrand factor type D - 350 28-32 20 WAP WA WAP (4-disulfide core) - 50 8 30 ZONAP ZP Zona pellucida domain 310 8/10 10 --- ► s ks/cs trans
FA58A
collagen -lik e Sei/Thr-rich keratiiVchondriotin sulfate binding
coiled coil m em brane
SWISS-PROT domain name ANAPHYLATOXIN
APPLE CIQ
C345C IC34
CADHERIN
CCP
C-TYPE LECTIl COL4C
FIBRILLAR COLLÀGENS CTCI
CUB
CYSTATIN-LII
CYTOKINE RECEPTORS N-T EGF-LIKE
F5/8 TYPE F5/8 TYP C FIBRINOGEN FIMAC FIBRONECTIN TYPE-I FIBRONECTIN TYPE-II FIBRONECTIN TYPE-III FOLLISTATIN-LIl FURIN-LIKE GLA HEMOPEXIN-LII IGFB/CTGF IG-LIKE IG-LIKE ^ IG-LIKE IG-LIKE KRINGLE 1
KUNITZ/BPTI TÎÎHIBITOR LAMININ DOMAIN TV
LAMININ EGF-LIJ LAMININ G-LII LAMININ N-TERMINAL LDL-RECEPTOR CLASS LDL-RECEPTOR YWTD LINK LRR
LRR N-FLr»i^i\j-1 .— .
LRR C-FLANKW [LO LY6/UPAR MAC/PERFORIi MAM NOTCH/LIN P-TYPE PKDl-LIKEl _
SAPOSINS-LIKE TYPE A SAPOSINS-LIKE TYPE B SEA
SOMATOMEDIN-B LIKE SRCR
TGFBP
t h y r o g lOBu l i n t y p e
TNFR-CYS TSP N-TERMING TSP TYPE-I VWFA VWFB VWFC VWFD WAP ZP
I
^I shortened lOOaa
Lam Nr
©
region
Modules in other proteins
Fetuin
cŸlfcŸYc7 K ininogen c y] [c y
T N F /N G F recep to r family
] H iS'rich glycoprotein
---C y st.,in s
1-3
Fiepatic receptor
Collecting
-@ ©
lA P p rep ro ^asm o ly sin[ 1 ^ » - C FBgQ FicoHn C D 5, C D 6, M 130, W C I-A
M ac-2, M A M A
di>" ^
speract re c e p to r
< fbg y
Scabrous protein
SR
CüXi5>|-Scavenger recep to r
sem inal fluid protein
P rosaposin/ Sgpl
m acrophage m annose recepto^
IGF type 2 receptor/C a-dep. m annose recep to r
□ n rz r
JIZJL
m
a
n
Pulm on. Surf.ass. prot. SP-B C a-dep. m annose D receptor
Plasma protein 11 V itronectin
lose u receptor
C helonianin
H em opexin
K allm ann's syndrom e protein
cadherin family
fa t tu m o r supressor
30
Rous sarcom a virus protein
plasm odium T R A P/SSP2
plasm odium
circum sporozoite proteins
Pea album in PA-2
Invertebr. Hgb linker chains
3
H ikaru genki
U N C -5
igXig
m m m L
vaccinia virus 35kd p rotein
som e in te g rin a chains tapew orm antigens
integrinPx chain
YLS8
Figure 1.3. Cartoon o f various mosaic proteins. For nomenclature o f domains see
Figure 1.2. (Taken from the EMBL Heidelberg World Wide Web Pages
www.emhl-heidelberg.de).
M odular proteins with sh ort in tracellu lar dom ains
Z P
Z P
Z P
Z p3
Zp2
] +
ZPxU ro m o d u lin
TGF re c . Ill
E n d o g lln
uPA R
Ly-€ I s p g -2
I lOOl
Figure 1.4. Cartoon o f multidomain proteins with short intracellular domains. For nomenclature o f domains see Figure 1.2. (Taken from the EMBL Heidelberg World
Modules in receptors
lOOaa
mm
G-CSF/1L3 receptors
LIF receptors
‘-I.‘oki.nej;eceptor family
Î IL6 receptor
“ ne-^lraradhesiott
molecules
axII/UFO receptor family
FGF, PDGF and other receptor families
EGF receptor family
«gj
Cys-rich
F3t)fE3
<£*)■
M-14
J g X * g X l g > C r a ^ g ) @ @ © ® - I —
c a rb o n ic a n h y d ra s t
300-900
Insulin receptor family
eph, eck, elk, her, ek4
Tie, Tek and _ re ja te ^ p r o te in s
cress kinase
se v e n le ss (Ros)
Dror, Ror1, Ror2
trk-like
D D R family
ret
Met, Ron, S ea
CD^5, PTR
DPTP99A, HPTR, D P '^ IO D , PT P etc.
DPTP
_ P m LAR, DLAR
_PTgt,PTR:
PTP-, PT%, phosphacan
Figure 1.5. Cartoon o f domains in receptors. For nomenclature o f domains see Figure
1.2. (Taken from the EMBL Heidelberg World Wide Web Pages
Selected enzym es flanked by mainly extracellular m odules
T h y ro x id peroxidase [ catalytic d o m ain
P ro stag la n d in sy n th ase lÉ I^ irrjP catalytic d o m ain [~
a glucosidase prec.
S u c ra s e iso m a lta se —
cataly tic d o m a in
isomaltase
A lkaline p h o sp h ata se P C cataly tic dom ain
S tro m ely sin family 1 catalytic dom^
C ollagenase IV fam ily ---1 c a t | [ ^ 0 | j ^ ca t, dom
T h y ro g io b u lin
acety lch o lin e esterase J C J C
fu rinin-fam ily — [ s u b t.- p t^
p ro te in Idnaal-M yosin light c h a in kinase (sm o o th m uscle)
T itin ( > 3 0 OOOaa!)
® - -^®®C5XS®<iiXSXiXS(S
I-b an d A -band
L uciferin 2-m onix)xygenase
c e ll u la r
iQcalUation
extracell.
ER
lysosom al
lum enal
extracell.
extracell.
extracell.
extracell
extracell
intracell.
— p ro te in k it
9
M 'b a n d
extracell.
cataly tic activity; the sam e co lo r does not im ply necessarily homology
Figure 1.6. Cartoon o f selected enzymes flanked mainly by extracellular domains. For nomenclature o f domains see Figure 1.2. (Taken from the EMBL Heidelberg World
Wide Web Pages www.emhl-heidelberg.de).
Modules in matrix molecules
Laminin Ae
Laminin B le
_ £ £ _____ Cartilage m atrix protein
[vS^—
UN C6/netrins L a m N t
Perlecan
UNC52
Rbrortectin
Th ro m b « p o n din s/C O M P U nk protein
F-spomlin ProperJin
♦♦♦♦♦♦
AggrecaivAfersican proteoglycan family
- I reeler " 2 ^ Reelin
SPARC Nidogen
Ascidüan nidogen-like |Tix>iein
-<0r-Figure 1.7. Cartoon o f domains in matrix molecules. For nomenclature o f domains see
Figure 1.2. (Taken from the EMBL Heidelberg World Wide Web Pages
www.embl-heidelberg.de).
Modules in vertebrate collagens
pf//;. a, P(/W ?
a , ( V I I 0 ' 0
-VA X VA
a j ( V I )
400
a ^ ( X V I ) , a i ( X I X ) ?
VA -►■vAmervA'
ffj , a2(VI) a i ( X V ) . ( X V I I I )
ai(VIII)
a, fx;
" ( 5 )
1000
( p r o - ) Oy ( V ) , a , ( X I )
1000
C Tc o l fD ?
[W O
-( p r o - ) (I), (II), a , ( I I I ) , V )
--- ► C ^ C ^
( X I I I )
ai-a„(IV)
i
_______ term inal noH'Collagenous
I I domains (>30aa) f o r which no
homology has been detected
6 0 0 shortened cllagenous
— helix which might
contain interruptions
Cty ( X V I I )
Figure 1.8. Cartoon o f domains in vertebrate collagens. For nomenclature o f domains
see Figure 1.2. (Taken from the EMBL Heidelberg World Wide Web Pages
www.emhl-heidelberg.de).
are probably only a small sub-set o f those that exist. M ore than 30 cytoplasmic domains,
have been described and the list is not exhaustive (Bork et al., 1996). The largest
fraction o f dom ains is extracellular and appears to have evolved w ith the radiation o f
invertebrates. M am m als appear to have the largest fraction o f extracellular proteins and
m ost o f them contain domains. Many o f the extracellular domains contain disulphide
bridges and thus cannot be located in the nucleus or cytoplasm. There are a few
m ultidom ain proteins w ith short intracellular domains (Figure 1.4).
1.3. E xtracellular dom ain and their biological role
A bout 60 abundant domains found in extracellular proteins are depicted in Figure
1.2. The lengths o f these domains vary from about 30 amino acids to over 300, the
larger ones possibly evolving by the duplication o f sets o f the o f smaller domains. The
various dom ains depicted in Figure 1.2 occur in functionally diverse proteins (Figures
1.3-1.10) and interact singly or in concert with others w ith a wide variety o f ligands,
including proteins, peptides and carbohydrates. W hile there often seems to be a
relatively unique role for a given dom ain in intracellular proteins [e.g. SH2 domains
bind phosphotyrosine peptides (Pawson, 1995)], this kind o f direct correspondence is
not always recognisable in domains. Only in a few cases a unique function has been
reported e.g. the carbohydrate-binding function o f (C-type lectin) CLECT or the
membrane-binding function o f y-carboxy-glutamate (GLA) dom ain found in coagulation
proteins (B ork et a l , 1996). For others, it becomes clear that their function varies in
different proteins e.g. the tenth Fn-III dom ain o f fibronectin is involved in cell binding
via an ROD sequence, whereas Fn-III domains in the insulin receptor are involved with
dimérisation. Apparently, different parts o f the domain surface can be used in different
situations to provide interaction sites often for other proteins. Some extracellular
dom ains m ight also have a purely structural role, thus allowing a m osaic protein to
present an interacting surface in an appropriate position, e.g. the N -term inal domains o f
factor H (FH) bind to intact C3b, while a second set o f domains, located in the m iddle
region o f FH, (SC R -6-SC R -18 inclusive), binds to the C3c fragment, and the C-terminal
domains located w ithin SC R -19 and SCR-20 binds to the C3d region (Jokiranta et a l,
1998). Two heparin binding domains in FH have been localised to SCR-7 and SCR-20
has been located in or near SC R -13 o f FH (Pangbum et a l, 1991). It is thought that the
synergistic action o f all these domains enable FH to perform differential control o f
com plem ent activation on activators and non-acticators o f the alternative pathway o f the
com plem ent (Chapter 5). Extracellular mosaic proteins are diverse (Figure 1.3), some
containing short intracellular domains (Figure 1.4). They are widely found as cytokine
receptors (Bazan, 1993; Figure 1.5) and in some cases, m osaic proteins play a clear role
in particular extracellular biological pathways (Figure 1.6), in cell adhesion proteins
(Barclay et a l, 1993) and in the extracellular matrix (Kreis & Vale, 1993 ; V enstrom &
Reichardt, 1993; Figures 1.7 and 1.8).
1.4. Blood C oagulation/Fibrinolysis and the C om plem ent Systems
The two best-studied pathways that involve m ultidom ain proteins are the blood
coagulation/fibrinolysis system and the complement system. The blood coagulation
cascade (Figure 1.9 and 1.10) is a host defence system that is initiated after blood vessel
injury (Patthy, 1993). It comprises alternative pathways in which certain plasm a
proenzym es are successively activated by cleaving the protease domain from the
N-term inal regulatory domains w ithin the m ultidom ain structure. It involves the
form ation o f complexes w ith non-proteolytic plasm a proteins and m embrane-associated
cofactors that ultimately leads to the formation o f a blood clot. D uring wound healing,
the clot is dissolved by proteases o f the fibrinolytic system. Throm bosis and fibrinolysis
are under m ultiple control. In addition to protease inhibitors, one anticoagulant pathway
involves activation o f protein C by throm bom odulin. A ctivated protein C then forms
a m em brane-associated complex w ith plasm a protein S that inactivates factor VII. The
level o f protein S is regulated by C4b binding protein, a regulator that also inhibits the
com plem ent system. The com plem ent cascade (Figure 1.10 and 1.11) is a defence
system against infectious agents and plays an im portant role in inflam m ation. Through
the form ation o f the m em brane attack complex (M AC), com plem ent is able to lyse
m em branes o f infectious organisms. The classical pathw ay is triggered by
immunoglobulins that recognise foreign organisms, the lectin pathw ay is triggered by
com plex carbohydrate, whereas the alternative pathway is triggered by a wide variety o f
com pounds and cell surfaces (Figure 1.12). A lthough the proteins involved are
Blood coagulation
intrinsic pathway
AP X AP X AP X A P
Prot.C
Prot.S
steroid bind, proteins
F V I l l
FA58A FA58A
e x trin s ic
pathway
FA58A FA58A
throm bom odulin
transdutam inase
F X I I I a F X I l I p prothrom bin
fibrinogen
' cc
a — I
I p —C FBG^
I y — - C F B G ^
---' fibrinolysis
tPA
uPA __
plasm inogen ^
apolipoprotein (a)
3 2
H G F/SF H GFL
lOOaa
H G F activator S e r P r
Figure 1.9. Cartoon o f the mosaic proteins involved in blood coagulation and
fibrinolysis with individual pathways highlighted by blocks. Some regulatory proteins and proteins with a similar modular architecture to proteins within the cascades are also blocked in the aqua at the lower part o f the diagram. For nomenclature o f domains see Figure 1.2. (Adapted from Bork et a i, 1996).
Complement system and regulators
h a p to g lo b in 2 fa c to r C h o rsesh o e c r a b
classical pathw ay
C I r
CIS
€ 4 — ► I
C 2 # # # O C I ^
— ^ —
“ I.
le ctin pathw ay
M B P
M A SP
!u@*B€cu
C 4 — ► j
C2 # # # 0 # ^ M E n 3 g |
a ltern ativ e pathw ay
propcrtiin
C3
€ 3
C5
1 m acro g lo b u lin family m
;
1 m acro g lo b u lin family lÂfi l T 3 3 i a
1
1
1 m acro g lo b u lin family 1 C345CJ
08
« # 3
lipocalin
M A C P F
M AC
p e rfo rin | [MACPF% ] # | |
Ly-6 I sp g -2
H
P2 g ly co p ro tein
u p d ated from FEES 307 (9 2 )4 4 -5 4 an d K BM R eid, personal co m m u n ic atio n
C 4B P
(h u m a n ) M C P
IL2
re c e p to r
F igure 1.10. Cartoon o f the complement cascade and some o f its regulators. Individual pathways are highlighted by blocks. Proteins with a similar modular architecture to proteins within the cascades are also displayed. For nomenclature o f domains see Figure 1.2. (Taken from Bork et al., 1996).
C lassical pathw ay activation
A ltern ative pa th w ay activation
0 3 0 4
Im m une com plexes
0 4 a 0 3 a
0 3 b 0 4 b
02
0 4 b 2 C 3 b B
M B P /M A S P M B P 'M A S P
0 3
B a 0 2 b
C om plex carbohydrates
0 3 a
0 3 b
0 5
C 4 b 3 b 2 a
C 5 a
0 5 b
0 6
0 7
0 8
0 9 ( 0 9 ( 0 9 ( 0 9 ( 0 9 0 9
/ 0 5 b 6 7 8 9 V M A O
F igure 1.12. The activation steps o f the complem ent system. The classical pathway (left) is triggered by immune complexes and the lectin pathway is triggered by complex carbohydrates (left), while the alternative pathway (right) is triggered by a wide variety o f compounds and cell surfaces. The number o f C9 molecules (n) within the C5b6789„ complex can vary between 1 and 18. Enzymatic cleavage is indicated as solid orange lines and the enzymatically active components are shaded orange. (Reproduced from Law and Reid, 1995).
o f both these cascades is not yet completely understood. Only approximate regulatory
functions such as Ca-dependent mem brane-binding can be assigned to specific domains
although assigning regulatory function to individual dom ains or groups o f domains is
increasing e.g. for FH, a specific biological function cannot be directly associated with
all domains, partly due to lack o f information and partly because some domains do not
function autonomously. N onetheless, structural inform ation is providing considerable
insight into dom ain function.
1.5. Some general observations on domains 1.5.1. Introduction
Generally speaking, structure determ ination o f sequence homologues, where
several structures have been determined for one particular domain, has demonstrated
considerable similarity in the core structure. Insertions and deletions are usually
accommodated only in surface loop regions, although some secondary structure elements
are subject to change. Homology m odelling o f one m em ber based on an atomic
structure from another is usually viable. This has been extensively studied in the case
o f the immunoglobulin family (Harpaz & Chothia, 1994). In all the know n structures o f
EGF and Fn-I domains, the observed structural changes betw een mem bers o f a family
occur mainly in the length o f the loops between P-strands. In contrast, proteins with
Ig-like topology not only have changes in their loop lengths but also have various strands
added to a core structure (Bork et al., 1994). D om ain w ith less regular secondary
structure {e.g. Kringle repeat (KR) and Fn-II) appear to have a higher proportion o f
conserved residues which stabilise their fold.
1.5.2. Conservation and variability o f disulphide bridges
M ost o f the extracellular domain contain disulphide bridges, w hose probable
roles are to increase the stability o f relatively small dom ains and to protect against
proteolysis. Structural analysis has suggested that a correlation m ay exist between the
size o f the hydrophobic core o f a domain and the num ber o f disulphide bridges which
stabilise its fold (Bork et a i, 1996). A lm ost all conserved cysteine residues in various
extracellular domains form disulphide bridges, m ostly intra-dom ain ones. Cysteine
domain by sequence analysis. W ith the exponential increase o f sequence data, more and
more exceptions to this rule have been identified. A w ell-know n exam ple is the
immunoglobulin (Ig) family that contains a subgroup that does not contain the otherwise
conserved disulphide bridge (W illiams & Barclay, 1988). The 3D structures o f several
cell surface proteins containing Ig-like dom ains clearly dem onstrate the switch o f
disulphide bridges between strands (Jones, 1993; Bork et al., 1994; W agner & Wyss,
1994). Dom ains o f the Fn-III superfamily usually do not contain disulphide bridges but
the structure o f the neuroglian protein o f D rosophila shows that they sometimes do
(Huber et al., 1994). Domain that contain the Fn-III consensus have been found in
CD45 and the numerous cysteines therein probably also form disulphide bridges. The
N -terminal domains o f various cytokine and related receptors are structurally very
similar to Fn-III domains and are possibly related in evolution. In the growth hormone
receptor, three disulphide bridges are formed w ithin its Fn-III domain, w hile sequence
related dom ains in other receptors only contain two. The location o f the cysteine
residues in related sequences also suggests different strand connections (Bork et a l,
1996). The exam ples above are a few from an exhaustive list.
In summary, disulphide bridges can stabilise certain conform ations and are thus
important and usually conserved structural features. H ow ever the loss, addition or
change o f position o f the S-S bridges frequently occurs. These changes m ight lead to
different stable topologies where the constraints on supporting hydrophobic core
residues m ight change, leading to the form ation o f structurally related domains that are
no longer detectable at the sequence level.
1.5.3. Com m on topological features o f different domains
As the domain size increases, the fraction o f core-stabilising disulphide bridges
decreases and the existence o f a hydrophobic core becomes more obvious. Another
striking feature o f extracellular domains is that many o f them are p-sheet and several
larger ones have very similar topology. This is especially the case w ith the Greek key
architecture o f the Ig, Fn-III, cytokine receptor N-term inal (CYTR) (Figure 1.1) and the
C aspase-activated DNase N-terminal (CAD) domain. A com parison was made o f 23
than 25% pairw ise residue identity (Bork et al., 1994). A structural core o f four
P-strands (B, C, E and F) was identified in all three types w ith three or five additional
strands (A, C , C", D and G) depending on the Ig-fold type (s, h, c and v). The structure
o f the additional p-strands is highly variable. Structure com parisons o f the different
domain types were carried out using a program that m axim ises a geometrical similarity
score. Analysis o f the pairwise structural similarity scores revealed three m ain structural
clusters. However, it was concluded that the presence o f the conserved structural core
did not imply a similar hydrophobicity pattern among the different sequence families.
Only strand F appeared to retain conserved hydrophobic features. It was observed that
there are often conserved aromatic positions in one or m ore strands o f the common core
although they do not correspond to equivalent positions in the topology. Loop lengths
vary in all positions between, and even within, sequence fam ilies o f the Ig-type. The
high degree o f structural flexibility outside the comm on core and the extreme variability
o f side-chain packing inside the core do not support a protein folding pathw ay common
to ail m em bers o f the structural class. M utation rates o f Ig-like dom ains in different
proteins vary considerably. Disulphide bridges, thought to contribute to structural
stability, were not invariant in num ber and location w ithin a subclass. In conclusion,
this kind o f analysis suggests that a com m on topology is achieved by fundamentally
different sequences. It is not known w hether different sequences have evolved from
each other or whether the fold was invented several tim es independently during
evolution. Three functionally related, but structural distinct protease inhibitors (cystatin,
Kunitz and Kazal) share some com m on topological features in that they all contain a
small antiparallel P-sheet with a surrounding a-helix. A s w ith m any protein folds, it
may not possible to discriminate divergent evolution from convergence to an
energetically stable fold.
1.5.4. D om ain assembly
The database for individual domain structures is extensive and knowledge about
the ways in w hich some domains are fitted together is growing. Only a limited set o f
protein folds exist (Orengo et a l, 1994) and this may confer a lim ited num ber o f ways
in w hich dom ains fit together. The ultim ate question in hand is w hether general rules
the geometry o f different extracellular modular proteins, com posed o f at least two
homologous domains o f know n structure (Chapter 5 and 6). In this analysis, individual
dom ain are treated as ellipsoid shapes and the geometry o f the double domain is
described by the rotational parameters linking one dom ain to the other. In general, it
seems that rules are hard to discern for the ways that dom ain fit together. There are
trends that can be noted e.g. tw ist angles o f around 130° for Ig domains, but it appears
that proteins can readily change the ways in w hich a given dom ain pair is oriented. The
structures o f the individual domains are predictable but the relative orientation o f a pair
can be changed by changing the length o f a linker peptide or a few surface residues near
the linker peptide. This means that a m ultidom ain protein structure can provide great
variety in the spatial position and type o f surface that can be presented. The relative
positions o f domains in a m ultidom ain protein can also be readily changed by
environmental changes (ligand, concentration, pH, etc.) thus providing a potential
regulation m echanism for function. Examples include the changes in angles observed
betw een the receptor Fn-111 domains o f the prolactin receptor and the growth hormone
receptor when growth hormone binds (Somers et al., 1994) or the SH3 and SH2 domain
rearrangement in intracellular signalling proteins induced by phosphorylation (Pawson,
1995).
1.6. Conclusions
In spite o f an increase in available sequences and structures, it is becom ing clear
that the total num ber o f extracellular dom ain types is probably lim ited to about 100 and
the structures o f many o f these are already known. W hat rem ains uncertain in many
cases is the ways in which m ultidom ain and mosaic proteins are assem bled and how they
present their appropriate surfaces for interaction w ith other proteins. The present level
o f structural inform ation is sufficient to construct, to a first approxim ation, 3D-models
for m any m ultidom ain proteins. This is leading to a new era in the determination o f
biological function since site-directed amino acid changes and dom ain deletion and
swapping experim ents can now be done in a m uch m ore rational way than was
previously possible. A new dimension in m ultidom ain research has been added w ith the
com pletion o f the genome sequencing program m es for several m ulticellular organisms.
proteins will enable a more complete comparative analysis. W ith the various genome
projects generating huge am ounts o f sequence data, database searches for possible
domains and m olecular modelling techniques will be very useful in determ ining putative
structure and functions o f the open reading frames. This should give unprecedented
inform ation about the phylogenetic questions concerning the spread o f domains. More
importantly, there is the possibility o f unravelling m any o f the functional networks in
w hich dom ains are found: perhaps unveil the ‘language’ o f domains. The purpose o f
this PhD thesis is to describe experimental studies o f elongated m ultidom ain proteins