Donika Ivanova
1, D, Rumiana Bakalova
2, 3, A, D–F, Dessisslava Lazarova
2,
Veselina Gadjeva
1, D, Zhivko Zhelev
1, A, E, FThe Impact of Reactive Oxygen Species
on Anticancer Therapeutic Strategies
Wpływ reaktywnych form tlenu na antyrakowe strategie terapeutyczne
1 Deparment of Medicinal Chemistry and Biochemistry, Medical Faculty, Trakia University, Stara Zagora,
Bulgaria
2 Medical Faculty, Sofia University, Sofia, Bulgaria
3 Molecular Imaging Center, National Institute of Radiological Sciences, Chiba, Japan
A – research concept and design; B – collection and/or assembly of data; C – data analysis and interpretation;
D – writing the article; E – critical revision of the article; F – final approval of article; G – other
Abstract
Over 50 years of experience in free radical biology and medicine shows that normal cells of healthy mammals are characterized by a low steady-state level of reactive oxygen species (ROS) and a constant (reference) level of reducing equivalents. A lasting increase of ROS above the critical level leads to permanent oxidative stress in the cells. This could cause genomic instability and mutations, which are responsible for adaptation of cells to oxidative stress and their survival in an oxidative environment. In turn, these events could provoke malignancy. It is widely accepted that the balance between ROS and reducing equivalents in cells and tissues determines their redox status. The evaluation of tissue redox status has great diagnostic potential in cancer, as well as prognostic potential for cancer therapy, and could significantly contribute to the planning of appropriate treatment and to increasing the patients’ quality of life. The conventional therapeutic strategy is based on drugs that increase ROS generation and induce apoptosis in cancer cells. However, this therapeutic approach has serious disadvantages: the expression of various toxic side effects in normal (non-cancer) tissues. The current review describes the basics of free radical biol-ogy in carcinogenesis. The authors emphasize the different redox status of normal and cancer cells, which permits the use of this parameter as a new therapeutic target. The authors also outline some directions for the development of promising therapeutic strategies based on the regulation of redox signaling using combined therapy. The review is intended for a broad readership – from non-specialists to researchers in the field of cancer biochemistry and pharmacy (Adv Clin Exp Med 2013, 22, 6, 899–908).
Key words: cancer; reactive oxygen species, redox signaling, therapy. Adv Clin Exp Med 2013, 22, 6, 899–908
ISSN 1899–5276
REVIEwS
© Copyright by wroclaw Medical University
Carcinogenesis is a multi-step process, accompa-nied by accumulated genetic alterations in the somat-ic cells. The targeted (initiated) cells in the primary cancer locus change their behavior. They are charac-terized by a selective growth advantage as a result of mutations in genes responsible for controlling cellu-lar proliferation and death. These mutations include the up-regulation of proto-oncogenes and down-reg-ulation of tumor suppressor genes [1, 2].
It is widely accepted that reactive oxygen spe-cies (ROS) are also among the main triggers and/ /or mediators of carcinogenesis. It is assumed that
oxidative stress contributes to the initiation of cel-lular malignancy and the progression of cancer by causing a genomic instability [3–6].
The Chemical Nature and
Major Endogenous Sources
of ROS
D. Ivanova et al.
900
1 or more unpaired electrons in their outer molec-ular orbitals (e.g., superoxide radical [O2–], nitric
oxide radical [NO.], hydroxyl radical [OH.] and
hy-droperoxyl radical [HOO.]); and 2) the non-radical
type, which have unpaired electrons, but are highly reactive and can be converted to the radical type of ROS (e.g., hydrogen peroxide [H2O2], ozone [O3],
trioxidan [HOOOH] and nitric oxide [NO])[7]. ROS are generated either by exogenous sourc-es such as pollutants, tobacco smoke, iron salts, ra-diation, etc., or by endogenous cellular sources. Cells produce ROS through multiple mechanisms. Major endogenous sources of ROS are 1) 4-ele-ctron reduction of oxygen in the mitochondria; 2) NADPH-dependent oxidases; 3) P-450-dependent monooxygenases; 4) xanthine oxidase; 5) lipoxy-genases, cyclooxylipoxy-genases, etc. Since mitochondria and NADPH-dependent oxidase complexes are the basic endogenous sources of ROS in carcino-genesis, the current article focuses mainly on these 2 factors (Fig. 1) [8, 9].
The mitochondrial electron-transport chain (ETC) consists of 4 multi-protein complexes (I–IV) embedded in the inner mitochondrial membrane. Complex I and complex II oxidize NADPH and FADH2 respectively, and transfer the resulting
electrons to ubiquinol, which carries the electrons to complex III. Complex III shunts the electrons across the intermembrane space to cytochrome c,
which brings the electrons to complex IV. Com-plex IV then uses the electrons to reduce oxygen to water. Complexes I, II, and III generate superox-ide radicals as a result of the electron flux through them. Complexes I and II generate ROS within the mitochondrial matrix, while complex III generates superoxide and releases it into either the inter-membrane space or the mitochondrial matrix. The superoxide formed can subsequently be converted to other ROS, e.g. hydrogen peroxide. Hydrogen peroxide is a lipophilic substance and easily pene-trates mitochondrial and plasmatic membranes, reaching the cytosol and extracellular environ-ment. Thus, if there is mitochondrial dysfunction accompanied by excessive generation of superox-ide and hydrogen peroxsuperox-ide, ROS can be found in the whole intracellular and extracellular space.
The other major endogenous source of ROS is the NADPH-oxidase pathway (Fig. 2) [10–12]. NADPH oxidases catalyze the reduction of oxy-gen with the production of superoxide, using NA-DPH as a reducing equivalent. The NANA-DPH ox-idase complex was initially found in phagocytes. NADPH oxidases are mainly expressed on the cell surface. The activation of NADPH oxidases occurs when the cytosolic components migrate to the cel-lular membrane. These enzymes generate super-oxide at the plasmatic membrane and release it into the extracellular space, where it is converted
into hydrogen peroxide. The extracellular hydro-gen peroxide can subsequently penetrate through the plasmatic membrane into the cytosol. Recent studies suggest that NADPH-oxidase generation of superoxide can also occur in endosomes and the endoplasmic reticulum [13]. The superoxide gene-rated in these intracellular compartments can be converted into hydrogen peroxide, which migrates into the cytosol and extracellular space.
Therefore, any dysfunction in the mitochon-dria and/or up-regulation and activation of the NADPH-oxidase complex can induce abnormal generation of ROS in somatic cells. Both events (mitochondrial dysfunction and up-regulation of the NADPH-oxidase complex) are observed in var-ious cancers and they have a major impact on the planning of therapeutic strategies. This hypothesis will be discussed bellow.
The Physiological Functions
and Harmful Effects of ROS
in Cells
Low levels of ROS can activate cellular prolife-ration or survival signaling pathways via kinase ac-tivation and/or phosphatase inhibition, as well as via regulation of proteinases, including matrix me-talloproteinases [14]. The most common mecha-nism of these regulatory processes is the interac-tion of ROS with cysteine residues, which results in the formation of disulfide bonds and the sub-sequent activation of signal transduction. In the context of carcinogenesis, it is important to note that ROS are considered to be regulators of the following major signaling mechanisms: extracellu-lar signal-regulated kinases (ERKs), mitogen-acti-vated protein kinases (MAPKs), phosphoinositide
D. Ivanova et al.
902
3-kinases (PI3Ks) and transcription factors such as hypoxia-inducible factors (HIFs). All of these in-tracellular components take part in cell prolifera-tion, growth and survival [3, 6, 14, 15].
The dysregulation of the MAPK-signaling path-way can lead to uncontrolled proliferation or per-manent cell cycle arrest [16]. These 2 processes are competitive and have opposite effects on cell surviv-al. The level of ROS in cells and their environment is a very important regulatory factor, which is in di-rect cross-talk with MAPK activity and determines which of these 2 events will occur [6, 16, 17].
A similar relationship exists between the level of ROS and the ERK-signaling pathway, which is also involved in cell cycle arrest [3, 16, 17]. ERK- -sustained activation of cyclin D1 expression is re-quired throughout the G1 phase in order to pro-ceed to the S phase of the cell cycle. Conversely, a high level of long-term ERK activation can pro-voke cell cycle arrest. It has been found that ROS inactivate the phosphatases responsible for the de-phosphorylation of ERK and thus ensure sustained ERK activation [3, 16, 17].
The PI3K-signaling pathway is necessary for a number of cellular processes, including cell growth, survival, proliferation and mobility. This pathway can be affected by the redox status of the cell. The best known direct target in the PI3K pathway for oxidants is the tumor suppressor gene phosphatase and tensin homolog (PTEN). Hydro-gen peroxide inactivates PTEN through the for-mation of a disulfide bond between the active site cysteine (Cys124) and a vicinal cysteine (Cys71) [3, 6, 18]. This process facilitates the subsequent activation of oncogenes.
High levels of ROS can directly induce oxida-tive damage in lipids, proteins and nucleic acids. ROS can participate in multistage carcinogenesis from initiation to malignant conversion, by caus-ing oxidative DNA damage and mutations in pro-to-oncogenes and tumor suppressor genes, and subsequent activation of signal transduction path-ways [1, 6, 14]. The p53 tumor suppressor gene and blc-2 (B-cell lymphoma 2) proto-oncogene are the most extensively studied genes known to mo-dulate cancer-related apoptosis [1, 6, 14].
At the initial stage of disease, cancer cells usu-ally exhibit genetic instability and a significant in-crease in ROS level due to a “vicious circle” [6]. For example, ROS induce genetic mutations (es-pecially in mitochondrial DNA), which subse-quently lead to metabolic dysfunction and addi-tional ROS generation. As a genomic guard, the p53 protein plays a major role in removing oxida-tive damage from nuclear DNA and mitochondri-al DNA (mtDNA). P53 prevents oxidative genet-ic mutations and genomgenet-ic instability. Moreover,
p53 is also a transcription factor that regulates the expression of many pro-oxidant and antioxidant genes. The down-regulation and loss of p53 is as-sociated with redox imbalance, increased produc-tion of ROS, mutagenesis and aggressive tumor growth. As Trachootham et al. wrote: “This is con-sistent with the high incidence of p53 mutation or loss of p53 function in over 50% of human can-cer” [6], and especially in advanced and early stage of cancer [14, 19].
ROS induce almost all forms of DNA damage, including modifications of the nucleotide bas-es, strand breakage and DNA protein cross-links, but the specific spectrum of products depends on the type of ROS. ROS type-specific mutations are involved in the genesis of cancer. ROS may also play a key role in the development of cancer by inducing and maintaining oncogenic phenotypes of cancer cells [6, 14, 19]. Currently, oxidative stress is widely accepted as a major contributor to carcinogenesis.
Antioxidant Defense
Systems and Carcinogenesis
The effects of ROS are balanced by antioxi-dant enzymes (e.g., superoxide dismutase [SOD], catalase, glutathione peroxidase, other peroxidas-es, etc.) and non-enzymic antioxidants (e.g., ascor-bate, tocopherols, tocotrienols, carotenoids, natu-ral flavonoids, melatonin, etc.) [7, 20].
Fig. 3 shows a schematic illustration of cellu-lar redox homeostasis and the effects of ROS sca-venging enzymes.
Fig. 3. Cellular redox homeostasis (according to Trachootham et al. [6]), Mito-ETC – mitochondrial electron-trans-port chain; SOD – superoxidedysmutase; GPx – glutathione peroxidase; GR – glutathione reductase; TRXox – oxi-dized thioredoxin; TRXred – reduced thioredoxin; GRXox – oxioxi-dized glutaredoxin; GRXred – reduced glutaredoxin; NOS – nitric oxide synthase; XO – xanthine oxidase; NOX – NADPH-oxidase
Catalase catalyzes the decomposition of hy-drogen peroxide to water and oxygen [7]. Cata-lase has one of the highest turnover rates for all enzymes: one molecule of enzyme can convert ap-proximately 6 m molecules of hydrogen peroxide to water and oxygen within a min [23]. The prog-nostic role of catalase in carcinogenesis is also dis-putable and currently it is not considered a valu-able therapeutic target.
It is widely accepted that glutathione peroxi-dase is the major redox-modulating enzyme in mammals. Along with other peroxidases, peroxi-redoxins, and thiol-reductases, glutathione per-oxidase regulates the pool of reduced glutathione (GSH) – one of the major reducing equivalents in cells and body fluids [6, 7]. Many phospho-pro-teins and proteinases with GSH-containing cys-teine residues undergo oxidation with the forma-tion of disulfide bonds (GSSG), which is a trigger for the activation of signal transduction pathways. The oxidation of GSH to GSSG is a reversible pro-cess. The disulphide reductases (also known as re-dox proteins – thiorere-doxin [TRX] and glutare-doxin [GRX]) convert the disulphides to dithiols within the target protein and thus restore the re-duced glutathione. An example of this redox-based
signaling is the activation of apoptosis signal-regu- lating kinase 1 (ASK1). ASK1 is bound to a re-duced TRX in its inactive form. ROS oxidize TRX, causing its disassociation from ASK1, which allows the auto-phosphorylation of ASK1 and its activa-tion [3, 24].
Various types of cancers are characterized by different expression of antioxidant enzymes, as well as by different levels of non-enzymatic oxidants. It is important to note that the total anti-oxidant capacity has to be considered in cancer di-agnostics and therapy.
ROS/Antioxidant Balance
in Normal Cells
and in Cancer Cells – Redox
Status as a Diagnostic Marker
and Therapeutic Target
D. Ivanova et al.
904
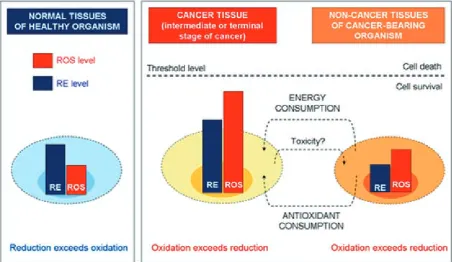
The cells and tissues of healthy mammals are cha-racterized by a low steady-state level of ROS and some constant level of reducing equivalents, while cancer cells are characterized by increased levels of ROS and reducing equivalents (Fig. 4) [25]. Cancer cells are also characterized by abnormal production of reducing equivalents (e.g., NADPH, NADH) as a result of accelerated glycolysis (the warburg ef-fect) and pentose phosphate cycle. However, these reducers are rapidly consumed to maintain accel-erated anabolism, which is necessary for cell pro-liferation and immortalization.
As Barrera wrote: “A moderate increase in ROS can promote cell proliferation and differ-entiation” [28]. However, extremely excessive amounts of ROS can cause irreversible oxidative damage to biomacromolecules, apoptosis and cell death [6]. Therefore, maintaining ROS homeosta-sis at low levels is crucial for normal cell survival, while a moderate enhancement of ROS is associat-ed with abnormal cancer cell growth and disrup-tion of redox homeostasis [6].
Prolonged operation of cells at abnormal steady-state levels of ROS provokes genetic mu-tations, which makes them well adapted to oxida-tive stress. This process is at the basis of malignant transformation. Cancer cells are usually characte-rized by increased antioxidant capacity, as noted in the authors’ earlier publication [20]. Cells that survive intrinsic oxidative stress mobilize a set of adaptive mechanisms, which not only activate ROS-scavenging systems to cope with the oxida-tive stress, but also inhibit apoptosis. As De Luca
et al. wrote: “Such adaptation contributes to malig-nant transformation, metastasis and resistance to anticancer drugs” [29].
Harris et al. reported that normal epithelial cells, exposed to low but continuous levels of ex-ogenous oxidants, become resistant to subsequent oxidative stress even at a higher level [30]. This ob-servation suggests that cells can adapt to survive under certain levels of oxidative stress. As Barre-ra wrote: “Those cells that survive oxidative stress are likely to have acquired adaptive mechanisms to counteract the potential toxic effects of elevated ROS and to promote cell-survival pathways” [28]. For example, Nonn et al. reported that HR as on-cogene-transformed cells, which exhibit increased superoxide and hydrogen peroxide levels, are al-so characterized by increased levels of antioxidants (e.g., peroxiredoxin-3 and thioredoxin peroxidase) in comparison with their non-cancer parental cells [31]. It seems likely that their enhanced anti-oxidant capability serves as a compensatory mech-anism to evade ROS-induced apoptosis. Thus, the abrogation of this adaptation mechanism could be an attractive strategy to preferentially affect cancer cells and may have promising therapeutic implications [6].
The authors’ recently published data on ex-perimental animals showed that reduction exceeds oxidation in the tissues of healthy organisms, while oxidation exceeds reduction in the tissues (both cancer tissues and non-cancer tissues) of cancer- -bearing organisms [25–27]. Moreover, the tissue redox status is very sensitive to cancer progression
and anti-cancer therapy [27]. These data suggest that tissue redox status could be a diagnostic mar-ker, a therapeutic target, and a hallmark for evalua-tion and for planning the therapeutic strategy.
Therapeutic Strategies
in Cancer Based on Redox
Signaling
The targeting of unique biochemical altera-tion in cancer cells might be a feasible approach to achieving therapeutic activity and selectivity and perhaps to prevent the development of drug resistance [6].
As Beck et al. wrote: “Agents such as arsenic trioxide, which impair the function of the mito-chondrial respiratory chain, are known to increase the production of superoxide” [32]. Radical inter-mediates are also formed by compounds known as “redox cyclers”, which may react with flavoprotein reductases such as cytochrome P450-reductase and NAD (P) H-dependent quinone oxidoreduc-tase (NQ01). Motexafin (an inhibitor of thiore-doxin reductase and ribonucleotide reductase) and anthracyclines (e.g., daunorubicin and doxorubi-cin – broad antibiotics used in conventional can-cer chemotherapy) are good examples of redox cy-clers [6, 33, 34]. Anthracyclines are redox-active because of their quinine-hydroquinone structure. They are widely used to treat various malignant tu-mors, but their clinical efficacy and use are often limited by side effects due to elevated ROS produc-tion in non-cancer tissues. These anticancer drugs are well-known generators of superoxide. The side effects of doxorubicin may also be due to intracel-lular chelation of iron, which may trigger a Fen-ton-type reaction and subsequent generation of highly reactive hydroxyl radicals [6, 33, 34].
Bleomycin and pharmorubicin are anticancer antibiotics that are most effective for treatment of lymphomas, certain types of squamous-cell carci-nomas and testicular carcicarci-nomas. Their side effects constitute one of the major obstacles to the use of these antibiotics as anticancer drugs: Pharmorubi-cin is cardiotoxic and bleomyPharmorubi-cin induces pulmo-nary toxicity as a result of increased generation of ROS not only in cancer cells, but also in non-can-cer cells and tissues [35].
Therapeutic selectivity is essential in cancer treatment. Some compounds exhibit a potent abi-lity to promote ROS generation predominantly in cancer cells and show promising anticancer activ-ity in vitro and in vivo [6].
As Shaaban et al.: “To maximally exploit the ROS-mediated cell-death mechanism as
a therapeutic strategy, it is possible to use a combi-nation of drugs” [36]. For example: 1) a drug that induces ROS generation in cancer cells, but not in normal cells; and 2) a drug that suppresses anti-oxidant capacity in cancer cells, but not in normal cells. Zhou et al. reported that a combination of the ROS-generating agent arsenic trioxide and SOD- -inhibitor 2-methoxyestradiol (2-ME) shows po-tent activity against primary chronic lymphocytic leukemia (CLL) and significantly increases the cy-totoxicity of 2-ME in CLL cells that were resistant to 2-ME alone [37]. It has also been established that a combination of arsenic trioxide and GSH depletion, mediated by ascorbic acid, is effective in the treatment of multiple myeloma [38, 39].
Beta-phenylethyl isothiocyanate (PEITC) is a natural compound known to increase intracel-lular ROS levels, preferentially killing ovarian epi-thelial cells, but not normal cells; injections of PEITC in mice grafted with Ras-transformed ovar-ian epithelial cells prolonged their survival [3] This suggests that modulating ROS levels is a promis-ing potential therapeutic strategy in cancer. These findings have been confirmed by the observation that mice grafted with an ovarian cancer cell line expressing a high Akt activity showed a reduction in tumor size after treatment with rapamycin and PEITC [3]. Rapamycin analogs are currently in use in clinical trials.
The presence of stable nitroxyl radical 2,2,6,6-tetramrthylpiperidine-1-oxyl (TEMPO) in an-titumor compounds such as 1-(2-chlororthyl)- -3-cyclohexyl-1-nitrosourea (CCNU) reduces their toxicity and increases radio-sensitizing prop-erties, imparting a beneficial influence on the an-tineoplastic properties of this drug. Nitroxide-la-beled analogs of CCNU showed lower toxicity and higher anticancer activity in experimental tumor models [35]. It has been reported that nitroxide- -labeled nitrosourea increases the cytotoxic effect of bleomycin and pharmorubicin in human hema-tological neoplasms, but not in normal lympho-cytes [35]. Nitroxide labeling could also decrease the toxic side effects of these antibiotics by sca- venging superoxide. Thus, the combination of bleomycin (at lower doses) with nitroxide-labeled nitrosourea would decrease bleomycin-induced pulmonary toxicity [35]. Several ROS-generat-ing chemical substances are currently undergoROS-generat-ing clinical trials as a part of a combined anti-cancer therapy.
D. Ivanova et al.
906
promising. As Kamada wrote: “CIRT is well-local-ized and superior-depth dose distribution of irra-diation and the concomitant local generation of ROS in addition to less repairable radiobiological effects in the cancer area. Since 1994, when the 1 st clinical study of cancer therapy with carbon ion beams was started, about 50 clinical studies have been completed safely and effectively. These stud-ies revealed that intractable cancers such as inop-erable bone and soft-tissue sarcomas can be cured safely in a shorter overall treatment time, as can cancers in the head, neck, lung, liver, prostate, and postoperative pelvic recurrence of rectal can-cer. The number of patients receiving CIRT has reached 6.000, and this anti-cancer therapy was approved as a highly advanced medical technology in 2003. Based on these experiences, a new-genera-tion beam delivery facilities such as a 3D scanning method with a pencil beam was developed and put into operation since May 2011” [40].
Currently there are serious limitations to the widespread application of CIRT because of the need for specific apparatus (e.g., heavy ion me- dical accelerators) and the high costs. Until CIRT becomes a conventional therapeutic technology, ef-forts are focused on improving existing, widely used ROS-modulated anti-cancer strategies such as con-ventional cytostatic chemotherapy and/or radiother-apy using X-rays. The ROS generated by antican-cer agents and radiotherapy are effective in killing cancer cells, but they can alter other cellular path-ways and to provoke various side effects in non-can-cer cells and tissues. The most serious side-effects observed in chemotherapy and radiotherapy, are nephrotoxicity, cardiotoxicity and ototoxicity [41].
The development of oxidative stress in the non-cancer tissues of a cancer-bearing organism is a serious problem for both chemotherapy and ra-diotherapy. Combining radiotherapy and/or stan-dard chemotherapy with antioxidants that increase the protection of non-cancer tissues against ox-idative stress should be explored as a therapeutic strategy [6]. However, it would conflict with ROS-mediated apoptosis and necrosis in cancer cells. As Maiti et al. wrote: “The clinical benefit of using antioxidant supplements along with chemotherapy and radiotherapy is highly debatable, and not con-clusive. Some of the clinical studies suggest that the antioxidant supplemented group had a worst sur-vival rate, than the group who did not use anti-oxidant supplements. Although in some cases, use of antioxidant has fewer side effects leading to less damage to normal tissues, but with a decrease in the overall survival rate. However, it is also be-lieved that antioxidant supplemented reduction of side-effects depends mainly on using specific anti-cancer drugs for certain anti-cancers” [41].
The reports of other groups also support this assertion [42, 43].
It is important to note that persistent ROS ge-neration as a result of chemo- and/or radiotherapy can also activate the antioxidant adaptive response system and thus increase the resistance of cancer cells, which will negatively affect therapeutic out-comes [44]. For example, radiotherapy leads direct-ly to the ionization of water, then to the generation of ROS, which are amplified by mitochondria, ge-nerating even larger quantities of ROS. Irradiation-induced ROS activate several proliferative and anti-apoptosis pathways (e.g., the MAPK pathway, the release of VEGF, the increase of survivin, etc.), re-sulting in cytoprotection [44–47]. Therefore, as Di-mova et al. wrote: “Antioxidant adaptive response system, activated by radiation, may be highly re-levant to tumor response during standard clinical dose-fractionated radiation therapy” [44].
References
[1] Kahlos K, Soini Y, Pakko P, Saily M, Linnainmaa K, Kinnula VL: Proliferation, apoptosis, and MnSOD in malig-nant mesothelioma. Int J Cancer 2000, 88, 37–42.
[2] Caldas C: Molecular assessment of cancer. Br Med J 1998, 316, 1360–1363.
[3] Weinberg F, Chandel NS: Reactive oxygen species – development signaling regulates cancer. Cell Mol Life Sci 2009, 66, 3663–3673.
[4] Weyemi U, Dupuy C: The emerging role of ROS-generating NADPH oxidase NOX4 in DNA-damage responses. Mutat Res 2012, 751, 77–81.
[5] Naka K, Muraguchi T, Hoshii T, Hirao A: Regulation of ROS and genomic stability in hematopoietic stem cells. Antioxid Redox Signal 2008, 10, 1883–1894.
[6] Trachootham D, Alexandre J, Huang P: Targeting cancer cells by ROS-mediated mechanisms: a radical therapeu-tic approach? Nat Rev Drug Discov 2009, 8, 579–591.
[7] Halliwell B, Gutteridge J: Free Radicals in Biology and Medicine, Oxford University Press, 4th Edition, 2007.
[8] Inoue M, Sato EF, Nishikawa M, Park A-M, Kira Y, Imada I, Utsumi K: Mitochondrial generation of ROS and its role in aerobic life. Curr Med Chem 2003, 10, 2495–2505.
[9] West AP, Shadel GS, Ghosh S: Mitochondria in innate immune responses. Nat Rev Immunol 2011, 11, 389–402.
[10] Weyemi U, Lagente-Chevallier O, Boufraqech M, Penois F, Courtin F, Caillou B, Talbot M, Dardalhon M, Ghuzlan AAl, Birdart J-M, Schlumberger M, Dupuy C: ROS-generating NADPH oxidase NOX4 is a critical mediator in oncogenic H-Ras-induced DNA damage and subsequent senescence. Oncogene 2012, 31, 1117–1129.
[11] Bedard K, Krause KH: The NOX family of ROS-generating NADPH oxidases: physiology and pathophysiology. Physiol Rev 2007, 87, 245–313.
[12] Chrissobolis S, Faraci FM: The role of oxidative stress and NADPH oxidase in cerebrovascular disease. Trends Mol Med 2008, 14, 495–502.
[13] Miller FJ, Chu X, Stanic B, Tian X, Sharma RV, Davisson RL, Lamb FS: A differential role for endocytosis in receptor-mediated activation of NOX1. Antioxid Redox Signal 2010, 12, 583–590.
[14] Trachootham D, Lu W, Ogasawara MA, Nilsa RD, Huang P: Redox regulation and cell survival. Antioxid Redox Signal 2008, 10, 1343–1374.
[15] Coleman WB, Tsongalis GJ: Molecular mechanisms of human carcinogenesis. In: Cancer: Cell Structures, Carcinogens and Genomic Instability, Bignold, L.P. (Ed.), Birkhauser Verlag, Switzerland 2006, 321.
[16] Meister M, Tomasovic A, Banning A, Tikkanen R: Mitogen-activated protein (MAP) kinase scaffolding proteins: A recount. Int J Mol Sci 2013, 14, 4854–4884.
[17] Verschoor M, Wilson LA, Singth G: Mechanisms associated with mitochondrial-generated ROS in cancer. Can J Physiol Pharmacol 2010, 88, 204–219.
[18] Chetram MA, Bethea DA, Odero-Marah VA, Don-Salu-Hewage AS, Jones KJ, Hinton CV: ROS-mediated acti-vation of Akt induced apoptosis via pVHL in prostate cancer cells. Mol Cell Biochem 2013, 376, 63–71.
[19] Suzuki K, Matsubara H: Recent advances in p53 research and cancer treatment. J Biomed Biotechnol 2011, art. 978312.
[20] Kagan V, Bakalova R, Karakashev P: Lipid peroxidation in tumor cells and tissues of tumor-bearing animals. In: “Membrane Lipid Oxidation”, Vigo-Pelfrey, C. (Ed.), CRC Press, Boca-Raton, Florida, Vol. III, 1990, 192–208.
[21] Kahlos K, Anttila S, Asikainen T, Kinnula K, Raivio KO, Mattson K, Linnainmaa K, Kinnula VL: Manganese superoxide dismutase in healthy human pleural mesothelium and in malignant pleural mesothelioma. Am J Respir Cell Mol Biol 1998, 18, 570–580.
[22] Amundson SA, Myers TG, Fornace JrAJ: Roles for p53 in growth arrest and apoptosis: putting on the brakes after genotoxic stress. Oncogene 1998, 17, 3287–3299.
[23] Maiti AK: Genetic determinants of oxidative stress-mediated sensitization of drug-resistant cancer cells. Int J Cancer 2012, 130, 1–9.
[24] Penney RB, Roy D: Thioredoxin-mediated redox regulation of resistance to endocrine therapy in breast cancer. Biochim Biophys Acta 2013, 1836, 60–79.
[25] Zhelev Z, Aoki I, Gadjeva V, Nikolova B, Bakalova R, Saga T: Tissue redox activity as a sensing platform for imaging of cancer based on nitroxide redox cycle. Eur J Cancer 2013, 49, 1467–1478.
[26] Zhelev Z, Gadjeva V, Aoki I, Bakalova R, Saga T: Cell-penetrating nitroxides as molecular sensors for imaging of cancer in vivo, based on tissue redox activity. Mol BioSystems 2012, 8, 2733–2740.
[27] Bakalova R, Zhelev Z, Aoki I, Saga T: Tissue redox activity as a hallmark of carcinogenesis: from early to terminal stage of cancer. Clin Cancer Res 2013, 19, 2503–2517.
[28] Barrera C: Oxidative stress and lipid peroxidation products in cancer progression and therapy. ISRN Oncology 2012, art. no. 137289.
[29] De Luca A, Sanna F, Sallese M, Ruggiero C, Grossi M, Sacchetta P, Rossi C, De Laurenzi V, Di Ilio C, Favaloro B: Methionine sulfoxide reductase A dawn-regulation in human breast cancer cells results in a more aggressive phenotype. Proc Natl Acad Sci USA 2010, 107, 18628–18633.
[30] Harris IS, Blaser H, Moreno J, Treloar AE, Gorrini C, Sasaki M, Mason JM, Knobbe CB, Rufini A, Halle M, Elia AJ, Wakeham A, Tremblay ML, Melino G, Done S, Mak TW: PTPN12 promotes resistance to oxidative stress and supports tumorogenesis by regulating FOXO signaling. Oncogene 2013, doi: 10.1038/onc.2013.24.
D. Ivanova et al.
908
[32] Beck R, Dejeans N, Glorieux C, Pedrosa RC, Vasquez D, Valderrama JA, Calderon PB, Verrax J: Molecular vhaperone Hsp90 as a target for oxidant-based anticancer therapy. Curr Med Chem 2011, 18, 2816–2825.
[33] Fong MY, Jin S, Rane M, Singh RK, Gupta R, Kakar SS: withaferin A synergizes the therapeutic effect of doxo-rubicin through ROS-mediated autophagy in ovarian cancer. PLoS One 2012, 7, e42265.
[34] Sterba M, Popelova O, Vavrova A, Jirkovsky E, Kovarikova P, Gersl V, Simunek T: Oxidative stress, redox sig-naling, and metal chelation in anthracycline cardiotoxicity and pharmacological cardioprotection. Antioxid Redox Signal 2013, 18, 899–929.
[35] Gadjeva V, Koldamova R: Spin-labeled 1-alkyl-1-nitrosourea synergists of antitumor antibiotics. Anti-Cancer Drug Design 2001, 16, 247–253.
[36] Shaaban S, Diestel R, Hinkelmann B, Muthukumar Y, Verma RP, Sasse F, Jacob C: Novel peptidomimetic com-pounds containing redox active chalcogens and quinones as potential anticancer agents. Eur J Med Chem 2012, 58, 192–205.
[37] Zhou Y, Hileman EO, Plunkett W, Keating MJ, Huang P: Free radical stress in chronic lymphocytic leukemia cells and its role in cellular sensitivity to ROS-generating anticancer agents. Blood 2003, 101, 4098–4104.
[38] Takahashi S: Combination therapy with arsenic trioxide for hematological malignancies. Anticancer Agents Med Chem 2010, 10, 504–510.
[39] Ramanatham B, Jan KY, Chen CH, Hour TC, Yu HJ, Pu YS: Resistance to paclitaxel is proportional to cellular total antioxidant capacity. Cancer Res 2005, 65, 8455–8460.
[40] Kamada T: Clinical evidence of particle beam therapy (carbon). Int J Clin Oncol 2012, 17, 85–88.
[41] Maiti AK: Elevate the ROS level to kill cancer cells during chemotherapy. Chemotherapy 2012, 1, 5.
[42] Lawenda BD, Kelly KM, Ladas EJ, Sagar SM, Vickers A, Blumberg JB: Should supplemental antioxidant admin-istration be avoided during chemotherapy and radiation therapy? J Natl Cancer Inst 2008, 100, 773–783.
[43] Block KI, Koch AC, Mead MN, Tothy PK, Newman RA, Gyllenhaal C: Impact of antioxidant supplementation on chemotherapeutic toxicity: a systematic review of the evidence from randomized controlled trials. Int J Cancer 2008, 123, 1227–1239.
[44] Dimova EG, Bryant PE, Chankova SG: “Adaptive response” – some underlying mechanisms and open questions. Genet Mol Biol 2008, 31.
[45] Dabrowski A, Boquslowicz C, Dabrowska M, Tribillo I, Gabryelewicz A: Reactive oxygen species activate MAPKs in pancreatic acinar cells. Pancreas 2000, 21, 376–384.
[46] Drigotas M, Affolter A, Mann WJ, Brieger J: Reactive oxygen species activation of MAPK pathway results in VEGF up-regulation as an undesired irradiation response. J Oral Pathol Med 2013, E-pub: March 11, doi: 10.1111/ jop.12056.
[47] Grdina DJ, Murley JS, Miller RC, Mauceri HJ, Sutton HG, Li JJ, Woloschak GE, Weichselbaum RR: A surviv-ing-associated adaptive response in radiation therapy. Cancer Res 2013, 73, 4418–4428.
Address for correspondence:
Zhivko Zhelev
Department of Medicinal Chemistry and Biochemistry Medical Faculty, Trakia University
11 Armeiska Stara Zagora 6000 Bulgaria
E-mail: [email protected]
Conflict of interest: None declared
![Fig. 1. Generation of ROS in the mitochondria (according to west et al. [9]), SOD – superoxidedys-mutase; GPx – glutathione peroxidase; CoQ – coen-zyme Q; Cyt c – cytochro-me c; VDAC – voltage dependent anion channel.](https://thumb-us.123doks.com/thumbv2/123dok_us/8770414.1756708/2.595.60.519.444.752/generation-mitochondria-according-superoxidedys-glutathione-peroxidase-cytochro-dependent.webp)
![Fig. 2. Generation of ROS by the NADPH-oxidase complex (according to Chrissobolis & Faraci [12]), NOX – NADPH-oxidase](https://thumb-us.123doks.com/thumbv2/123dok_us/8770414.1756708/3.595.74.464.69.495/generation-nadph-oxidase-complex-according-chrissobolis-faraci-oxidase.webp)
![Fig. 3. Cellular redox homeostasis (according to Trachootham et al. [6]), Mito-ETC – mitochondrial electron-trans-port chain; SOD – superoxidedysmutase; GPx – glutathione peroxidase; GR – glutathione reductase; TRXox – oxi-dized thioredoxin; TRXred – reduced thioredoxin; GRXox – oxidized glutaredoxin; GRXred – reduced glutaredoxin; NOS – nitric oxide synthase; XO – xanthine oxidase; NOX – NADPH-oxidase](https://thumb-us.123doks.com/thumbv2/123dok_us/8770414.1756708/5.595.88.508.63.374/homeostasis-trachootham-mitochondrial-superoxidedysmutase-glutathione-thioredoxin-glutaredoxin-glutaredoxin.webp)