ISSN 2319-7625 (Online)
(An International Research Journal), www.chemistry-journal.org
Crystal Structure & Hirshfeld Surface of (
S
)-1-Phenylethane-
1, 2-diammonium Dichloride Hemihydrate
P. Yowan Jeba Raj1, T. Vidhyasagar1 and K. Rajeswari2
1Department of Chemistry,
Annamalai University, Annamalainagar - 608 002., INDIA. 2PG & Research Department of Chemistry,
Government College for Women (A), Kumbakonam - 612 001, INDIA. email: [email protected]
(Received on: November 6, Accepted: November 16, 2017)
ABSTRACT
The crystal structure of the monochiral diamine with the molecular formula C8H14Cl2N2O0.50 is discussed. The title compound crystallizes in monoclinic space
group, P 21/c, Z = 4. The unit cell dimensions are a = 15.2070(8) Å, b = 5.8410(3) Å, c = 12.5920(6) Å, α = 90°, β = 101.7220(10)° and γ =90°. The crystal size is 0.30 x 0.25 x 0.25 mm. The final R-factor for the compound is 3.48%. The dihedral angle of N2-C7-C8-N1 175.01°, reveals the anti disposition of two amino groups in the molecule. The atomic inter-contacts were computed and visualized using Crystal Explorer (3.1).
Keywords: monochiral (S)-diamine, XRD, atomic inter-contacts, CCDC: 1008701.
INTRODUCTION
Diamines are the promising candidates for the generation of significant new platin type anti-cancer drugs (chemotherapeutics)1,2 due to their stereochemical2 dispositions and they are useful templates in the field of asymmetric synthesis.3,4 The conformations adopted and 3D spatial arrangements of groups play the vital role for the biological activity viz.,
chemotherapeutic activities. Hence, our previous work5 and the present work interested in the establishment of structure of such diamine are significant, which contributes towards their utility in future.
EXPERIMENTAL
Synthesis & crystallization
non-regioselective Schmidt reaction with NaN3 (0.025 mmol) resulted in the formation of diazepanones, which on acid hydrolysis (6N HCl) produced a sandal-yellow solid mixture of both (R)- and (S)-isomers, which on optimized chiral transformation (using L(+) –Tartaric acid) end up with distinct (R)- and (S)-isomers, separately. They were recrystallized from ethanol, as pure white crystals by slow evaporation technique. The single crystal of the title compound achieved was used for XRD studies.
Data collection and Structure refinement
Single crystal ray diffraction data was collected on a Bruker AXS single crystal X-ray diffractometer using MoKα radiation and a SMART APEX CCD detector. Data was collected with ω scan width of 0.3°. A total of 606 frames were collected in each of three different settings of ϕ (0, 90 and 180) keeping the sample to detector distance at 6.03 cm and the 2θ value was also fixed at –25°. The data was reduced by SAINTPLUS9 and an empirical absorption correction was applied using the package SADABS9 The structure was solved and refined using SHELXTL.9 Molecular and packing diagrams were generated by ORTEP3210 and CAMERON11 present in the WINGX12 program suite. The geometric calculations were done by PARST95.13 The refinement converged to a final R-factor of 0.0348. The crystal data has been deposited at Cambridge Crystallographic Data Centre [CCDC: 1008701].
Computational Details
The molecular interactions, were viewed with the help of mercury 3.8 software program package.14 Hirshfeld surface analysis15 was carried out using Crystal explorer 3.1 software program.16
RESULT AND DISCUSSION Crystal structure
The title compound crystallizes in the space group, P 21/c with Z = 4. Molecular structure and ORTEP of the title compound is presented in Fig. 1. Crystal data and refinement parameters for the compound were given in Table 1. The geometrical parameters concerning both the conformation and configuration (significant dihedrals) were presented in Table 2. The dihedral angle between N2 and N1 atoms (N2-C7-C8-N1) is 175.01°, reveals the anti disposition of two amino groups in the molecule.
Packing diagram & Molecular interactions
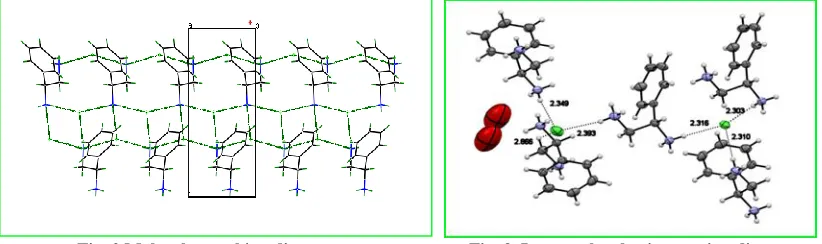
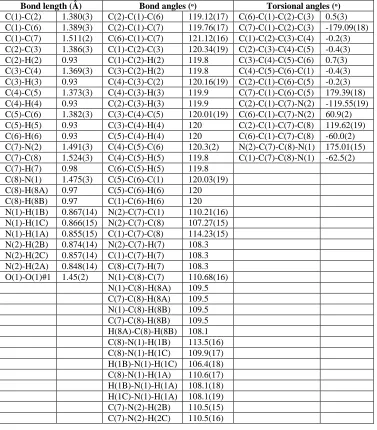
The packing diagram for the compound was displayed in Fig. 2. The molecular interactions between the repeating units were visualized using Mercury software. Both inter and intra-molecular interactions viz., hydrogen bonding, ionic interactions, van der Waals interactions, polarized interactions, etc., stabilize the molecule. The combined intra and inter-molecular interactions were displayed in Fig. 3. The hydrogen bonding & inter-molecular interactions (distances (Å) and angles (ᵒ)) were presented in Table 3.
Fig. 2 Molecular packing diagram Fig. 3. Inter-molecular interaction diagram
Table 1 Crystal data and structure refinement details
CCDC Number 1008701
Crystal data Values
Empirical formula C8 H14 Cl2 N2 O0.50
Formula weight 217.11
Temperature 293(2) K
Wavelength 0.71073 Å
Crystal system, space group Monoclinic, P21/c Unit cell dimensions:
Cell lengths a = 15.2070(8) Å b = 5.8410(3) Å c = 12.5920(6) Å
Cell angles α = 90ᵒ β = 101.7220(10)ᵒ γ = 90ᵒ
Volume 1095.15(10) Å3
Z, Calculated density 4, 1.317 Mg/m3
Absorption coefficient 0.552 mm-1
F(000) 456
Habit, Colour Block, Colourless
Crystal size 0.30 x 0.25 x 0.25 mm
Data collection Values
Theta range for data collection 2.74 to 26.00 deg.
Limiting indices -18 ≤ h ≤ 18, -7 ≤ k ≤ 7, -14 ≤ l ≤ 15 Reflections collected / unique 11355 / 2147 [R(int) = 0.0406] Completeness to theta = 26.00 100.0 %
Absorption correction Semi-empirical from equivalents Max. and min. transmission 0.8792 and 0.8501
Data / restraints / parameters 2147 / 12 / 142
Refinement parameters Values
Goodness-of-fit on F2 1.051
Final R indices [I>2sigma(I)] R1 = 0.0348, wR2 = 0.0900
R indices (all data) R1 = 0.0428, wR2 = 0.0943
R-factor (%) 3.48
Largest diff. peak and hole 0.521 and -0.272 eA-3
Table 2. Selected Bond lengths (Å), Bond angles (ᵒ) and Torsion angles (ᵒ)
Bond length (Å) Bond angles (ᵒ) Torsional angles (ᵒ)
C(1)-C(2) 1.380(3) C(2)-C(1)-C(6) 119.12(17) C(6)-C(1)-C(2)-C(3) 0.5(3) C(1)-C(6) 1.389(3) C(2)-C(1)-C(7) 119.76(17) C(7)-C(1)-C(2)-C(3) -179.09(18) C(1)-C(7) 1.511(2) C(6)-C(1)-C(7) 121.12(16) C(1)-C(2)-C(3)-C(4) -0.2(3) C(2)-C(3) 1.386(3) C(1)-C(2)-C(3) 120.34(19) C(2)-C(3)-C(4)-C(5) -0.4(3) C(2)-H(2) 0.93 C(1)-C(2)-H(2) 119.8 C(3)-C(4)-C(5)-C(6) 0.7(3) C(3)-C(4) 1.369(3) C(3)-C(2)-H(2) 119.8 C(4)-C(5)-C(6)-C(1) -0.4(3) C(3)-H(3) 0.93 C(4)-C(3)-C(2) 120.16(19) C(2)-C(1)-C(6)-C(5) -0.2(3) C(4)-C(5) 1.373(3) C(4)-C(3)-H(3) 119.9 C(7)-C(1)-C(6)-C(5) 179.39(18) C(4)-H(4) 0.93 C(2)-C(3)-H(3) 119.9 C(2)-C(1)-C(7)-N(2) -119.55(19) C(5)-C(6) 1.382(3) C(3)-C(4)-C(5) 120.01(19) C(6)-C(1)-C(7)-N(2) 60.9(2) C(5)-H(5) 0.93 C(3)-C(4)-H(4) 120 C(2)-C(1)-C(7)-C(8) 119.62(19) C(6)-H(6) 0.93 C(5)-C(4)-H(4) 120 C(6)-C(1)-C(7)-C(8) -60.0(2) C(7)-N(2) 1.491(3) C(4)-C(5)-C(6) 120.3(2) N(2)-C(7)-C(8)-N(1) 175.01(15) C(7)-C(8) 1.524(3) C(4)-C(5)-H(5) 119.8 C(1)-C(7)-C(8)-N(1) -62.5(2) C(7)-H(7) 0.98 C(6)-C(5)-H(5) 119.8
C(8)-N(1) 1.475(3) C(5)-C(6)-C(1) 120.03(19) C(8)-H(8A) 0.97 C(5)-C(6)-H(6) 120 C(8)-H(8B) 0.97 C(1)-C(6)-H(6) 120 N(1)-H(1B) 0.867(14) N(2)-C(7)-C(1) 110.21(16) N(1)-H(1C) 0.866(15) N(2)-C(7)-C(8) 107.27(15) N(1)-H(1A) 0.855(15) C(1)-C(7)-C(8) 114.23(15) N(2)-H(2B) 0.874(14) N(2)-C(7)-H(7) 108.3 N(2)-H(2C) 0.857(14) C(1)-C(7)-H(7) 108.3 N(2)-H(2A) 0.848(14) C(8)-C(7)-H(7) 108.3 O(1)-O(1)#1 1.45(2) N(1)-C(8)-C(7) 110.68(16)
H(2B)-N(2)-H(2C) 107.2(18) C(7)-N(2)-H(2A) 111.2(18) H(2B)-N(2)-H(2A) 107.5(18) H(2C)-N(2)-H(2A) 109.9(19)
* Symmetry transformations used to generate equivalent atoms: #1 -x+2,-y+2,-z.
Table 3. Hydrogen bonds & inter-molecular interactions
D-H...A d(D-H) d(H...A) d(D...A) <(DHA) C(8)-H(8A)...Cl(1)#2 0.97 2.91 3.623(2) 131.6 C(8)-H(8B)...Cl(2)#3 0.97 2.87 3.528(2) 126.3 N(2)-H(2B)...Cl(1)#2 0.874(14) 2.302(16) 3.1289(19) 157.7(19) N(1)-H(1B)...Cl(1)#4 0.867(14) 2.310(16) 3.1591(18) 166(2) N(2)-H(2C)...Cl(1) 0.857(14) 2.314(16) 3.1105(18) 154.8(19) N(2)-H(2A)...Cl(2)#5 0.848(14) 2.348(17) 3.1682(18) 163(2) N(1)-H(1C)...Cl(2) 0.866(15) 2.392(16) 3.2161(19) 159(2) N(1)-H(1A)...Cl(2)#6 0.855(15) 2.392(16) 3.204(2) 159(2)
* Symmetry transformations used to generate equivalent atoms:
#1 -x+2,-y+2,-z; #2 x,y+1,z; #3 -x+2,y-1/2,-z+1/2 #4 x,-y-1/2,z-1/2; #5 x,-y+1/2,z+1/2; #6 x,y-1,z.
Crystal Explorer theoretical predictions
The crystal explorer computations were carried out at Crystal Explorer 3.1 software program.14,15 and the computations are helpful in understanding different axial views, crystal voids, atomic inter-contacts on Hirshfeld surfaces & additional Hirshfeld properties.
Atomic inter-contacts on Hirshfeld surface
The atomic inter-contacts generated for the crystal structure of the title compound, using Crystal Explorer 3.1 software package is presented in Fig. 4. The red-glowing spheres in the Hirshfelds surface indicate the intermolecular contacts shorter than the van der Waals interaction while the blue regions indicate the long range contacts.
Hirshfeld surface mapped with basic properties
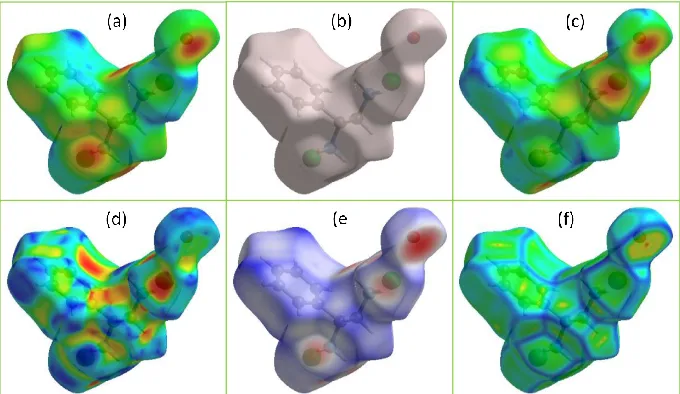
There are different basic surface properties that can be mapped over the Hirshfeld surfaces and those predict semi-theoretical molecular interaction surfaces. The mapped Hirshfeld surfaces with basic properties, viz., di, de, dnorm, shape index and curvedness were displayed in Fig. 5.
Fig. 5 Hirshfeld surface mapped with basic properties viz., (a) di, (b) none, (c) de, (d) shape index, (e) dnorm and (f) curvedness.
CONCLUSIONS
The optical-pure monochiral diamine, (S)-1-Phenylethane-1,2-diammonium dichloride hemihydrate was crystallized with the unit cell dimensions as: a ≠ b ≠ c and belongs to P 21/c in a monoclinic system and observed dihedral angles reveal the anti disposition of two amino groups in the molecule. The atomic inter-contacts were studied using computational programs viz., Crystal Explorer (3.1). This report will support the understanding of structure of one more medicinally significant monochiral diamine.
ACKNOWLEDGEMENT
Authors thank SAIF, IIT-Madras for the support in single crystal data collection and gratefully acknowledge UGC for financial support under MRP scheme.
REFERENCES
1. Abu-Surrah AS; Kettunen M, Current Med. Chem., 3, 1337-1357 (2006).
Fig. 5 Hirshfeld surface mapped with basic properties viz., (a) di, (b) none, (c) de, (d) shape index,
2. Dufrasne F; Galanski M Curr. Pharm. Design., 13, 2781-2194 (2007).
3. Kim H; Nguyen Y; Yen CPH; Chagal L; Lough A J; Kim BM; Chin J, J. Am. Chem. Soc.,
130, 12184-12191 (2008).
4. Seo MS; Kim K; Kim H, Chem. Commun., 49, 11623-11625 (2013).
5. Yowan Jeba Raj P; Vidhyasagar T; Rajeswari K, Int. J. TechnoChem Res., 2(3), 189-201 (2016).
6. Noller CR; Baliah V, J. Am. Chem. Soc., 70(11), 3853-3855 (1948). 7. Thennarasu S; Perumal PT, Molecules, 7, 487-493 (2002).
8. Froentjes W; Dijkema KM, Recueil, 62(11), 723-728 (1943).
9. Brucker. SMART, SAINT, XPREP AND SHELXTL, Brucker AXS Inc., Madison, Wiscosin, USA, (1998).
10. Farrugia LJ, J. Appl. Cryst., 20, 565 (1997).
11. Watkin DM; Pearce L; Prout CK, CAMERON. Chemical Crystallography Laboratory, University of Oxford, England, (1993).
12. Farrugia LJ, J. Appl. Cryst.,32, 837-838 (1999). 13. Nardelli M, J. Appl. Cryst.28, 659 (1995). 14. Mercury 3.8 for windows-32bit.
15. Crystal Explorer 3.1 for Windows-Intel-32bit.