0095-1137/05/$08.00⫹0 doi:10.1128/JCM.43.11.5601–5613.2005
Copyright © 2005, American Society for Microbiology. All Rights Reserved.
Population Structure and Properties of
Candida albicans
, as
Determined by Multilocus Sequence Typing†
Arianna Tavanti,
1Amanda D. Davidson,
1Mark J. Fordyce,
1Neil A. R. Gow,
1Martin C. J. Maiden,
2and Frank C. Odds
1*
Institute of Medical Sciences, University of Aberdeen, Aberdeen AB25 2ZD,1and The Peter Medawar
Building for Pathogen Research and Department of Zoology, University of Oxford, OX1 3SY Oxford,2United Kingdom
Received 10 May 2005/Returned for modification 24 July 2005/Accepted 21 August 2005
We submitted a panel of 416 isolates ofCandida albicansfrom separate sources to multilocus sequence typing
(MLST). The data generated determined a population structure in which four major clades of closely related isolates were delineated, together with eight minor clades comprising five or more isolates. By Fisher’s exact test, a statistically significant association was found between particular clades and the anatomical source, geographical source, ABC genotype, decade of isolation, and homozygosity versus heterozygosity at the mating type-like locus (MTL) of the isolates in the clade. However, these associations may have been influenced by confounding variables, since in a univariate analysis of variance, only the clade associations with ABC type and anatomical source emerged as statistically significant, providing the first indication of possible differences
betweenC. albicansstrain type clades and their propensity to infect or colonize different anatomical locations.
There were no significant differences between clades with respect to distributions of isolates resistant to fluconazole, itraconazole, or flucytosine. However, the majority of flucytosine-resistant isolates belonged to clade 1, and these isolates, but not flucytosine-resistant isolates in other clades, bore a unique mutation in the
FUR1gene that probably accounts for their resistance. A significantly higher proportion of isolates resistant
to fluconazole, itraconazole, and flucytosine were homozygous at the MTL, suggesting that antifungal pressure may trigger a common mechanism that leads both to resistance and to MTL homozygosity. The utility of MLST for determining clade assignments of clinical isolates will form the basis for strain selection for future research intoC. albicansvirulence.
Differentiation of microbial isolates by sequencing a small sample of unrelated housekeeping genes has become estab-lished as a reliable and effective method for typing strains of many bacteria (9, 50). Such multilocus sequence typing (MLST) is highly reproducible, and data can be archived in Web-based databases accessible to all users. For the fungal pathogenCandida albicans, an MLST system based on seven DNA fragments was developed as an optimal consensus for typing strains within the species (6) following two earlier pro-posed systems (5, 48). BecauseC. albicansis a diploid organ-ism, sequence data contain heterozygous as well as homozy-gous sites, adding an extra discriminatory feature to MLST for this species.
Among other approaches to strain typing of C. albicans, DNA fingerprinting based on the moderately repetitive se-quence Ca3 has been widely used. By this method,C. albicans
populations were shown to comprise five major clades of closely related strain types (46), including clades enriched in isolates from Europe (36) and South Africa (2, 3). Resistance ofC. albicansto flucytosine in vitro was found to be a property restricted almost entirely to isolates from a singleC. albicans
clade as determined by Ca3 fingerprinting (37), and the sole
mechanism of resistance to flucytosine within this clade was the mutation of a single nucleotide in theFUR1gene (14).
Typing by a combination of restriction fragment polymor-phisms and randomly isolated polymorphic DNA also showed geographical enrichment of groups of related isolates (10), and “ABC typing,” based on the size and number of internally transcribed spacer regions (ITS1) in DNA encoding rRNA showed a higher proportion of flucytosine-resistant isolates among type A strains (26).
We have now typed more than 400 isolates ofC. albicansby MLST. Our data show how MLST reveals a population struc-ture with four major clades of related strain types plus at least eight minor clades. It confirms the predominant association of flucytosine resistance with one major clade and reveals associ-ations between clades and their geographical, anatomic, and temporal origins.
MATERIALS AND METHODS
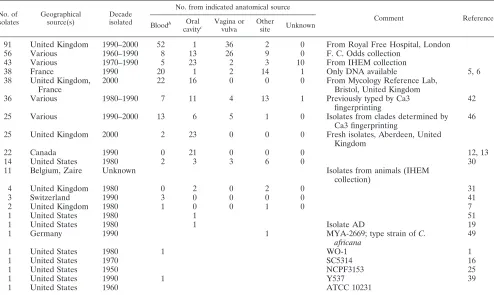
C. albicansisolates.We typed 416 isolates from a large range of sources. Each isolate was from a separate source. Table 1 summarizes the details of the panel of isolates tested. Data for isolates described by Bougnoux et al. in their MLST work (5) were included in the present study; for these isolates, only extracted DNA had been provided by Marie-Elisabeth Bougnoux and Christophe d’Enfert for MLST (6), so we could not conduct antifungal susceptibility tests with these isolates. A set of 25 isolates was provided by David R. Soll (Table 1); these comprised five isolates from each of the five majorC. albicansclades as deter-mined by Ca3 fingerprinting (46) and served as a reference for cross-validation of typing methodologies. Isolate WO1, the original “white-opaque switcher” now undergoing full-genome sequencing, was also originally supplied by D. R. Soll. A set of 36 isolates was provided by Jan Schmid; 35 of these were a subset of isolates that had been typed by Ca3 fingerprinting (42, 43). Another set of 22
* Corresponding author. Mailing address: Institute of Medical Sci-ences, University of Aberdeen, Aberdeen AB25 2ZD, United King-dom. Phone and Fax: 44 1224 555828. E-mail: [email protected].
† Supplemental material for this article can be found at http://jcm .asm.org/.
5601
on May 15, 2020 by guest
http://jcm.asm.org/
clinical isolates was provided by James B. Anderson from his group’s study of population genomics of fluconazole resistance inC. albicans(12, 13). Other notable isolates included SC5314, which was used for whole-genome sequencing ofC. albicans(21) and is the wild-type isolate used widely for gene disruption research (16); a representative from the set of 16 serial isolates with increasing fluconazole resistance from a patient infected with human immunodeficiency virus (HIV) (38, 51), provided by Theodore White; isolate NCPF3153, often referred to as 3153A and used in many laboratories; and isolate MYA-2669, the type strain of the putative speciesCandida africana(49), purchased from the American Type Culture Collection. A large set of isolates was provided from the collection of the Belgian Institute for Hygiene, Epidemiology, and Medicine (IHEM) through the collaboration of Nicole Nolard and Franc¸oise Symoens; some of these were subcultures originally obtained from the senior author’s collection. Three isolates that had been used to study mechanisms of fluconazole resistance (41) were originally contributed by Dominique Sanglard.
Recent clinical isolates ofC. albicanswere provided by Elizabeth M. Johnson (Mycology Reference Laboratory, Bristol, United Kingdom) and Christopher C. Kibbler (Royal Free Hospital, London, United Kingdom) from their diagnostic mycology laboratories. Fresh oral commensal isolates ofC. albicanswere ob-tained from healthy undergraduate student volunteers in Aberdeen, United Kingdom. The remainder of the isolates typed came from the senior author’s collection ofC. albicansisolates, which was started in the 1970s. This collection, originally stored under distilled water (29), was transferred twice in its entirety, once for an investigation of the prevalence of the speciesC. dubliniensisin the collection (32). Some of these isolates had been previously described in epide-miological studies ofC. albicansbased on phenotypic typing tests (30, 31). The quality of the collection was confirmed by the near-identity of MLST data for several isolates from the same source (details not shown).
Isolates were maintained on Sabouraud agar (Oxoid, Basingstoke, United Kingdom) for the duration of this study.
MLST.Sequences of bases in the gene fragmentsAAT1a,ACC1,ADP1,MPIb,
SYA1,VPS13, andZWF1bwere determined as previously described (6, 48). Isolates were designated as genotype A, B, or C on the basis of PCR for rRNA
genes (26), and the status of the mating-type-like locus (MTL) in each strain was assessed as heterozygous (a/␣), homozygous (a/a), or homozygous (␣/␣) by PCR as previously described (48).
Sequencing of other genes.GenesFCY22,FCA1, andFUR1encode, respec-tively, cytosine permease, cytosine deaminase, and uracil phosphoribosyltrans-ferase in the pathway for conversion of flucytosine to the active antifungal 5-fluorouracil inC. albicans(14). For selectedC. albicansisolates, the DNA sequences of these genes were determined by the same method as that used for MLST (above). ForFCY22, the forward primers used were FCY22-1 (5⬘AACA TAATCTGTAGTTATATCTC), FCY22-2 (ATTATTGGAAGCATTCCAGTG 3⬘), FCY22-3 (5⬘TCCAACTGAAGCTGGTAATG3⬘), and FCY22-4 (TGCCA TTCAATGTATATGGAG), and the reverse primer was 5⬘TAACAGGGCAG AATGAATCG3⬘. ForFCA1, the forward primers were FCA1-1 (5⬘AATTCTC TTTCAATTCTTCTTG3⬘) and FCA1-2 (5⬘TCACCATGTAGTATGTGTAC 3⬘), with the reverse primer being 5⬘TTGAACAAGATGATGATGTTG3⬘. For
FUR1, the forward primers were FUR1-1 (5⬘ATAATGGAGCATCTCGCAAC 3⬘) and FUR1-2 (5⬘TAGCTGAAGATATCAGTGAAC3⬘), with the reverse primer being 5⬘TGTTGTACATATAATCCTATAAG3⬘.
Susceptibility testing.MICs of fluconazole, flucytosine, and itraconazole were determined by microdilution tests by a previously described method and spec-trophotometric endpoints (33) that give MIC results compatible with those obtained by the NCCLS reference method (27). Isolates with fluconazole MICs of 16g/ml or higher, flucytosine MICs of 8g/ml or higher, or itraconazole MICs of 0.25g/ml or higher were classed as resistant to these agents (these breakpoints indicate full susceptibility to the agents concerned [27]).
Data analysis.Sequence data were analyzed by scrutiny of capillary sequencer output chromatograms with DNASTAR Seqman software. Each unique (dip-loid) sequence for each of the seven DNA fragments was given a separate identifying number. Each unique combination of seven sequences was assigned a separate diploid sequence type (DST) number. The sequence and DST iden-tifiers were compiled on a central database (http://test1.mlst.net/).
[image:2.585.47.541.81.376.2]The eBURST package (http://eburst.mlst.net/ [15]) was used to determine putative relationships between isolates. WithC. albicansdata, this software scans
TABLE 1. Summary of 416C. albicansisolates tested by MLST and their originsa
No. of isolates
Geographical source(s)
Decade isolated
No. from indicated anatomical source
Comment Reference
Bloodb Oral
cavityc
Vagina or vulva
Other
site Unknown
91 United Kingdom 1990–2000 52 1 36 2 0 From Royal Free Hospital, London
56 Various 1960–1990 8 13 26 9 0 F. C. Odds collection
43 Various 1970–1990 5 23 2 3 10 From IHEM collection
38 France 1990 20 1 2 14 1 Only DNA available 5, 6
38 United Kingdom, France
2000 22 16 0 0 0 From Mycology Reference Lab,
Bristol, United Kingdom
36 Various 1980–1990 7 11 4 13 1 Previously typed by Ca3
fingerprinting
42
25 Various 1990–2000 13 6 5 1 0 Isolates from clades determined by
Ca3 fingerprinting
46
25 United Kingdom 2000 2 23 0 0 0 Fresh isolates, Aberdeen, United
Kingdom
22 Canada 1990 0 21 0 0 0 12, 13
14 United States 1980 2 3 3 6 0 30
11 Belgium, Zaire Unknown Isolates from animals (IHEM
collection)
4 United Kingdom 1980 0 2 0 2 0 31
3 Switzerland 1990 3 0 0 0 0 41
2 United Kingdom 1980 1 0 0 1 0 7
1 United States 1980 1 51
1 United States 1980 1 Isolate AD 19
1 Germany 1990 1 MYA-2669; type strain ofC.
africana
49
1 United States 1980 1 WO-1 1
1 United States 1970 SC5314 16
1 United States 1950 NCPF3153 25
1 United States 1990 1 Y537 39
1 United States 1960 ATCC 10231
a
A full list of isolates and their details are available in the supplementary material.
b
Includes other sterile, deep tissue sites.
c
Includes samples designated as from sputum, throat, and tongue.
on May 15, 2020 by guest
http://jcm.asm.org/
allele sequences from pairs of isolates and records isolates as related when six of the seven sequences are identical between a pair. The eBURST algorithm places all related isolates into clonal complexes and, where possible, predicts the found-ing, or ancestral DST of each complex. The output is a display of the most parsimonious patterns of descent of each DST from the ancestral type.
Phylogenetic analyses by unweighted pair group method with arithmetic av-erages (UPGMA) and neighbor-joining algorithms were conducted with MEGA version 2.1 (22) applied to modified sequence data. The analyses were based only on the results for polymorphic bases to maximize their power to discriminate between isolates. The data set of only the variable bases was suitable for pair-wise-difference analysis, which has been used previously withC. albicansMLST (5, 48). To obtain sequences that could be handled by the MEGA software, which is not programmed to analyze heterozygous code data, the following procedure was used. The results for the variable sites from the seven gene fragments sequenced were concatenated into a single sequence. For any pair of isolates, each with a diploid genome, the base at each polymorphic site could be homozygous and identical between the isolates, heterozygous and identical, homozygous and different, or heterozygous in one isolate and homozygous in another. For example, the sequencing result (a IUPAC single-letter code) for a given base across a set of strains might appear as A, T, or W (⫽A⫹T). Data from the polymorphic sites from the sevenC. albicansalleles were therefore conjoined into a single sequence, and then each base in the sequence was rewritten twice for a homozygous (A, C, G, or T) datum or once each for the two component bases for a heterozygous (K, M, R, S, W, or Y) datum. These revised sequences could then be analyzed to generate dendrograms in MEGA 2.1. In this form, the analysis was the functional equivalent of scoring a pair of results as 1 for homozygous or heterozygous identical data, 0 for homozygous different data, and 0.5 when one polymorphic site had a heterozygous result and the other a homozygous result and then creating a difference matrix. Use of the modified sequences, rather than of difference matrices, enabled calculation of statistical significance for the cluster nodes by bootstrapping with 1,000 replications.
Distributions of properties betweenC. albicansclades were determined sta-tistically by Fisher’s exact test. Results were considered significant for aPof ⬍0.05. A univariate analysis of variance (ANOVA) was used to determine associations between clades and A, B, or C type (with 1, 2, and 3 corresponding to A, B, and C, respectively); MTL status (with 1, 2, and 3 corresponding toa/␣,
a/a, and␣/␣, respectively); fluconazole, itraconazole, and flucytosine susceptibil-ities (with 1 indicating susceptibility and 2 indicating resistance); decades of isolation (1 being a year prior to 1990, 2 being a year in the 1990s, and 3 being a year in the 2000s); geographical origin (1 being the United Kingdom, 2 being another European country, 3 being North America, and 4 being another coun-try); and anatomical origin (1 being blood or other sterile site, 2 being the oropharynx, 3 being the vagina, and 4 being another location). Isolates from animals were excluded from this analysis.
Discriminatory power was calculated according to Hunter (20).
RESULTS
Reproducibility and sequencing errors.A total of 13
differ-entC. albicansisolates was submitted for MLST in duplicate on different occasions, and the person conducting the sequenc-ing was unaware of their identities. The seven genes sequenced yielded a total of 109 variable sites. Of the 13 isolates tested in duplicate, three yielded different DSTs. In two cases, the dif-ference resulted from a single-base homozygous/heterozygous difference in one of the seven genes; in the third there were homozygous/heterozygous discrepancies at two sites in a single gene. Thus, in a total of 1,417 variable sites tested in duplicate, a sequencing error arose on four (0.28%) occasions, indicating a reproducibility of 99.72%. Sequence differences greater than three homozygous/heterozygous discrepancies between iso-lates were therefore assumed to indicate true interstrain dif-ferences.
Statistics ofC. albicansMLST.The 416 isolates yielded 351
distinct DSTs, with a discriminatory power index of 0.9996. Among the seven fragments sequenced,ZWF1gave the high-est discriminatory ratio, yielding 84 different sequences from just 10 polymorphic sites, followed bySYA1(70 alleles from 14
variable sites), VPS13 (103 alleles from 21 variable sites),
AAT1a(57 alleles from 13 variable sites), ACC1 (37 alleles from 10 variable sites), ADP1 (46 alleles from 18 variable sites), andMPIb(36 alleles from 21 variable sites).
Details of the alleles and DSTs for each isolate are shown in the full version of Table S1 in the supplemental material, together with the geographical and anatomical sources of the isolates. Insufficient DNA for sample BougnCP10 precluded determination of ABC and MTL type for this isolate. Among the remaining 415 isolates, 281 (67.7%) were type A, 96 (23.1%) were type B, and 38 (9.2%) were type C. The great majority of isolates (371, or 89.4%) were heterozygous at the MTL, while 27 isolates (6.5%) were of homozygous MTL type ␣/␣and 17 isolates (4.1%) were of typea/a.
Population structure of 416 unrelated C. albicansisolates.
MLST data allow for different approaches to phylogenetic analysis of various degrees of sophistication. We utilized sev-eral approaches, with an emphasis on methods based on pair differences at polymorphic nucleotide sites and on gross allele sequence differences in an effort to determine robust subspe-cific clades that will allow easy assignment of future isolates to the population structure.
eBURST analysis of the set of 416C. albicansisolates from unique sources revealed 18 clonal clusters, with 5 being based on six or more DSTs (Fig. 1). Cluster 1, the largest, comprised 105 isolates (25.2% of all isolates), with DST 69 as the putative ancestral type. Clonal cluster 2, the second largest, comprised 38 isolates and 27 DSTs and no putative founding DST (type 155 was represented by 7 isolates but did not emerge as a likely ancestor for the cluster). Clonal cluster 3 comprised 28 isolates and 17 DSTs, with type 124 being the predicted founding type. Cluster 4 comprised eight isolates and seven DSTs, with type 305 being the likely ancestral DST, and cluster 5 comprised seven isolates and six DSTs, with no putative ancestral type. The remaining clonal clusters consisted maximally of six iso-lates.
Of the 416 isolates analyzed by the eBURST software, 228 (54.8%) were assigned to clonal clusters, thus leaving almost half of the isolates with singleton status (Fig. 1).
Unlike the eBURST analysis, a UPGMA pairwise-difference dendrogram for the 416 separate-source isolates (Fig. 2) took no account of the gene fragment in which sequence polymor-phisms arose and therefore provided a different perspective on relationships between isolates: isolates with small numbers of differences in the concatenated sequences for variable sites across all fragments sequenced were regarded as similar by UPGMA analysis, even if the polymorphic sites were located on separate fragments. Comparison of the clustering in the UPGMA dendrogram with the eBURST clonal clusters (Fig. 2) showed clonal clusters 11 and 17 and several eBURST singletons dispersed within the broad range of isolates in clonal cluster 1. Similarly, isolates in clonal clusters 5, 10, and 13 were mixed with each other and with several eBURST singletons by UPGMA analysis.
Although the UPGMA dendrogram for the concatenated variable sequences from the 416 separate-source isolates clearly suggested patterns of clustering between the isolates, bootstrap values for cluster nodes were generally low except for a few pairs of very closely related isolates. The following procedure was therefore used to determine which isolates were
on May 15, 2020 by guest
http://jcm.asm.org/
FIG.
1.
Modified
eBURST
“snapshot”
for
416
C.
albicans
isolates
from
unique
sources.
The
figure
is
redrawn
from
the
original
output
of
the
eBURST
analysis
to
eliminate
overlapping
lines
and
numbers.
The
diameter
of
each
circular
spot
is
proportional
to
the
number
of
isolates
with
the
same
DST
(indicated
b
y
the
number
adjacent
to
the
spot).
Lines
join
DSTs
that
dif
fer
in
just
one
of
the
seven
fragments
sequenced
for
MLST
and
thus
indicate
a
hierarchical
relatio
nship
between
DSTs.
Where
a
putative
ancestral
DST
was
determined
for
a
cluster,
the
DST
number
has
been
marked
with
an
asterisk.
Each
clonal
cluster
of
relat
ed
isolates
is
numbered
in
boldface
type.
Lengths
of
lines
joining
DSTs
are
of
no
relevance.
on May 15, 2020 by guest
http://jcm.asm.org/
on May 15, 2020 by guest
http://jcm.asm.org/
on May 15, 2020 by guest
http://jcm.asm.org/
most likely to belong to clusters of highly related strains. The information from Ca3 typing, particularly the results for iso-lates belonging to known Ca3 clades (46), was considered in conjunction with clonal cluster information to determine a cutoff point that could be used to define UPGMA clades and outlying singletons.
All five of the isolates from clade I as determined by Ca3 typing (46) clustered within a large group of isolates closely related by UPGMA (Fig. 2); four of the five belonged to clonal cluster 1 by eBURST analysis. Similarly, 19 of the 21 isolates designated types A1 and A2 by Ca3 fingerprinting and desig-nated by neighbor-joining statistics (42, 43) appeared in the same large UPGMA cluster that was broadly delimited by eBURST clonal cluster 1. All 155 isolates from AM2003/0046 to AM2004/0049 in Fig. 2 were therefore provisionally assigned to clade 1 as demarcated by MLST. The five Ca3 clade II isolates and both Ca3 group B isolates correlated with a well-delineated UPGMA cluster (Fig. 2); four of these seven iso-lates appeared in eBURST clonal cluster 2. Isoiso-lates from AM2004/0030 to IHEM17110 in Fig. 2 were therefore provi-sionally assigned to MLST clade 2. Similarly, the five isolates from Ca3 clade SA clustered together in the UPGMA dendro-gram, with four of the five in clonal cluster 3. However, the UPGMA cluster designated MLST clade 4 in Fig. 2 (isolates HUN68 to IHEM16731), though appearing reasonably homo-geneous, contained a subcluster comprising two of the Ca3 clade E isolates, and all of the members of clonal cluster 15. The five isolates from Ca3 clade III emerged in clonal clusters 9 or 10 or as singletons by eBURST analysis, but they clustered together in the UPGMA dendrogram in a group of isolates with the same overall level of similarity as those in MLST clades 1, 2, and 4. Isolates AM2003/0082 to b30998/5 were therefore provisionally considered to belong to MLST clade 3. It was noted that isolates previously designated as group C by neighbor-joining analysis of Ca3 typing (42, 43) did not show obvious affinities with eBURST- or UPGMA-defined clusters. The provisional assignment of isolates to clades 1 to 4 indi-cated a maximum dissimilarity of 10 by pairwise-nucleotide difference analysis as calculated with the MEGA software. The following definition of a clade was therefore used: a cluster of at least five isolates with a dissimilarity not exceeding 10 by pairwise-difference analysis. By this definition, 348 (83.7%) of the 416 isolates could be assigned to 12 clades, as specified in Fig. 2. Reanalysis of the MLST variable-site data only for the 348 isolates that matched the clade definition confirmed the validity of the clade assignments in Fig. 2. The bootstrap values for the nodes defining clades were 85% (clade 1), 88% (clade 2), 87% (clade 3), 86% (clade 4), and 98 or 99% for the minor clades. The exception was clade 5, for which the node boot-strap value was only 64%, while the values for the node defin-ing the pair of isolates 81/078 and AM2003/0084 and the node for the remaining four isolates nominated for clade 5 were 97% and 99%, respectively. These results suggest that clade 5 is a less robust grouping of isolates than the other clades and may need to be revised in future as furtherC. albicansisolates are added to the MLST database.
To examine the population structure ofC. albicansclades by an alternative statistical approach the MLST data for the 416 isolates of separate origins were reanalyzed by determination of p-distance with the unrooted neighbor-joining algorithm in
FIG. 2. UPGMA pairwise-dif ference dendrogram for 416 C. albicans isolates from separate sources. The dendrogram is broken into three sections, a to c, to fit the printed page. The complete dendrogram is shown at a much reduced vertical scale at the bottom right, with indications of the points at which the tree has been split. For each isolat e, its clonal cluster (or singleton status) as determined by eBURST analysis is indicated, as is the result of Ca3 typing for isolates provided by D. R. Soll (Cl is the Soll clade result by UPGMA anal ysis) or Schmid (Gp is the Schmid group result by neighbor-joining analysis). The assignment of isolates to clades, as indicated in the third data column, was made by consideration of the eBURST and Ca3 data in conjunction with UPGMA bootstrap values as described in the text.
on May 15, 2020 by guest
http://jcm.asm.org/
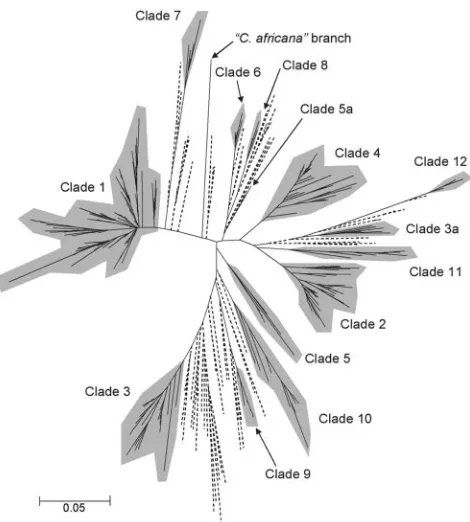
the MEGA2 software package. The result (Fig. 3) confirmed most of the isolate clusters determined by pairwise-distance UPGMA analysis (Fig. 2) and reiterated that the majority of the isolates studied could be assigned to one of four major clades. However, the neighbor-joining algorithm also produced some differences in the structure from the UPGMA analysis.
In Fig. 3, the three isolates that included the type strain of
the putative species C. africana, while still forming a well-separated branch in the tree, nevertheless appeared less dis-tant from other isolates than in the UPGMA dendrogram (Fig. 2). In the neighbor-joining tree, isolates 81/078 and AM2003/ 0084 clustered separately from the remainder of isolates classed as clade 5 in Fig. 2 (the position of the pair is indicated as clade 5a in Fig. 3), reemphasizing the previous conclusion that clade 5 is less robust than most others. The population structure by neighbor joining also removed isolates C82, AM2003/0065, BougnCP11, 73/025, and J981315 as a group from the rest of clade 3 in Fig. 2 to a new position shown as clade 3a in Fig. 3. The clade assignments in Fig. 2 were used for subsequent analyses of clade properties; however, it is self-evident that the most robust definition ofC. albicans clades must await a larger future database of MLST results and pos-sibly further approaches to statistical analyses.
Isolates and clusters of notable provenance.Three isolates,
AM2003/0025, AM2003/0032, and MYA-2669, the type strain of the putative speciesC. africana, were indistinguishable by MLST but clustered with very low similarity to the other iso-lates in Fig. 2, confirming that MYA-2669 differs substantially from otherC. albicansisolates. All three of these indistinguish-able isolates came from genital swabs.
Isolate SC5314, which was used for the first whole-genome sequencing project withC. albicans (21), fell within clade 1. Isolate WO-1, which has been extensively used in research into phenotypic switching and mating inC. albicans(45), appeared as a singleton between clades 8 and 4 in Fig. 2.
[image:8.585.46.281.66.327.2]Within clade 1, a group of four isolates—IHEM20416, J942149, IHEM20414, and IHEM20417—that were extremely similar by MLST was notable because all four were resistant to fluconazole, and the first three were also resistant to itracon-azole. These three were␣/␣at the MTL (Table 1). All four of the isolates in this subcluster came from the mouths of AIDS patients in Germany. A similar subcluster of four closely re-lated isolates within cluster 5, already noted above by the high significance of the bootstrap value for the cluster node, in-cluded three isolates resistant to both azole antifungal agents and three isolates that werea/aat the MTL. All four isolates came from the mouths of AIDS patients in France.
FIG. 3. Population structure of 416 isolates ofC. albicans from separate sources, as determined by p-distance (22) in a neighbor-joining analysis. The tree is displayed in radial format to clarify the relative positions of putative clades of related isolates, which are in-dicated with gray shading. The clade numbers correspond to those in Fig. 2; note that a minority of isolates in clades 3 and 5 were separated in the neighbor-joining tree labeled 3a and 5a, respectively. Branches to singleton isolates without clade affinities are shown with dashed lines.
TABLE 2. Main properties of clades ofC. albicansisolates
Clade No. of isolates
% of ITS type: MTL homozygous
(%)
% Isolated in: % Isolated % Isolated from: % Resistant toa:
A B C Europe North America Other pre-1990 1990s Blood Oropharynx Vagina FCZ ITZ 5FC
1 155 95.5 1.9 1.9 14.9 58.7 32.3 8.4 20.5 58.7 25.2 33.8 23.2 8.5 6.4 10.6
2 55 98.2 1.8 0.0 5.5 74.5 10.9 14.5 17.0 38.2 35.2 37.0 18.5 3.8 3.8 1.9
3 30 3.3 93.3 3.3 16.7 56.7 36.7 6.7 10.3 63.3 40.0 20.0 30.0 20.0 4.0 4.0
4 45 22.2 35.6 42.2 4.4 71.1 11.1 17.8 16.3 26.7 41.9 32.6 18.6 10.0 7.5 0.0
5 6 0.0 100.0 0.0 50.0 100.0 0.0 0.0 16.7 0.0 0.0 80.0 20.0 50.0 50.0 16.7
6 9 0.0 100.0 0.0 22.2 88.9 11.1 0.0 0.0 55.6 0.0 100.0 0.0 33.3 0.0 11.1
7 5 100.0 0.0 0.0 20.0 60.0 20.0 20.0 60.0 20.0 50.0 25.0 25.0 0.0 0.0 0.0
8 5 60.0 0.0 40.0 0.0 60.0 0.0 40.0 25.0 20.0 60.0 0.0 20.0 0.0 0.0 0.0
9 10 70.0 30.0 0.0 0.0 30.0 20.0 50.0 12.5 60.0 0.0 50.0 20.0 0.0 0.0 0.0
10 13 15.4 69.2 15.4 0.0 92.3 0.0 7.7 12.5 23.1 16.7 16.7 16.7 0.0 0.0 0.0
11 9 88.9 0.0 11.1 0.0 88.9 0.0 11.1 0.0 44.4 55.6 33.3 11.1 0.0 12.5 0.0
12 6 33.3 33.3 33.3 16.7 83.3 0.0 16.7 0.0 83.3 33.3 50.0 0.0 16.7 16.7 16.7
Sb 68 60.3 27.9 11.8 5.9 75.0 16.2 8.8 20.3 42.6 48.5 24.2 12.1 7.4 5.9 4.4
aFCZ, fluconazole; ITZ, itraconazole; 5FC, flucytosine. b S, singletons.
on May 15, 2020 by guest
http://jcm.asm.org/
[image:8.585.40.544.551.708.2]C. albicansclades: properties and clinical relevance.Table 2 summarizes the properties of the 12 individual C. albicans
clades and of singleton isolates. The numbers of isolates in each of clades 5 to 12 was too small to permit reliable statistical analysis; these isolates were therefore grouped together as “minor clades” for the calculations detailed below.
Clades 1 and 2 comprised almost entirely isolates ofITS1
type A, while clade 3 isolates were almost entirely type B. Clades 5 and 6 isolates were all type B, and clade 7 isolates were all type A. Only clade 4 had a preponderance of type C isolates, but this clade also contained type A and type B iso-lates. The distributions of A, B, and C types between clades obviously differed significantly (Fisher’s exact test,P⬍0.0001). There were 44 (10.6%) of 415 typeable isolates with ho-mozygous alleles at the MTL. Just over half of these isolates (29) were in clade 1. The distribution of isolates homozygous at the MTL differed significantly between clades (by Fisher’s ex-act test,Pwas 0.016; the test was applied to numbers of isolates that werea/␣, a/a, or ␣/␣ [Table 1]). Minor clades 8 to 11 contained no examples of MTL homozygous isolates, while a high proportion of isolates in clades 5 to 7 were homozygous at the MTL (Table 2).
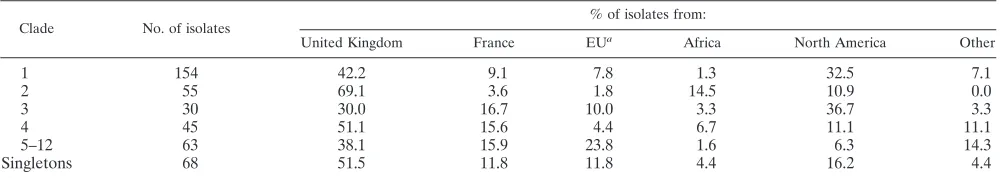
Except for clade 9, the majority of isolates in each clade came from sources within Europe (information on geographical origin was available for 415 of the 416 isolates in Fig. 2). The 6 isolates in clade 5 were all of European origin, and there were no North American isolates among the 28 that made up clades 10 to 12. Table 3 breaks down the geographical-origin data more finely, but with minor clades treated as a single group. Statistical analysis of the distribution of isolate sources by clade as shown in Table 3 indicated a highly significant association of clade with geograph-ical source (Fisher’s exact test,P⬍0.001). Even when the data were analyzed with French and other EU isolates combined into a single group and African isolates included with “other” geo-graphical sources, the differential distribution of sources within
each clade remained statistically significant (P⬍0.001). It should particularly be noted that, among the 45 isolates in clade 4, equiv-alent to the “South African” clade based on Ca3 fingerprinting (2), only 3 isolates came from the African continent. The majority of African isolates in the present study belonged to clade 2.
Information on the anatomical source of isolates was avail-able for 406 of the 416 isolates in Fig. 2. There were 11 isolates from animals, so analysis of relations between anatomical sources and clades were based on 395 isolates (Table 4). Clades 5, 6, and 9 contained no isolates from blood cultures (Table 2), and the proportion of isolates from blood and other sterile sites in clade 1 was lower than in the other major clades. The distribution of isolates from blood, the oropharynx, the vagina, and other sites varied between clades at a level that reached statistical significance (Fisher’s exact test,P⫽0.045). The clade distribution of the 11 isolates from animals dif-fered considerably from that of human isolates. Five of the isolates were singletons, four were in clade 10, and one each belonged to clades 1 and 9. This differential distribution was highly significant (Fisher’s exact test,P⫽0.003). Since 10 of the 11 animal isolates originated in Belgium, their clade dis-tribution was compared separately with that of the 11 human isolates that came from Belgian patients only (5 in clade 1, 2 in clade 11, and 1 each in clades 2, 3, 4, 6, 9, and 12). Even with clades 5 to 12 treated as a single group, the distribution of animal isolates differed significantly from that of Belgian hu-man isolates (P⫽0.02).
[image:9.585.43.543.81.170.2]For most of the clades, between 10% and 20% of the isolates were first cultured before 1990 (Table 2). In clades 1 to 3, 6, 7, and 9, isolates first cultured before 1990 were represented and most of the isolates were first cultured before the year 2000, while clades 4, 5, 8 and 10 to 12 bore evidence of possibly more recent origins (Table 2). The distribution of isolates of differ-ent ages differed significantly between the clades (by Fisher’s exact test,Pwas 0.003; analysis done with clades 5 to 12 treated
TABLE 3. Geographical origins ofC. albicansisolates in different MLST clades
Clade No. of isolates
% of isolates from:
United Kingdom France EUa Africa North America Other
1 154 42.2 9.1 7.8 1.3 32.5 7.1
2 55 69.1 3.6 1.8 14.5 10.9 0.0
3 30 30.0 16.7 10.0 3.3 36.7 3.3
4 45 51.1 15.6 4.4 6.7 11.1 11.1
5–12 63 38.1 15.9 23.8 1.6 6.3 14.3
Singletons 68 51.5 11.8 11.8 4.4 16.2 4.4
a
[image:9.585.42.541.625.716.2]EU, European Union, excluding the United Kingdom and France.
TABLE 4. Anatomical origins ofC. albicansisolates in different MLST clades
Clade No. of isolates % of isolates from:
Blooda Oropharynx Vagina Other
1 150 25.3 34.0 23.3 17.3
2 55 36.4 36.4 18.2 9.1
3 30 40.0 20.0 30.0 10.0
4 44 43.2 31.8 18.2 6.8
5–12 55 27.3 45.5 14.5 12.7
Singletons 61 52.5 26.2 13.1 8.2
a Includes data for six isolates from other normally sterile sites.
on May 15, 2020 by guest
http://jcm.asm.org/
isolates as a single group, and dates of isolation segregated as pre-1990, 1990s, and 2000s).
A high proportion of the five isolates in clade 5 was resistant to azole antifungal agents, while clades 7 to 11 contained no isolates resistant to any of the three agents tested (Table 2). However, the overall differences in clade distributions of the 378 isolates with susceptibility data available were not signifi-cant for those that were resistant to fluconazole (Fisher’s exact test,P⫽0.289), itraconazole (P⫽0.94), or flucytosine (P⫽
0.105). Other significant associations were, however, apparent for the antifungal resistant isolates. Fifteen (65%) of the 23 flucytosine-resistant isolates were members of clade 1. In every one of these isolates, flucytosine resistance was associated with a homozygous allele pair encoding cysteine at position 101 in the FUR1gene. In a random selection of 14 flucytosine-sus-ceptible isolates from clade 1, the equivalent position was either homozygous for arginine or heterozygous (cysteine/ar-ginine). The eight flucytosine-resistant isolates from other clades and seven randomly selected susceptible isolates from other clades were all homozygous, encoding arginine at posi-tion 101, except for one flucytosine-resistant isolate for which noFUR1sequence was obtained despite three attempts. No common mutation was found in theFUR1,FCY22, orFCA1
gene sequences of these non-clade-1, flucytosine-resistant iso-lates that might account for their resistance to the drug. Among the 35 fluconazole-resistant isolates, 17 (48.6%) were homozygous at the MTL. The same was true for 10 (41.7%) of the 24 itraconazole-resistant isolates and for 7 (21.2%) of the 23 flucytosine-resistant isolates. The prevalence of MTL ho-mozygosity among the corresponding susceptible isolates was 7.3, 9.0, and 9.9%, respectively (by Fisher’s exact test,Pwas ⬍0.001,⬍0.001, and equal to 0.004, respectively). There was no significant association between itraconazole or flucytosine resistance and ABC type, but an unusually high proportion (40%) of 35 isolates resistant to fluconazole were ABC type B (Fisher’s exact test,P⫽0.013).
Twenty-eight of the 35 fluconazole-resistant isolates came from the oropharynx; all of these were from AIDS patients. For 109 oropharyngeal isolates, the HIV status of the patient source was known. The distribution of oral isolates from HIV-positive versus HIV-negative patients differed significantly be-tween clades, as follows. For 48 HIV-negative patients, 26.8% were in clade 1, 17.9% in clade 2, 0.0% in clade 3, 17.9% in clade 4, and 23.2% in minor clades, and 14.3% were singletons. For 61 HIV-positive patients, the corresponding data were 50.7%, 9.0%, 9.0%, 6.0%, 16.4%, and 9.0% (Fisher’s exact test,
P⬍0.001).
The individual data comparisons made by Fisher’s exact test can be vulnerable to biases arising from uneven distributions of contributing variables other than the variable under analysis. A univariate ANOVA was done to determine the associations between properties and sources of isolates and their clade distribution. Animal isolates were excluded. The ANOVA was repeated with individual clades and singletons as the depen-dent variable and also with the minor clades included as a single variable. With individual clades analyzed, ABC type emerged as the most significant variable (P⬍0.001) and an-atomical source as the next most significant variable (P ⫽
0.027). With minor clades grouped, the correspondingPvalues were⬍0.001 for ABC type and 0.065 for anatomical source.
MTL status, fluconazole, itraconazole, and flucytosine resis-tance, decade of isolation, and geographical source were not significant clade-related variables by ANOVA.
DISCUSSION
This study has generated a considerable amount of data on the population structure of isolates of C. albicans and the relationship between clades of isolates and their properties of clinical relevance. We have provided evidence that different clades ofC. albicansmay differ significantly in the proportions of isolates from blood, the oropharynx, the vagina, and other sites. While, on the one hand, it is beyond dispute that the immune status of the host is the major factor determining the ability of aC. albicansto invade human tissues (8, 28), our data provide the first-ever clear suggestion that particular strain type clusters within the species may differ in their proportions of isolates from disseminated or mucosal infections. It is espe-cially notable that, while the proportions of several properties of the isolates studied varied significantly between clades in single-factor tests, only ABC type and anatomical origin emerged as significant factors relating to clade number by univariate ANOVA. This result lends particular emphasis to the likely importance of an association between clades and putative virulence properties within the species ofC. albicans. Assignation of population structures and clade boundaries to a set of isolates cannot be done entirely objectively. The clade assignments in Fig. 2 take account of three types of information: the UPGMA dendrogram, results of eBURST analysis, and the results of Ca3 oligonucleotide fingerprinting for several of the isolates. Use of neighbor-joining statistics in place of pairwise-difference analyses further redistributed some isolates between clades (Fig. 3). We intend to evaluate a wider range of statistical approaches to clade designation, in-cluding Bayesian statistics, when our database of MLST results is at least twice as large as that reported in this study. For the time being, we consider pairwise-difference analyses to be the simplest and most useful approach for clade assignments in the population of isolates at hand.
The eBURST results were helpful in demarcating potential clades, but not definitive: many examples occurred where iso-lates that coclustered by Ca3 typing or by MLST analyzed by pair differences were designated as singletons by the eBURST test. It may be that the nature of sequence variation in C. albicansand the diploid genome of the species make eBURST analyses less relevant than they are to haploid bacteria (50). In common with others (4), we found an excellent correlation between UPGMA clusters for MLST and Ca3 fingerprinting data. Clades I, II, III, and SA as currently delimited by Ca3 typing (46) matched clades 1 to 4, respectively, as defined by MLST in the present study. Only MLST clade E failed to emerge as a single group of isolates as determined by MLST; the 5 representatives of Ca3 clade E split into two clusters by UPGMA analysis of MLST data (Fig. 2).
The very clear demarcation of geographical origin of strains determined by Ca3 typing as described previously (46) was not confirmed in the present study. Our panel of isolates was dominated by isolates of European origin, particularly by iso-lates from the United Kingdom, while the panel of isoiso-lates studied by Ca3 typing (36, 46) was dominated by isolates from
on May 15, 2020 by guest
http://jcm.asm.org/
South Africa. In common with the Ca3 studies, we found sig-nificant interclade differences in proportions of various geo-graphical origins, but the relationships between clade and or-igin were not the same, almost certainly because of the large differences in proportions of isolates from different sources. Most notably, MLST clade 4, which corresponded to Ca3 clade SA, contained only three isolates of African origin, while most of the African isolates in our study clustered in clade 2. We have therefore chosen to refer to clades only by numbers until the MLST database contains a sufficiently large representation of isolates from all parts of the globe to allow more objective determination of possible geographic evolutionary origins of clades. The statistical significance of geographical variations between MLST clades determined by Fisher’s exact test was not confirmed in the ANOVA, perhaps showing the effect of biases from differences in anatomical origin, date of isolation, etc. A larger MLST database will allow for statistical analyses of geographical origin based on, for example, only blood iso-lates or only oral isoiso-lates. It is to be expected that individualC. albicansclades may be enriched with isolates from a particular geographical source, and several previous studies by other DNA typing methods have concluded that geographical en-richment ofC. albicansclades is a real phenomenon (10, 23, 44), even to the extent of regional differences within the Amer-ican continent (36, 46).
However, there seems also to have been sufficient mixing of isolates, possibly because of international migrations, to blur the geographical distinctions, and a microsatellite-based typing survey found genetic homogeneity of isolates in four different locations in the United States (24).
One unequivocal conclusion that we can draw is that C. albicansisolates from animals are distributed differently from human isolates in the MLST population structure. The differ-ences in the present study, which are self-evident even without the high statistical support, confirm a similarly large difference in distribution of strains from birds compared with human isolates by MLST done with a slightly different gene set (4).
The speciesC. africanawas erected to delineateC. albicans
isolates with atypical carbohydrate assimilation patterns (49). All the isolates with the characteristics of C. africana were cultured from male or female genitalia, and most came from patients in Africa. TheC. africanatype strain MYA-2669 (from the penis of a German patient) clustered indistinguishably with two United Kingdom vaginal isolates by MLST, so this type continues to be associated exclusively with isolates of genital origin. Although the three isolates were highly distinct from the remainder of the panel we typed by MLST in the UPGMA dendrogram (Fig. 2), they were less distinct in an unrooted neighbor-joining tree (Fig. 3). The fact that all seven of the gene fragments that we used for MLST easily gave PCR prod-ucts with MYA-2669 contrasts with the situation that we en-countered when we attempted to typeC. parapsilosisisolates by MLST and ultimately proposed two new species to account for isolates that gave PCR products with only a minority of the gene set tested (47). In common with others (17), we think the case for separate species status forC. africanais not confirmed by the results of the present study.
Our data concerning the antifungal susceptibility of our panel of isolates confirms and extends existing information. Like Pujol et al. (37), we found that a majority of
flucytosine-resistant isolates belonged to clade 1. Since almost all clade 1 isolates are also ABC type A, this conclusion corroborates the work of McCullough et al. (26), who also found a significantly higher proportion of flucytosine-resistant isolates among type A isolates. All clade 1 isolates with flucytosine resistance en-coded arginine at position 101 in theFUR1gene product, as previously reported by others (14, 18). This mutation will pre-sumably retard or prevent the conversion of flucytosine to fungicidal 5-fluorouracil within the fungal cells. Flucytosine-susceptible isolates in clade 1 did not encode arginine at po-sition 101, nor did resistant and susceptible isolates from other clades. This finding of an apparent clade-specific molecular resistance mechanism to an antifungal agent opens the possi-bility for development of a molecular diagnostic test to predict flucytosine resistance and raises the expectation that further research may elucidate clade-consistent molecular bases for resistance to other agents. Although we did not uncover any self-evident associations betweenC. albicansclades and resis-tance to azole antifungal agents, the numbers of resistant iso-lates available to us was relatively small, and we have not investigated the molecular basis of resistance in these isolates. It should be noted that all of the isolates tested against flucon-azole, itraconflucon-azole, and flucytosine in this study were also tested for caspofungin susceptibility, but none showed resis-tance to this agent, so this result was not detailed.
It is notable that most of the azole-resistant isolates of C. albicans in our panel were oral isolates from HIV-positive patients. A recent, very large, global surveillance study shows a very low prevalence of fluconazole resistance amongC. albi-cansisolates since the introduction of highly active antiretro-viral therapy (HAART) in 1996 (34). In the absence of a substantial future failure of efficacy of HAART, it is likely to prove difficult to obtain substantial numbers of azole-resistant clinical isolates of C. albicans for further investigation. The strong association that we saw between homozygosity at theC. albicansmating type locus and fluconazole resistance was de-scribed previously (40). Our data extend this association to itraconazole and even to flucytosine, which exerts its antifungal effects by a mechanism different from that of the azoles. Pre-vious experimentation showed that engineering homozygosity at theC. albicansMTL does not lead per se to the develop-ment of fluconazole resistance (35). The association that we describe between MTL homozygosity and resistance to several antifungals therefore suggests that a common event, perhaps a change in expression of a transcription factor (11), is respon-sible both for resistance development and MTL homozygosity in wild-type isolates.
An important consideration to emerge from the determina-tion ofC. albicanspopulation structure concerns the choice of strains used for experimental work with the fungus. To date, almost all molecular research withC. albicans has been done with the single clade 1 isolate SC5314 and its Ura–derivatives,
in the absence of positive selectable markers for molecular genetic work. Now that at least one positive selectable marker has been developed forC. albicans(52), our delineation of the four major clades of the species, and a preliminary demonstra-tion of potential clade-related differences in the propensity to cause infection, form a basis for rational selection of a wider diversity of strains for future research in the field.
on May 15, 2020 by guest
http://jcm.asm.org/
ACKNOWLEDGMENTS
This study was supported by a grant from the Wellcome Trust. We gratefully acknowledge the many colleagues who have supplied us over many years with the isolates we submitted to MLST. We are particularly grateful to E. M. Johnson, C. C. Kibbler, N. Nolard, F. Symoens (at the IHEM collection), J. B. Anderson, D. R. Soll, and J. Schmid for supplying us with well-pedigreed isolates for this MLST project and to Marie-Elisabeth Bougnoux and Christophe d’Enfert for DNA from their panel of isolates.
REFERENCES
1.Anderson, J. M., and D. R. Soll.1987. The unique phenotype of opaque cells in the “white-opaque transition” inCandida albicans. J. Bacteriol.169:5579– 5588.
2.Blignaut, E., C. Pujol, S. Lockhart, S. Joly, and D. R. Soll. 2002. Ca3 fingerprinting ofCandida albicansisolates from human immunodeficiency virus-positive and healthy individuals reveals a new clade in South Africa. J. Clin. Microbiol.40:826–836.
3.Blignaut, E., C. Pujol, S. Lockhart, S. Joly, and D. R. Soll.2002. A new clade ofCandida albicansamong South African oral yeast isolates. J. Dent. Res.
81:2231.
4.Bougnoux, M.-E., A. D. M., S. Morand, M. The´raud, B. G. Spratt, and C. d’Enfert.2004. Multilocus sequence typing ofCandida albicans: strategies, data exchange and applications. Infect. Genet. Evol.4:243–252.
5.Bougnoux, M.-E., S. Morand, and C. d’Enfert.2002. Usefulness of multilo-cus sequence typing for characterization of clinical isolates ofCandida albi-cans. J. Clin. Microbiol.40:1290–1297.
6.Bougnoux, M. E., A. Tavanti, C. Bouchier, N. A. R. Gow, A. Magnier, A. D. Davidson, M. C. J. Maiden, C. d’Enfert, and F. C. Odds.2003. Collaborative consensus for optimized multilocus sequence typing ofCandida albicans. J. Clin. Microbiol.41:5265–5266.
7.Burnie, J. P., F. C. Odds, W. Lee, C. Webster, and J. D. Williams.1985. Outbreak of systemicCandida albicansin intensive care unit caused by cross infection. Brit. Med. J.290:746–748.
8.Calderone, R. A.2002. Candida and candidiasis. ASM Press, Washington, D.C. 9.Chan, M. S., M. C. J. Maiden, and B. G. Spratt.2001. Database-driven multi locus sequence typing (MLST) of bacterial pathogens. Bioinformatics17:
1077–1083.
10.Clemons, K. V., F. Feroze, K. Holmberg, and D. A. Stevens.1997. Compar-ative analysis of genetic variability amongCandida albicansisolates from different geographic locales by three genotypic methods. J. Clin. Microbiol.
35:1332–1336.
11.Coste, A. T., M. Karababa, F. Ischer, J. Bille, and D. Sanglard.2004.TAC1, transcriptional activator ofCDRgenes, is a new transcription factor involved in the regulation ofCandida albicansABC transportersCDR1andCDR2. Eukaryot. Cell3:1639–1652.
12.Cowen, L. E., A. Nantel, M. S. Whiteway, D. Y. Thomas, D. C. Tessier, L. M. Kohn, and J. B. Anderson.2002. Population genomics of drug resistance in
Candida albicans. Proc. Natl. Acad. Sci. USA99:9284–9289.
13.Cowen, L. E., D. Sanglard, D. Calabrese, C. Sirjusingh, J. B. Anderson, and L. M. Kohn.2000. Evolution of drug resistance in experimental populations ofCandida albicans. J. Bacteriol.182:1515–1522.
14.Dodgson, A. R., K. J. Dodgson, C. Pujol, M. A. Pfaller, and D. R. Soll.2004. Clade-specific flucytosine resistance is due to a single nucleotide change in theFUR1gene ofCandida albicans. Antimicrob. Agents Chemother.48:
2223–2227.
15.Feil, E. J., B. C. Li, D. M. Aanensen, W. P. Hanage, and B. G. Spratt.2004. eBURST: inferring patterns of evolutionary descent among clusters of re-lated bacterial genotypes from multilocus sequence typing data. J. Bacteriol.
186:1518–1530.
16.Fonzi, W., and M. Irwin.1993. Isogenic strain construction and gene map-ping inCandida albicans. Genetics134:717–728.
17.Forche, A., G. Scho¨nian, Y. Gra¨ser, R. Vilgalys, and T. G. Mitchell.1999. Genetic structure of typical and atypical populations ofCandida albicans
from Africa. Fungal Genet. Biol.28:107–125.
18.Hope, W. W., L. Tabernero, D. W. Denning, and M. J. Anderson.2004. Molecular mechanisms of primary resistance to flucytosine inCandida albi-cans. Antimicrob. Agents Chemother.78:4377–4386.
19.Horsburgh, C. R., and C. H. Kirkpatrick. 1983. Long-term therapy of chronic mucocutaneous candidiasis with ketoconazole: experience with twenty-one patients. Am. J. Med.74:23–29.
20.Hunter, P. R.1990. Reproducibility and indices of discriminatory power of microbial typing methods. J. Clin. Microbiol.28:1903–1905.
21.Jones, T., N. A. Federspiel, H. Chibana, J. Dungan, S. Kalman, B. B. Magee, G. Newport, Y. R. Thorstenson, N. Agabian, P. T. Magee, R. W. Davis, and S. Scherer.2004. The diploid genome sequence ofCandida albicans. Proc. Natl. Acad. Sci. USA101:7329–7334.
22.Kumar, S., K. Tamura, I. B. Jakobsen, and M. Nei.2001. MEGA2: Molec-ular Evolutionary Genetics Analysis software. Bioinformatics17:1244–1245. 23.Lott, T. J., and M. M. Effat.2001. Evidence for a more recently evolved clade
within aCandida albicansNorth American population. Microbiology147:
1687–1692.
24.Lott, T. J., R. E. Fundyga, M. E. Brandt, L. H. Harrison, A. N. Sofair, R. A. Hajjeh, and D. W. Warnock.2003. Stability of allelic frequencies and distri-butions ofCandida albicansmicrosatellite loci from US population-based surveillance isolates. J. Clin. Microbiol.41:1316–1321.
25.Mackenzie, D. W. R., and F. C. Odds.1991. Non-identity and authentication of two major reference strains ofCandida albicans. J. Med. Vet. Mycol.
29:255–261.
26.McCullough, M. J., K. V. Clemons, and D. A. Stevens.1999. Molecular and phenotypic characterization of genotypicCandida albicanssubgroups and comparison withCandida dubliniensisandCandida stellatoidea. J. Clin. Mi-crobiol.37:417–421.
27.NCCLS.2002. Reference method for broth dilution antifungal susceptibility testing of yeasts; approved standard, 2nd ed., vol. 17(9). NCCLS, Wayne, Pa. 28.Odds, F. C.1988. Candida and candidosis, 2nd ed. Bailliere Tindall, London,
United Kingdom.
29.Odds, F. C.1991. Long-term preservation of pathogenic yeasts under dis-tilled water. J. Med. Vet. Mycol.29:413–415.
30.Odds, F. C., A. B. Abbott, R. L. Stiller, H. J. Scholer, A. Polak, and D. A. Stevens.1983. Analysis ofCandida albicansphenotypes from different geo-graphical and anatomical sources. J. Clin. Microbiol.18:849–857. 31.Odds, F. C., C. C. Kibbler, E. Walker, A. Bhamra, H. G. Prentice, and P.
Noone.1989. Carriage ofCandidaspecies andC. albicansbiotypes in patients undergoing chemotherapy or bone marrow transplantation for haematologi-cal disease. J. Clin. Pathol.42:1259–1266.
32.Odds, F. C., L. Van Nuffel, and G. Dams.1998. Prevalence ofCandida dubliniensisisolates in a yeast stock collection. J. Clin. Microbiol.36:2869– 2873.
33.Odds, F. C., L. Vranckx, and F. Woestenborghs.1995. Antifungal suscepti-bility testing of yeasts: evaluation of technical variables for test automation. Antimicrob. Agents Chemother.39:2051–2060.
34.Pfaller, M. A., and D. J. Diekema.2004. Twelve years of fluconazole in clinical practice: global trends in species distribution and fluconazole sus-ceptibility of bloodstream isolates of Candida. Clin. Microbiol. Infect.
10(Suppl. 1):11–23.
35.Pujol, C., S. A. Messer, M. Pfaller, and D. R. Soll.2003. Drug resistance is not directly affected by mating type locus zygosity inCandida albicans. Antimicrob Agents Chemother.47:1207–1212.
36.Pujol, C., M. Pfaller, and D. R. Soll.2002. Ca3 fingerprinting ofCandida albicansbloodstream isolates from the United States, Canada, South Amer-ica, and Europe reveals a European clade. J. Clin. Microbiol.40:2729–2740. 37.Pujol, C., M. A. Pfaller, and D. R. Soll. 2004. Flucytosine resistance is restricted to a single genetic clade ofCandida albicans. Antimicrob. Agents Chemother.48:262–266.
38.Redding, S., J. Smith, G. Farinacci, M. Rinaldi, A. Fothergill, J. Rhinechal-berg, and M. Pfaller.1994. Resistance ofCandida albicansto fluconazole during treatment of oropharyngeal candidiasis in a patient with AIDS— documentation by in vitro susceptibility testing and DNA subtype analysis. Clin. Infect. Dis.18:240–242.
39.Rex, J. H., C. R. Cooper, W. G. Merz, J. N. Galgiani, and E. J. Anaissie.1995. Detection of amphotericin B-resistant Candida isolates in a broth-based system. Antimicrob. Agents Chemother.39:906–909.
40.Rustad, T. R., D. A. Stevens, M. A. Pfaller, and T. C. White.2002. Homozy-gosity at theCandida albicansMTL locus associated with azole resistance. Microbiology148:1061–1072.
41.Sanglard, D., K. Kuchler, F. Ischer, J. L. Pagani, M. Monod, and J. Bille.
1995. Mechanisms of resistance to azole antifungal agents inCandida albi-cansisolates from AIDS patients involve specific multidrug transporters. Antimicrob. Agents Chemother.39:2378–2386.
42.Schmid, J., S. Herd, P. R. Hunter, R. D. Cannon, M. S. M. Yasin, S. Samad, M. Carr, D. Parr, W. McKinney, M. Schousboe, B. Harris, R. Ikram, M. Harris, A. Restrepo, G. Hoyos, and K. P. Singh. 1999. Evidence for a general-purpose genotype inCandida albicans, highly prevalent in multiple geographical regions, patient types and types of infection. Microbiology
145:2405–2413.
43.Schmid, J., P. R. Hunter, G. C. White, A. K. Nand, and R. D. Cannon.1995. Physiological traits associated with success ofCandida albicansstrains as commensal colonizers and pathogens. J. Clin. Microbiol.33:2920–2926. 44.Schonian, G., A. Forche, H. J. Tietz, M. Muller, Y. Graser, R. Vilgalys, T. G.
Mitchell, and W. Presber.2000. Genetic structure of geographically different populations ofCandida albicans. Mycoses43:51–56.
45.Soll, D. R.2004. Mating-type locus homozygosis, phenotypic switching and mating: a unique sequence of dependencies inCandida albicans. Bioessays
26:10–20.
46.Soll, D. R., and C. Pujol.2003.Candida albicansclades. FEMS Immunol. Med. Microbiol.39:1–7.
47.Tavanti, A., A. D. Davidson, N. A. R. Gow, M. C. J. Maiden, and F. C. Odds.
2005.Candida orthopsilosisandCandida metapsilosisspp. nov. to replace
Candida parapsilosisgroups II and III. J. Clin. Microbiol.43:284–292.
on May 15, 2020 by guest
http://jcm.asm.org/
48.Tavanti, A., N. A. R. Gow, S. Senesi, M. C. J. Maiden, and F. C. Odds.2003. optimization and validation of multilocus sequence typing forCandida albi-cans. J. Clin. Microbiol.41:3765–3776.
49.Tietz, H. J., M. Hopp, A. Schmalreck, W. Sterry, and V. Czaika.2001.
Candida africanasp. nov., a new human pathogen or a variant ofCandida albicans? Mycoses44:437–445.
50.Urwin, R., and M. C. J. Maiden.2003. Multi-locus sequence typing: a tool for global epidemiology. Trends Microbiol.11:479–487.
51.White, T. C.1997. Increased mRNA levels ofERG16, CDR, andMDR1
correlate with increases in azole resistance inCandida albicansisolates from a patient infected with human immunodeficiency virus. Antimicrob. Agents Chemother.41:1482–1487.
52.Wirsching, S., S. Michel, and J. Morschhauser.2000. Targeted gene disrup-tion inCandida albicanswild-type strains: the role of theMDR1gene in fluconazole resistance of clinicalCandida albicansisolates. Mol. Microbiol.
36:856–865.