Flight Mass Spectrum of
Staphylococcus aureus
Identifies Mutations
That Allow Differentiation of the Main Clonal Lineages
Michaele Josten,aMarion Reif,aChristiane Szekat,aNahed Al-Sabti,aTerry Roemer,bKatrin Sparbier,cMarkus Kostrzewa,c Holger Rohde,dHans-Georg Sahl,aGabriele Bierbauma
University of Bonn, Institute of Medical Microbiology, Immunology, and Parasitology, Bonn, Germanya; Merck & Co., Infectious Diseases, Kenilworth, New Jersey, USAb; Bruker Daltonik GmbH, Bremen, Germanyc; University Clinic Hamburg-Eppendorf, Institute for Medical Microbiology, Virology and Hygiene, Hamburg, Germanyd
Nosocomial infections involving epidemic methicillin-resistantStaphylococcus aureus(MRSA) strains are a serious problem in many countries. In order to analyze outbreaks, the infectious isolates have to be typed; however, most molecular methods are expensive or labor-intensive. Here, we evaluated matrix-assisted laser desorption ionization–time of flight mass spectrometry (MALDI-TOF MS) of cell extracts for the molecular characterization ofS. aureusstrains. The peak patterns of 401 MRSA and methicillin-susceptibleS. aureus(MSSA) strains, including clinical and laboratory strains, were analyzed. Database searches indicated the peptides that were represented by the corresponding peaks in the spectra. The identities of the peptides were con-firmed by the sequencing of mutants, the expression of antisense RNA fragments that resulted in the knockdown of the peptide of interest and the concomitant loss of the signal, or tandem MALDI-TOF MS (MALDI-TOF/TOF MS). It was shown that the signals derive mainly from stress proteins and ribosomal proteins. Peak shifts that differentiate the mainS. aureusclonal com-plexes CC5, CC22, CC8, CC45, CC30, and CC1 correlate to point mutations in the respective genes. Retrospective typing of an MRSA outbreak showed that it is possible to differentiate unrelated MSSA, MRSA, and borderline resistantS. aureus(BORSA) strains isolated from health care workers. In conclusion, this method allows for the detection of the epidemic lineages ofS. au-reusduring species identification by MALDI-TOF MS analysis.
S
taphylococcus aureuscauses a wide range of hospital infections.Often, these infections involve epidemic methicillin-resistant
S. aureus(MRSA) strains that are transferred by health care
work-ers to patients. In order to detect outbreaks that are caused by epidemic strains, the clinical isolates have to be typed. Multilocus sequence typing (MLST) relies on the sequence analysis of house-keeping genes and is used to allocate the strains to sequence types (ST), which can be grouped into clonal complexes (CC). This method has provided a detailed insight into the population struc-ture of MRSA and methicillin-susceptible S. aureus (MSSA) strains (1). Finer discrimination is achieved by pulsed-field gel electrophoresis (PFGE) andspatyping (2). All of these methods need additional experimentation.
In matrix-assisted laser desorption ionization–time of flight mass spectrometry (MALDI-TOF MS), different spectra or signa-tures of cell extracts (3,4,5,6) or whole cells (7,8,9,10,11,12) could be identified for different strains or groups of strains. An increasing number of laboratories use MALDI-TOF MS for the identification ofS. aureus. However, the differences in the signa-tures of the strains have not been evaluated so far, because the discriminatory threshold of the software used in clinical settings is set up to assign the isolate to a species. To this end, more subtle differences are ignored. Another reason is that, so far, MALDI-TOF MS of whole bacterial cells has been employed in a heuristic manner, and for most species, the identities of the compounds that are detected in the measurements are unknown. Thus, the spectra are not well understood, and the variations in the signa-tures cannot be interpreted.
In principle, two spectra might differ by signal intensity, loss of a signal, or by the shift of a signal. Variations in signal intensity are probably caused by expression differences, which might be
di-rectly correlated with culture conditions and, therefore, do not give unambiguous information about a genotype of the strain. The loss of a signal is caused by the total failure to express a protein or peptide, which in turn might indicate a mutation causing a frameshift or stop codon, but might also depend on culture con-ditions, mutations of regulatory factors, or sample preparation. Thus, there is no clear-cut correlation between the loss of a signal and a genotype. In contrast, peak shifts (i.e., loss of a signal cou-pled to the appearance of a new signal, both of which are corre-lated to the same peptide) correspond to point mutations in the genes of the peptides detected in the analysis; the mutation leads to an amino acid exchange that alters the molecular weight of the corresponding gene product.
In order to characterize the clonal lineages ofS. aureusin the MALDI-TOF MS, this work was aimed at the identification of the peptides that are detected in the spectra and that show mass vari-ations between the clonal complexes ofS. aureus. To this end, we analyzed the spectra of 401S. aureusstrains, concentrating on peak shifts, and we correlated these peak shifts to mutations, i.e., the genotypes of the strains. The results showed that the signals in MALDI-TOF MS correspond to (i) more- or less-conserved housekeeping proteins, e.g., the ribosomal proteins, and (ii) other
Received25 February 2013 Returned for modification21 March 2013
Accepted27 March 2013
Published ahead of print3 April 2013
Address correspondence to Gabriele Bierbaum, [email protected]. Copyright © 2013, American Society for Microbiology. All Rights Reserved.
doi:10.1128/JCM.00518-13
on May 16, 2020 by guest
http://jcm.asm.org/
peptides, e.g., stress proteins and low-molecular-weight toxins, and that these peptides sometimes appear in multiple variants that allow for the detection of subgroups of strains. These differences may serve to identify the clonal lineage of an isolate directly by MALDI-TOF MS analysis.
MATERIALS AND METHODS
Bacterial strains.The analyzed bacterial strains included 7 well-charac-terized laboratory MSSA and 14 MRSA type strains (Table 1) that cover the most relevant clonal lineages from Europe and northern America, 247 clinical MRSA strains, 38 strains isolated during an outbreak, and 95 clin-ical MSSA strains. In the first phase, the algorithm for the discrimination of clonal lineages was set up with the type strain panel and 111 clinical strains that showed the typical phage or PFGE patterns of the German endemic epidemic clones (Rhine-Hesse epidemic clone [t002 and t003, CC5], the Barnim epidemic clone [CC22], or the Berlin epidemic clone [CC45]). In the second phase, all endemic epidemic isolates with typical phage patterns were excluded from the study, and only 136 strains that showed deviating autonomous phage patterns or were nontypeable by phages were included in the analysis in a blind fashion and werespatyped later (13). PFGE of 38 outbreak isolates and CC22 isolates was performed as described previously (14).
MALDI-TOF MS measurements.In order to mimic routine condi-tions, one or several uniform colonies from fresh Columbia blood agar plates (Becton-Dickinson, Heidelberg, Germany) were resuspended into 300l of distilled water (Baker, Deventer, Netherlands). After the addi-tion of 900l ethanol (Baker), the extraction tube (Eppendorf, Hamburg, Germany) was centrifuged for 2 min at 20,000⫻g(2K15; Sigma, Os-terode, Germany), the supernatant was discarded, and the bacterial pellet was resuspended in 50l of 70% formic acid. Before centrifuging again, 50l acetonitrile was added. One microliter of the supernatant was placed onto a ground steel MALDI-TOF MS target plate and allowed to dry at room temperature. Subsequently, each sample was overlaid with 2l of matrix (saturated solution of␣-cyano-4-hydroxycinnamic acid in 50% acetonitrile-2.5% trifluoroacetic acid [TFA]) and air dried at room tem-perature. Two MS profile spectra were obtained for each strain on a Biflex III (Bruker Daltonik GmbH, Bremen, Germany) in the linear positive mode. Data analysis was performed using the flexAnalysis software (Bruker Daltonik GmbH).
[image:2.585.39.551.77.473.2]For cleaning, the ground steel target plates were brushed in 50% meth-anol, immersed in an ultrasonic bath for 30 min, then were brushed again using 50% methanol, immersed in acetone, and air dried. After this clean-ing procedure, the purity of the target plates was checked by reflected light microscopy.
TABLE 1Strains and constructs used in the project
Strain or antisense expt host Epidemic type (spagene) or plasmid Reference, source, or NCBI accession no. of genome sequence
MRSA strains
S. aureusN315 Japan-New York ST5 (t002) NC_002745
S. aureusMu50 Japan-New York ST5 (t002) NC_002758
S. aureusCOL ST250 NC_002951
S. aureusLT2981-04 ST225 (t003) PFGE Rhine-Hesse CP001844
S. aureusSanger 252 ST30 BX571856
S. aureusUSA100 ST5 (t002) NARSA: NRS382
S. aureusLT3391-02 ST5 (t002) PFGE Rhine-Hesse German Reference Center for Staphylococci, Wernigerode, Germany S. aureusLT1450-94 Northern German, Iberian ST247 German Reference Center for Staphylococci, Wernigerode, Germany S. aureusLT825-96 Berlin ST45 German Reference Center for Staphylococci, Wernigerode, Germany S. aureusLT831-96 Southern German ST228 German Reference Center for Staphylococci, Wernigerode, Germany S. aureusLT635-93 Vienna ST239 German Reference Center for Staphylococci, Wernigerode, Germany S. aureusLT1000-93 Hannover ST254 German Reference Center for Staphylococci, Wernigerode, Germany S. aureusLT2321-01 Barnim EMRSA15 ST22 German Reference Center for Staphylococci, Wernigerode, Germany S. aureusLT2040-05 USA300 ST8 German Reference Center for Staphylococci, Wernigerode, Germany
111 MRSA strains Clinical MRSA isolates This study
136 MRSA strains Clinical MRSA isolates (blind group) This study
38 outbreak strains Mainly MRSA 14
MSSA strains
S. aureusNCTC8325 (RN1) Laboratory MSSA,rsbU-negative ST8, t211 NC_007795 S. aureusNCTC8325-4
(RN0450)
NCTC8325 after curation of phages, rsbU-negative
NARSA: NRS135
S. aureusSH1000 S. aureusNCTC 8325-4rsbUrepaired 24
S. aureusRN4220 Restriction-negative derivative of NCTC8325-4 NARSA: NRS144
S. aureusReynolds MSSA ST25 NARSA: NRS102
S. aureusCowan I (S11) MSSA ST30 NCTC: NCTC 8530
S. aureusNewman MSSA ST254 AP009351
95 MSSA strains Clinical MSSA isolates This study
Antisense expt host
S. aureusRN4220 pEPSA5-AS-SAS078 This study
S. aureusSanger 252 pEPSA5-AS-SAR1012 This study
S. aureusRN4220,S. aureus N315
pEPSA5-AS-SA0772 This study
S. aureusRN4220 pEPSA5-AS-SA2039 This study
S. aureusRN4220 pEPSA5-AS-SA1414 This study
S. aureusRN4220 pEPSA5-AS-SA1305 This study
on May 16, 2020 by guest
http://jcm.asm.org/
In order to exclude the possibility that medium variations in blood agar charges would influence the peak shifts, the extraction and measure-ment of the primary set of type strains were repeated after 6 months.
Identification of peaks.Masses measured during MALDI-TOF MS were converted into theoretical calculated masses that contain the N-ter-minal Met, Val, or Leu, or theN-formyl-Met residue. Peak identities were then queried using the AureusDB database (http://aureusdb.biologie.uni -greifswald.de/) and the Swiss-Prot database using the TagIdent tool (http://web.expasy.org/tagident/).
The identities of the peaks were confirmed by sequencing strains that showed peak shifts; the primers used and strains sequenced are listed in
Table 2(Seqlab, Göttingen, Germany). Point mutations were translated into the resulting amino acid sequence, and the predicted shift in molec-ular weight was compared to that of the measured shift. Peak shifts that were encountered in genome-sequenced strains were correlated to se-quence variations using the NCBI database or the database of the Sanger Institute (http://www.sanger.ac.uk/cgi-bin/blast/submitblast/s_aureus). Alternatively, employingS. aureusNCTC 8325 as a template, a short frag-ment (about 100 bp) of the corresponding open reading frame (ORF) was amplified by PCR (for the primers used, seeTable 2) carrying EcoRI and XbaI cleavage sites. These fragments were cloned into the pEPSA5 vector (15) usingEscherichia coliJM83. The plasmids were then shuttled toS. aureusRN4220 and, if necessary, toS. aureusN315 (for genes under SigB control) or MRSA Sanger 252 by electroporation (16). The strains were cultured on lysogeny broth (LB) agar plates containing 12.5, 25, or 50 mM
xylose and 34 mg/liter chloramphenicol, or in LB broth (optical density of
ⱕ1.5). Subsequently, the MALDI-TOF MS spectra were compared. The following controls were analyzed on the same day after growth on the same batch of LB containing 34 mg/liter chloramphenicol: the clone car-rying the empty vector pEPSA5 grown in the absence of xylose, the clone with the empty vector grown in the presence of xylose, and the clone harboring the recombinant plasmid with the antisense fragment grown in the absence of xylose. The method that was chosen for the confirmation of each peak is presented inTable 3.
The peak atm/z3,875 was analyzed by a fractionated elution from hydrophobic microbeads, followed by liquid chromatography matrix-as-sisted laser desorption ionization–time of flight/time of flight (LC-MALDI-TOF/TOF) MS analysis. Bacteria were lysed according to the MALDI Biotyper standard protocol. After centrifugation, the cell-free su-pernatant was diluted with 0.1% TFA to reduce the acetonitrile concen-tration to 10%. Subsequently, the supernatant was incubated with mag-netic HIC C8 beads (Bruker Daltonik GmbH). The beads were washed with 40% acetonitrile-0.1% TFA, and bound compounds were eluted with 60% acetonitrile-0.1% TFA. After drying in a vacuum centrifuge, the pellet was redissolved in 20% acetonitrile-0.1% TFA and directly sub-jected to LC-MALDI-TOF/TOF MS (17,18).
[image:3.585.41.547.79.444.2]Determination of sensitivity and specificity.These values are based on the double spectra of 261 MRSA strains that werespatypeable, includ-ing the type strains and SigB-negative strains. With the exception of the SAR1012 peak and the GraC truncation, only the presence of true peak TABLE 2Primers used in this study
Target gene Primer Primer sequence S. aureusstrain(s) used for sequencing
Primers used for cloning of antisense products
rpmJ(SAS078) SAS078for AAATCTAGA AAGACCATCAGTAAAACCTATTTGC
SAS078rev AAAGAATTC CCTTGTCTTTGTTTGTGTTTTGG
SAR1012 SAR1012for AAAGAATTC TGTTTATTATCTGTCGGATGTGA
SAR1012rev AAATCTAGA CGATGCGTCAATTCATTAAAAG
SA0772 SA0772for AAATCTAGA GGCAGACGAAAGTAAATTTGAA
SA0772rev AAAGAATTC TTACTCGCCTTTGTTACCTTTT
rpmC(SA2039) SA2039for AAATCTAGA GCTAAGGAAATTAGAGACTTAACCAC
SA2039rev AAAGAATTC TTTGTTCAATTTCTCTTTCACGA
rpsT(SA1414) SA1414for AAATCTAGA AAAAAGCTGAAGCACGCAAC
SA1414rev AAAGAATTC CGGTCAGCTTTGTTTGAATG
hup(SA1305) SA1305for AAATCTAGA ATCAATGCAGTTGCAGAGCA
SA1305rev AAAGAATTC TGAATGCTGGAACTTTACTTGC
Primers used for PCR and sequencing of genes
hld(SAOUHSC_02260) SAOUHSC_02260for TGGTTATTAAGTTGGGATGGC LT282-12
SAOUHSC_02260rev TTAAGAAAAATTGGAAAATAAATGC
graF(SAS030) forSAS030 GTTTGTAAACTTATGTCAATAGG LT2321-01, LT1261-11
revSAS030 AAAATAATTTCGGCGTGTTTCC
SAS049 forSAS049 AAACTAAGTGAAAGTATGTATCC Cowan, Reynolds, MSSA 19590-11
revSAS049 AGGTTTCACTATGGCAGACG
rpmF(SAS033) SAS033for TTTTTCGTTTATAATCTAAGGAGGAT LT243-12
SAS033rev CAATCATTTTAAGCGGGACTT
rpmD(SA2030) forSAOUHSC_02493 CTTTACGTGAACCTTCTGCC LT1270-11
revSAOUHSC_02493 TGAAGATGTTGCGAAATTACG
SA1452 SA1452for CCATAAAACTCATATTAATCGCTCCT LT98-12, MSSA A796-12, MSSA 1444-12
SA1452rev TTTAGCGAACCTCCTTAGTGGT
graC(SAS044) forSAOUHSC_01362 TTTGAGTTTTGGTATCCGAGG LT181-12, LT183-12, LT1784-05, LT825-96
revSAOUHSC_01362 GGTCAATACATAGAACATCCC
SA2039 forSA2039 CTTTACCTACATAAACTTTACG MSSA A2065-12, LT637-12
revSA2039 GTAAAACGTGAGGAATTGGG
rpsP(SA1081) forSA1081 TCTTTACAAACATTCATACACC LT159-12
revSA1081 TAACATGTGATATTCCCAACG
on May 16, 2020 by guest
http://jcm.asm.org/
shifts was evaluated as a positive test result. A peak shift was defined as the loss of the commonly observed signal in both spectra and the simultane-ous detection of the variant peptide in at least one spectrum. All other possible outcomes were evaluated as negative test results: (i) the absence of the peak shift (i.e., detection of the commonly observed peptide in at least one spectrum), (ii) the failure to detect any of the signals, or (iii) the detection of both signals.
RESULTS
Identification of peptide signals.Previously, different peak pat-terns for the different clonal groups ofS. aureuswere observed in cell extracts (3,5), and therefore, we adhered to this method. The cells were extracted from Columbia blood agar plates that had been incubated overnight, and every clinical sample was measured only twice to mimic routine conditions. After tentative identifica-tion of the proteins via the databases, their identities were con-firmed by the sequencing of mutants, if such strains were available or, in one case, by MALDI-TOF/TOF MS. Otherwise, a fragment of the gene was cloned in antisense direction into the vector pEPSA5 (15), shuttled fromE. coliintoS. aureusRN4220,S.
au-reusN315, orS. aureusSanger 252, and the expression of antisense RNA was induced by the addition of xylose. The identity of the peak was confirmed if MALDI-TOF MS analysis showed the de-pletion of the protein in question in the presence of xylose. As an example, the depletion of the DNA binding protein HU after the expression of antisense RNA is shown inFig. 1. When the signals were strong enough, this method worked well with nonessential proteins and also the nonessential ribosomal proteins. A knock-down of a group of essential ribosomal proteins available through the pEPSA5 antisense library (15) was not possible, as it led to growth arrest and had a general effect on the protein levels (data not shown). Therefore, those essential ribosomal proteins that did not show peak shifts (i.e., mutations) were assumed to be not useful for typing and were not followed further. All confirmed signals and their isoforms are listed inTable 3.
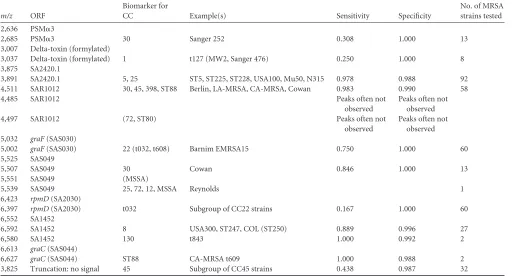
[image:4.585.41.551.79.446.2]Overview of theS. aureusspectrum.The predominant peaks of theS. aureusspectrum were constituted by SA0772 (m/z6,889 [value for doubly charged peptide, 3,445]), which represents a protein of the general stress response, the hypothetical protein TABLE 3Peptides that can be assigned to mass signals and the mutations that lead to peak shifts
m/z ORF
Reference no. or type
of confirmationa IEP sigBb Peptide sequencee
2,201 PSM␣4 19 9.70 fMAIVGTIIKIIKAIIDIFAK
2,289 PSM␣1 9.70 fMGIIAGIIKVIKSLIEQFTGK
2,307 PSM␣2 10.00 fMGIIAGIIKFIKGLIEKFTGK
2,636 PSM␣3 9.52 fMEFVAKLFKFFKDLLGKFLGNN
2,685 PSM␣3 fMEFVAKLFKFFKDLLGKFLGNY
3,007 Delta-toxin 20 8.19 fMAQDIISTIGDLVKWIIDTVNKFTKK
3,037 Delta-toxin fMAQDIISTISDLVKWIIDTVNKFTKK
3,875 SA2420.1 m 9.60 fMKKLAVILTLVGGLYFAFKKYQERVNQAPNIEY
3,891 SA2420.1 fMKKLAVILTLVGGLYYAFKKYQERVNQAPNIEY
4,306 rpmJ(SAS078) k 10.32 MKVRPSVKPICEKCKVIKRKGKVMVICENPKHKQRQG
4,511 SAR1012 k 11.24 fMRQFIKRIVKTILVGYVIKFIRNKLSGKSSHPTDNKHK
4,485c
SAR1012 fMRQFIKRTVKTILVGYVIKFIRNKLSGKSSHPTDNKHN
4,497d SAR1012 fMRQFIKRIVKTILVGYVIKFIRNKLSGKSSHPTDNKHN 5,032 graF(SAS030) 5.05 y SNENQNKKAAEKAKEVEEKLKDKKEEKTEDINQTKQDIQDTLN
5,032 graF(SAS030) s SNENQNKKATEKAKEVEEKLKDKKEVKTEDINQTKQDIQDTLN
5,002 graF(SAS030) s SNENQNKKAAEKAKEVEEKLKDKKEVKTEDINQTKQDIQDTLN
5,525 SAS049 5.07 y SFMDKAKDAVEKFKNSDNEQVKNVKDKINEYTGSNNEEKKENEDKEK
5,507 SAS049 s SFIDKAKDAVEKFKNSDNEQVKNVKDKINEYTGSNNEEKKENEDKEK
5,551 SAS049 s SFMDKAKDAVEKFKNSDNEQVKNVKDKINEYTGLNNEEKKENEDKEK
5,539 SAS049 s SFMDKAKDAVEKFKNSDNEQVKNVKDKINEYTGSNNEEKKEKEDKEK
6,354 rpmF(SAS033) 10.05 AVPKRRTSKTRKNKRRTHFKISVPGMTECPNCGEYKLSHRVCKNCGSYNGEEVAAK
6,412 rpmF(SAS033) s AVPKRRTSKTRKNKRRTHFKISVPDMTECPNCGEYKLSHRVCKNCGSYNGEEVAAK
6,423 rpmD(SA2030) 10.11 AKLQITLTRSVIGRPETQRKTVEALGLKKTNSSVVVEDNPAIRGQINKVKHLVTVEEK
6,397 rpmD(SA2030) s AKLQITLTRSVIGRPETQRKTVEALGLKKTNSSVVVEDNAAIRGQINKVKHLVTVEEK
6,552 SA1452 6.51 y ADESKFDQFKGNVKETVGNVTDNKELEKEGQQDKATGKAKEVVENAKNKITDAIDKLKK
6,592 SA1452 s ADESKFDQFKGNVKETVGNVTDNKELEKEGQQDKVIGKAKEVVENAKNKITDAIDKLKK
6,580 SA1452 s ADESKFDQFKGNVKETVGNVTDNKELEKEGQQDKATGKAKEVVENAKNKITDAIDKLRK
6,568 SA1452 s ADESKFDQFKGNVKETVGNVTDNKELEKEGQQDKATGKAKEVVENAKNKITDSIDKLKK
6,613 graC(SAS044) 6.11 PIVNVKLLEGRSDEQLKNLVSEVTDAVEKTTGANRQAIHVVIEEMKPNHYGVAGVRKSDQ
6,627 graC(SAS044) s PIVNVKLLEGRSDEQLKKLVSEVTDAVEKTTGANRQAIHVVIEEMKPNHYGVAGVRKSDQ
6,641 graC(SAS044) s PIVNVKLLEGRSDEQLKNLVSEVTDVVEKTTGANRQAIHVVIEEMKPNHYGVAGVRKSDQ
3,825c
graC(SAS044) s PIVNVKLLEGRSDEQLKNLVSEVTDAVEKTTGANRstop
6,889 SA0772 k 5.16 y ADESKFEQAKGNVKETVGNVTDNKNLENEGKEDKASGKAKEFVENAKEKATDFIDKVKGNKGE
8,091 rpmC(SA2039) k 9.52 MKAKEIRDLTTSEIEEQIKSSKEELFNLRFQLATGQLEETARIRTVRKTIARLKTVAREREIEQSKANQ
8,019 rpmC(SA2039) s MKAKEIRDLTTSEIEEQIKSSKEELFNLRFQLATGQLEETARIRTVRKTIARLKTVARERGIEQSKANQ
8,061 rpmC(SA2039) s MKAKEIRDLTTSEIEEQIKSSKEELFNLRFQLATGQLEETARIRAVRKTIARLKTVAREREIEQSKANQ
8,891 rpsT(SA1414) k 10.50 ANIKSAIKRVKTTEKAEARNISQKSAMRTAVKNAKTAVSNNADNKNELVSLAVKLVDKAAQSNL IHSNKADRIKSQLMTANK
9,627 hup(SA1305) k 9.52 MNKTDLINAVAEQADLTKKEAGSAVDAVFESIQNSLAKGEKVQLIGFGNFEVRERAARKGRNPQT GKEIDIPASKVPAFKAGKALKDAVK
10,105 rpsP(SA1081) 9.93 AVKIRLTRLGSKRNPFYRIVVADARSPRDGRIIEQIGTYNPTSANAPEIKVDEALALKWLNDGAKPT
DTVHNILSKEGIMKKFDEQKKAK
10,153 rpsP(SA1081) s AVKIRLTRLGSKRNPFYRIVVADARSPRDGRIIEQIGTYNPTSANAPEIKVDEALALKWLNDGAKPT
YTVHNILSKEGIMKKFDEQKKAK
a
The identities of the peaks were confirmed by knockdown (k), sequencing of strains with peak shifts (s), or MALDI-TOF/TOF (m). bTranscription of the peak is dependent on sigma factor B (y).
c
The peptide variant was usually not observed. dThe peptide variant was sometimes observed. e
Boldface letters signify variant amino acids.
on May 16, 2020 by guest
http://jcm.asm.org/
SAS049 (m/z5,525 [value for doubly charged peptide, 2,763]), and the DNA binding protein HU (SA1305) (m/z9,627 [value for doubly charged peptide, 4,814]) (Fig. 2A). The first two peaks have been observed in cell extracts before (4).
The second group of signals represented ribosomal proteins, which constituteⱕ50% of the cellular proteins (Table 3). A third group of signals in the low molecular weight range was formed by excreted peptides, the phenol-soluble modulins (PSMs), and the delta-toxin double peak, which had already been identified (19, 20). In cell extracts, PSM␣2 (m/z2,307) and PSM␣3 (m/z2,636) were predominant. The delta-toxin atm/z3,007 (formylated ver-sion) is one of the peaks that was frequently discovered in the early attempts at mass spectrometry of intactS. aureuscells (7,9,10, 21). Most useful for typing was the fourth group of signals, which comprised the stress or hypothetical proteins that are present in multiple isoforms.
Effect of inactivation of sigma factor B. Transcription of SAS049 and SA0772 is dependent on the activity of the secondary stress transcription factor sigma B (SigB) and its activator protein, the serine phosphatase RsbU, inS. aureus(22,23). SixteensigB- or
rsbU-negative strains were present in our collection of 261 MRSA strains (6%). Here, the signals atm/z6,889 (value for doubly charged peptide, 3,445) andm/z5,525 (value for doubly charged peptide, 2,763), as well as some smaller signals, were missing (sigB
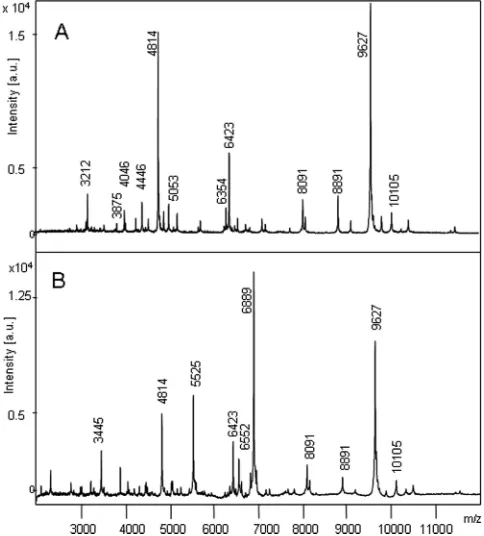
mutants) or they were very low (rsbUmutants). In the analyses of
these strains, the DNA binding protein HU represented the two strongest signals.Figure 2shows the spectra of anrsbU-negative laboratory strain (S. aureus8325-4) and itsrsbU-repaired mutant
S. aureusSH1000 strain (24).
Differences in the spectra of the large MRSA clonal groups. Generally, the peak patterns that had been reported previously (3, 5) could be assigned to peak shifts in several proteins and might serve to distinguish the major MRSA lineages (Table 4) using the type strain panel (21 strains) and 111 typed clinical MRSA strains. However, some minor peaks described previously (3) were not detected under the experimental conditions chosen here. The measurements of the type strains were repeated after 6 months and confirmed that the peak shifts had not been induced by vari-ations of the media used for the cultivation of the cells.
The most commonspatypes of CC22 (Barnim epidemic strain, EMRSA15) are distinguished by an E27V exchange in the protein GraF. One strain did not show this mass shift and, upon sequenc-ing, a secondary A11T exchange, which reverted the molecular mass tom/z5,032, was revealed. In some strains, the peak was not discovered at all; only the neighboring double-charged signal of the ribosomal protein RpsP atm/z5,053 was visible. However, the simultaneous absence of the major SigB-transcribed peaks indi-cated that these strains showed no or low activity of SigB. Analysis of the promoter sequence of SAS030 by DBTBS (http://dbtbs.hgc .jp/) confirmed that there is a well-conserved SigB consensus mo-FIG 1Identification of a mass signal by antisense technology. Antisense RNA
against SA1305, which encodes the DNA binding protein HU with a calculated molecular mass of 9,626 Da, was expressed using the vector pEPSA5 carrying the structural gene of the xylose repressor and a xylose-inducible promoter. Shown are the spectra of fourS. aureusRN4220 clones in LB (34 mg/liter chloramphenicol) harboring the empty vector pEPSA5 grown in the absence of xylose (A), pEPSA5 grown in the presence of 50 mM xylose (B), the recom-binant plasmid, pEPSA5-AS-SA1305, grown in the absence of xylose (C), and pEPSA5-AS-SA1305 grown in the presence of 50 mM xylose (D). Here, the loss of the signal atm/z9,627 was observed only when the expression of antisense RNA was induced by the addition of xylose, thus proving that this peak repre-sents the DNA binding protein HU. a.u., arbitrary units.
FIG 2Overview of the MALDI-TOF mass spectra of two closely relatedS. aureuslaboratory strains,S. aureusNCTC 8325-4 (A) andS. aureusNCTC SH1000 (B).S. aureusNCTC 8325-4 displays only a very weak activity of the transcription factor sigma B; therefore, its spectrum features the DNA binding protein HU atm/z9,627 and ribosomal proteins (seeTable 3). In contrast, the spectrum ofS. aureusNCTC SH1000 is dominated by additional signals that derive from proteins that are transcribed by the stress sigma factor B (m/z6,889 [value for doubly charged peptide, 3,445] [SA0772],m/z5,525 [SAS049],m/z 6,552 [SA1452]). Such a spectrum with higher complexity was present in about 94% of the isolates.
on May 16, 2020 by guest
http://jcm.asm.org/
[image:5.585.45.279.63.315.2] [image:5.585.300.542.68.336.2]tive (GTTT-N15-GGGTAA) in the promoter sequence of this
gene. Furthermore, some rarespatypes were encountered (t223 t309, t016, t7752, t2970) that did not show this peak shift. A sub-group of CC22 strains was further characterized by a variant of the ribosomal protein L30 (SA2030). Using PFGE typing, these strains were indistinguishable from the other CC22 isolates (data not shown); i.e., in this case, typing by MALDI-TOF showed a higher level of discrimination than did PFGE typing.
The CC5 clonal complex constitutes a group of frequent hos-pital-acquired MRSA (HA-MRSA) strains and harbors an F16Y exchange in the protein SA2420.1. This small ORF is not anno-tated in most genomes, and its gene product retained the N-ter-minal formylmethionyl (fMet) residue, which shows that the sec-ond possible ATG serves as a start codon. This peptide variant is also found in sequenced CC5 strains (JH1, JH9, N315, Mu50, ED98, etc.) and in strain Reynolds, an ST25 MSSA; however, our CC25 strains were also characterized by a second peak shift in SAS049.
The CC8 clonal complex that is prevalent in hospitals in Eu-rope and as community-associated MRSA (CA-MRSA) in the United States was characterized by a peak shift in the SigB-depen-dent CsbD-like stress protein SA1452 that is caused by an AT¡VI exchange in positions 36 and 37. The presence of this peak atm/z
6,592 in the extracts of USA300 isolates has been reported previ-ously (6) and BLAST searches showed its presence in the se-quenced ST8 USA300_FPR3757 and USA300_TCH1516 strains. An exception is found in the laboratory strains of the NCTC 8325 MSSA lineage; after the repair ofrsbUin strainS. aureusSH1000 (24), the signal still amounted to m/z6,552 (value for doubly charged peptide, 3,277), which also corresponds to the published genome sequence. In some rare cases, an additional signal at about
m/z6,580 was detected in unrelated strains; therefore, this peak is only discriminatory if the signal atm/z6,552 is not detected.
ST239 strains arose from a recombination event in which 557 kb covering the origin of replication was replaced by CC30 DNA in a CC8 strain (or vice versa) (1). The type strain appeared as CC8 in MALDI-TOF MS, since no CC30 specific marker (see below) is located on the transferred 557-kb fragment. BLAST searches con-firmed the conservation of the exchange in SA1452 in the se-quenced ST239 strainsS. aureus TW20, T0131, JKD6008, and ATCC BAA-39. In contrast, thespagene is on the CC30-derived fragment, so the strains possessed the t030spatype.
The CC30 complex could be identified by a unique peak shift of the hypothetical protein SAS049 tom/z5,507; furthermore, it contained amino acid exchanges in PSM␣3 and the hypothetical protein SAR1012 (the correct ORF that uses the second available start codon is annotated in the genome ofS. aureusstrain Sanger 252). The MALDI signal of the latter peptide was only strong if the C-terminal N38K exchange was present as in, e.g., strain Sanger 252. The other variants of the peptide were frequently not detect-able in the analyses or gave very weak signals.
CC45 strains, which also constitute a group of hospital-ac-quired MRSA strains, also possessed them/z4,511 signal caused by SAR1012. In a subgroup of these strains that showed the typical strain “Berlin” phage pattern, loss of them/z6,613 signal corre-sponding to GraC was observed; this was caused by a premature stop codon.
The ST80 and ST88 CA-MRSA lineages currently circulate in Europe. ST80 strains were solely characterized by a shift in SAR1012, leading to a mass ofm/z4,497, which yields a rather weak signal. BLAST searches in NCBI showed that this profile is also kept in the sequencedS. aureus11819-97 strain. ST88 CA-TABLE 4Distribution of peak shifts and sensitivity and specificity of the markers
m/z ORF
Biomarker for
CC Example(s) Sensitivity Specificity
No. of MRSA strains tested
2,636 PSM␣3
2,685 PSM␣3 30 Sanger 252 0.308 1.000 13
3,007 Delta-toxin (formylated)
3,037 Delta-toxin (formylated) 1 t127 (MW2, Sanger 476) 0.250 1.000 8
3,875 SA2420.1
3,891 SA2420.1 5, 25 ST5, ST225, ST228, USA100, Mu50, N315 0.978 0.988 92
4,511 SAR1012 30, 45, 398, ST88 Berlin, LA-MRSA, CA-MRSA, Cowan 0.983 0.990 58
4,485 SAR1012 Peaks often not
observed
Peaks often not observed
4,497 SAR1012 (72, ST80) Peaks often not
observed
Peaks often not observed 5,032 graF(SAS030)
5,002 graF(SAS030) 22 (t032, t608) Barnim EMRSA15 0.750 1.000 60
5,525 SAS049
5,507 SAS049 30 Cowan 0.846 1.000 13
5,551 SAS049 (MSSA)
5,539 SAS049 25, 72, 12, MSSA Reynolds 1
6,423 rpmD(SA2030)
6,397 rpmD(SA2030) t032 Subgroup of CC22 strains 0.167 1.000 60
6,552 SA1452
6,592 SA1452 8 USA300, ST247, COL (ST250) 0.889 0.996 27
6,580 SA1452 130 t843 1.000 0.992 2
6,613 graC(SAS044)
6,627 graC(SAS044) ST88 CA-MRSA t609 1.000 0.988 2
3,825 Truncation: no signal 45 Subgroup of CC45 strains 0.438 0.987 32
on May 16, 2020 by guest
http://jcm.asm.org/
[image:6.585.40.552.79.355.2]MRSA and the CC398 livestock-associated MRSA (LA-MRSA) strains also gave them/z4,511 signal corresponding to SAR1012, which is also conserved in the sequencedS. aureusST398 (acces-sion no. NC_017333.1) andS. aureus08BA02176 (accession no. NC_CP003808.1) strains. Our two ST88 isolates contained an N19K exchange in SAS044, leading to a shift tom/z6,627. Two German CC130spat843 strains, which are the most common isolates harboring the newmecCvariant gene (25), which is not detected by conventional PCR, were characterized by a shift of SA1452 to m/z 6,580. This single-nucleotide polymorphism (SNP) is also present in two sequenced CC130 MSSA isolates
from ovine mastitis (accession no. AEUQ00000000 and
AEUR00000000) but was not observed in the sequencedS. aureus
LGA251 strain, which belongs to another clonal lineage (ST425, t6300) (accession no. NC_017349.1).
The sensitivities and specificities of the signals are listed in Table 4. After the method had been set up with the type strain panel and 111 clinical MRSA strains, another group of 136 clinical MRSA strains, which showed unusual or no phage patterns, was typed in a blind fashion. Five strains were singletons with rarespa
groups, and 81% of the remaining 131 strains could be assigned to the correct clonal complexes (CC1, CC5, CC8, CC22, CC30, CC130, t843, ST88), or at least to the group that is distinguished by the presence of them/z4,511 signal and contains the CC45 and CC398 isolates.
Spectra of MSSA.Ninety-five MSSA strains were also charac-terized. Since all MRSA lineages have MSSA ancestors, it was not surprising that the strains with profiles corresponding to MRSA clonal groups CC5, CC8 (e.g., strain Newman), CC45, CC25 (e.g., strain Reynolds), CC30 (e.g., strain Cowan), and ST80 could be detected; however, their frequency differed. For example, whereas only three CC30 and no CC25 MRSA strains could be isolated, MSSA strains harboring the biomarkers of these groups occurred at higher frequencies of about 10% or 5%, respectively. Unique signatures that were found only in MSSA strains were the S35L exchange in SAS049, leading to them/z5,551 signal, and the com-bination of them/z4,511 signal and them/z6,568 mass signal.
Outbreak situation.In order to evaluate the method further, 38 strains isolated during an outbreak of ST225 MRSA (14) were retrospectively characterized by MALDI-TOF MS. Thirty-three isolates were assigned to CC5 and one MSSA strain, one unrelated MRSA strain, and one BORSA strain isolated from health care workers were correctly identified as unrelated strains. The CC5 biomarkerm/z3,891 was shifted tom/z3,875 in the MRSA and MSSA isolates, and the BORSA strain contained the A27V ex-change in GraC. The remaining two strains were unclear. Al-though they showed the biomarker for CC5 and their PFGE pro-files were identical to that of ST225, they were characterized by another deviating peak.
DISCUSSION
This study shows that MALDI-TOF MS presents a rapid technol-ogy for discrimination of the differentS. aureusclonal lineages. In a previous work withS. aureusextracts by Wolters et al. (3), the peaks atm/z3,876, 4,511, 5,002, 5,032, 5,508, 5,524, 6,591, and 6,612 were also observed using Biotyper 2.0 and a collection of human isolates. Employing a Voyager-DE STR MALDI-TOF mass spectrometer and a collection of animal isolates, Böhme et al. (5) described peaks and peak shifts (indicated by slashes) atm/z
2,637/2,686, 3,008, 3,875/3,890, 4,483/4,496/4,510, and 5,506/
5,523. The fact that the same signals were identified by three dif-ferent groups confirms that these peaks are detectable with differ-ent instrumdiffer-ents and differdiffer-ent strain collections.
In addition to describing peak shifts and assigning them to clonal groups, we identified the peptides that are detected during mass spectrometry and showed that the observed peak shifts rep-resent SNPs in the genomes of different strains. The identification of the signals demonstrated that the ribosomal proteins show little variability. The ribosomal protein S20 RpsT was assigned to the
m/z4,446 signal. Since the precision of linear MALDI-TOF MS measurements is limited, with a potential mass deviation of⬎500 ppm with external calibration, RpsT might be identical to the signal atm/z4,448 that was erroneously postulated to signify the production of the Panton-Valentine leukocidin in ST80 CA-MRSA (11,26).
The most variable group of signals was formed by small hypo-thetical nonessential proteins without known function or stress proteins, which appear in multiple isoforms. The majority of the signals comprised peptides with high isoelectric points (IEP); however, some peptides with an IEP of around 5 were also found. Reports onE. coli andPseudomonasalso identified cold shock proteins, the DNA binding protein HU, ribosomal proteins, and other peptides displaying a high basicity and high abundance in the cell in MALDI-TOF MS analysis (27,28). Several of the pep-tides retained the N-terminal fMet or Met residue; with two ex-ceptions, these peptides belonged to the PSM group. The peptides starting with Met were characterized by an amino acid with a large side chain in position 2 that has been shown to inhibit the methi-onine peptidase ofE. coli(29).
The calculation of the sensitivities and specificities (Table 4) of the different signals showed that the overall specificity of the sig-nals was always⬎0.98. However, the sensitivities of the signals varied strongly between 0.98 and 0.16, reflecting either the level of conservation of the mutations in the clonal lineages or a diversity of expression levels of the genes in theS. aureuspopulation. Mu-tations that were only conserved in certain subtypes are repre-sented by the variant of the L30 ribosomal protein in CC22 strains or the truncation of GraC in CC45. Similarly, the CC22 biomarker was not conserved in allspatypes of this clonal complex. The low-molecular-weight toxins (PSM␣3, delta-toxin) were not de-tected in all strains. It has been noted before that these small cy-tolytic peptides are under the direct control of the accessory gene regulator (agr) system (30,31), and it has been reported recently that the expression ofmecAin strains that harbor a type II staph-ylococcal cassette chromosomemecelement (SCCmec) interferes with the functionality of this system (32). Furthermore, three of the markers that distinguish the clonal complexes CC22, CC8, and CC30 are under SigB control and are not expressed in SigB-nega-tive strains (22). On Columbia blood agar,rsbU- orsigB-negative colonies are detected by their white color, since staphyloxanthin biosynthesis is dependent on SigB (22), and strong hemolysis (33). Upon analysis of the MALDI spectra, the loss of activity of SigB is easily inferred from the absence of the large SigB-dependent sig-nals atm/z6,889 andm/z5,525 and, therefore, should be included in the interpretation of the data. However, if any of the signals with low sensitivity were detected, they indicated the lineage of the strain with a high specificity.
In the hospital, the transmission of MRSA between patients or between patients and staff is a frequent problem. Preliminary data show that the peak shifts are also visible using a bioMérieux Vitek
on May 16, 2020 by guest
http://jcm.asm.org/
mass spectrometer and experiments that aim at the transfer of this method to the clinically used procedure and instrumentation are in progress. Visual comparison of the chromatograms is possible, preferably not by using printouts but instead the computer soft-ware provided by the supplier; however, as a future perspective, the evaluation of these data should be implemented into the soft-ware programs of clinical instruments, thereby enabling the direct identification of the clonal lineages of the MRSA strains. If both strains belong to the same clonal lineage and no further peak shifts are visible in the analyses, more-sensitive genotyping methods should be employed, e.g.,spatyping, random PCR methods, or PFGE. However, if the isolates in question belong to different lineages, a nosocomial transmission can be excluded after the first MALDI-TOF MS analysis that serves for species identification. Thus, better evaluation of the data that are obtained during the primary MALDI-TOF MS analysis might save costs and labor that are associated with further typing procedures.
In conclusion, we demonstrate that after the identification of the peptides that form the signals detected in the MALDI-TOF MS spectra ofS. aureus, it is possible to understand the spectra and their variations better and to assign a lineage to many strains. Additionally, the SigB-dependent products, PSMs, and delta toxin provide quick information on SigB function and, even more im-portantly, on the virulence of the strains (32).
ACKNOWLEDGMENTS
We appreciate the expert technical assistance of K. Hermanns, F. Stewart, M. Gajdiss, and A. Zweynert.
We gratefully acknowledge funding by the Bonfor program of the Medical Faculty of the University of Bonn and continued support by the German Research Foundation.
We made use of thespatyping website (http://www.spaserver.ridom .de/) that was developed by Ridom GmbH and curated by SeqNet.org (http://www.SeqNet.org/).
M. Kostrzewa and K. Sparbier are employed by Daltonik GmbH, Bre-men, Germany.
REFERENCES
1.Robinson DA, Enright MC.2003. Evolutionary models of the emergence of methicillin-resistantStaphylococcus aureus.Antimicrob. Agents Che-mother.47:3926 –3934.
2.Cookson BD, Robinson DA, Monk AB, Murchan S, Deplano A, de Ryck R, Struelens MJ, Scheel C, Fussing V, Salmenlinna S, Vuopio-Varkila J, Cuny C, Witte W, Tassios PT, Legakis NJ, van Leeuwen W, van Belkum A, Vindel A, Garaizar J, Haeggman S, Olsson-Liljequist B, Ransjo U, Muller-Premru M, Hryniewicz W, Rossney A, O’Connell B, Short BD, Thomas J, O’Hanlon S, Enright MC. 2007. Evaluation of molecular typing methods in characterizing a European collection of epidemic me-thicillin-resistantStaphylococcus aureusstrains: the HARMONY collec-tion. J. Clin. Microbiol.45:1830 –1837.
3.Wolters M, Rohde H, Maier T, Belmar-Campos C, Franke G, Scherpe S, Aepfelbacher M, Christner M.2011. MALDI-TOF MS fingerprinting allows for discrimination of major methicillin-resistantStaphylococcus au-reuslineages. Int. J. Med. Microbiol.301:64 – 68.
4.Bernardo K, Pakulat N, Macht M, Krut O, Seifert H, Fleer S, Hunger F, Krönke M.2002. Identification and discrimination ofStaphylococcus au-reus strains using matrix-assisted laser desorption/ionization-time of flight mass spectrometry. Proteomics2:747–753.
5.Böhme K, Morandi S, Cremonesi P, Fernández N, Barros-Velázquez IJ, Castiglioni B, Brasca M, Canas B, Calo-Mata P.2012. Characterization ofStaphylococcus aureusstrains isolated from Italian dairy products by MALDI-TOF mass fingerprinting. Electrophoresis33:2355–2364. 6.Boggs SR, Cazares LH, Drake R.2012. Characterization of a
Staphylo-coccus aureusUSA300 protein signature using matrix-assisted laser de-sorption/ionization time-of-flight mass spectrometry. J. Med. Microbiol. 61:640 – 644.
7.Walker J, Fox AJ, Edwards-Jones V, Gordon DB.2002. Intact cell mass spectrometry (ICMS) used to type methicillin-resistantStaphylococcus au-reus: media effects and inter-laboratory reproducibility. J. Microbiol. Methods48:117–126.
8.Edwards-Jones V, Claydon MA, Evason DJ, Walker J, Fox AJ, Gordon DB.2000. Rapid discrimination between methicillin-sensitive and meth-icillin-resistantStaphylococcus aureusby intact cell mass spectrometry. J. Med. Microbiol.49:295–300.
9.Du Z, Yang R, Guo Z, Song Y, Wang J.2002. Identification of Staphy-lococcus aureusand determination of its methicillin resistance by matrix-assisted laser desorption/ionization time-of-flight mass spectrometry. Anal. Chem.74:5487–5491.
10. Majcherczyk PA, McKenna T, Moreillon P, Vaudaux P. 2006. The discriminatory power of MALDI-TOF mass spectrometry to differentiate between isogenic teicoplanin-susceptible and teicoplanin-resistant strains of methicillin-resistantStaphylococcus aureus.FEMS Microbiol. Lett.255: 233–239.
11. Bittar F, Ouchenane Z, Smati F, Raoult D, Rolain JM.2009. MALDI-TOF-MS for rapid detection of staphylococcal Panton-Valentine leukoci-din. Int. J. Antimicrob. Agents34:467– 470.
12. Lu JJ, Tsai FJ, Ho CM, Liu YC, Chen CJ.2012. Peptide biomarker discovery for identification of methicillin-resistant and vancomycin-intermediateStaphylococcus aureusstrains by MALDI-TOF. Anal. Chem. 84:5685–5692.
13. Harmsen D, Claus H, Witte W, Rothgänger J, Claus H, Turnwald D, Vogel U.2003. Typing of methicillin-resistantStaphylococcus aureusin a university hospital setting by using novel software forsparepeat determi-nation and database management. J. Clin. Microbiol.41:5442–5448. 14. Heinrich N, Mueller A, Bartmann P, Simon A, Bierbaum G, Engelhart
S.2011. Successful management of an MRSA outbreak in a neonatal in-tensive care unit. Eur. J. Clin. Microbiol. Infect. Dis.30:909 –913. 15. Forsyth RA, Haselbeck RJ, Ohlsen KL, Yamamoto RT, Xu H, Trawick
JD, Wall D, Wang L, Brown-Driver V, Froelich JM, C KG, King P, McCarthy M, Malone C, Misiner B, Robbins D, Tan Z, Zhu ZY, Carr G, Mosca DA, Zamudio C, Foulkes JG, Zyskind JW.2002. A genome-wide strategy for the identification of essential genes inStaphylococcus aureus.Mol. Microbiol.43:1387–1400.
16. Augustin J, Götz F.1990. Transformation ofStaphylococcus epidermidis and other staphylococcal species with plasmid DNA by electroporation. FEMS Microbiol. Lett.54:203–207.
17. Maltman DJ, Brand S, Belau E, Paape R, Suckau D, Przyborski SA. 2011. Top-down label-free LC-MALDI analysis of the peptidome during neural progenitor cell differentiation reveals complexity in cytoskeletal protein dynamics and identifies progenitor cell markers. Proteomics11: 3992– 4006.
18. Neubert H, Bonnert TP, Rumpel K, Hunt BT, Henle ES, James IT. 2008. Label-free detection of differential protein expression by LC/ MALDI mass spectrometry. J. Proteome Res.7:2270 –2279.
19. Gonzalez DJ, Okumura CY, Hollands A, Kersten R, Akong-Moore K, Pence MA, Malone CL, Derieux J, Moore BS, Horswill AR, Dixon JE, Dorrestein PC, Nizet V.2012. Novel phenol-soluble modulin derivatives in community-associated methicillin-resistantStaphylococcus aureus identified through imaging mass spectrometry. J. Biol. Chem.287:13889 – 13898.
20. Gagnaire J, Dauwalder O, Boisset S, Khau D, Freydière AM, Ader F, Bes M, Lina G, Tristan A, Reverdy ME, Marchand A, Geissmann T, Benito Y, Durand G, Charrier JP, Etienne J, Welker M, Van Belkum A, Vandenesch F.2012. Detection ofStaphylococcus aureusdelta-toxin pro-duction by whole-cell MALDI-TOF mass spectrometry. PLoS One 7:e40660. doi:10.1371/journal.pone.0040660.
21. Rajakaruna L, Hallas G, Molenaar L, Dare D, Sutton H, Encheva V, Culak R, Innes I, Ball G, Sefton AM, Eydmann M, Kearns AM, Shah HN.2009. High throughput identification of clinical isolates of Staphylo-coccus aureususing MALDI-TOF-MS of intact cells. Infect. Genet. Evol. 9:507–513.
22. Bischoff M, Dunman P, Kormanec J, Macapagal D, Murphy E, Mounts W, Berger-Bächi B, Projan S.2004. Microarray-based analysis of the Staphylococcus aureussigmaB regulon. J. Bacteriol.186:4085– 4099. 23. Giachino P, Engelmann S, Bischoff M.2001. Sigma(B) activity depends
on RsbU inStaphylococcus aureus.J. Bacteriol.183:1843–1852. 24. Horsburgh MJ, Aish JL, White IJ, Shaw L, Lithgow JK, Foster SJ.2002.
sigmaB modulates virulence determinant expression and stress resistance:
on May 16, 2020 by guest
http://jcm.asm.org/
characterization of a functionalrsbUstrain derived fromStaphylococcus aureus8325-4. J. Bacteriol.184:5457–5467.
25. Garcia-Álvarez L, Holden MTG, Lindsay H, Webb CR, Brown DFJ, Curran MD, Walpole E, Brooks K, Pickard DJ, Teale C, Parkhill J, Bentley SD, Edwards GF, Girvan EK, Kearns AM, Pichon B, Hill RLR, Larsen AR, Skov RL, Peacock SJ, Maskell DJ, Holmes MA. 2011. Meticillin-resistantStaphylococcus aureuswith a novelmecAhomologue in human and bovine populations in the UK and Denmark: a descriptive study. Lancet Infect. Dis.11:595– 603.
26. Szabados F, Becker K, von Eiff C, Kaase M, Gatermann S.2011. The matrix-assisted laser desorption/ionisation time-of-flight mass spectrom-etry (MALDI-TOF MS)-based protein peaks of 4448 and 5302 Da are not associated with the presence of Panton-Valentine leukocidin. Int. J. Med. Microbiol.301:58 – 63.
27. Ryzhov V, Fenselau C.2001. Characterization of the protein subset de-sorbed by MALDI from whole bacterial cells. Anal. Chem.73:746 –750. 28. Holland RD, Duffy CR, Rafii F, Sutherland JB, Heinze TM, Holder CL,
Voorhees KJ, Lay JO, Jr.1999. Identification of bacterial proteins ob-served in MALDI TOF mass spectra from whole cells. Anal. Chem.71: 3226 –3230.
29. Ben-Bassat A, Bauer K, Chang SY, Myambo K, Boosman A, Chang S. 1987. Processing of the initiation methionine from proteins: properties of
theEscherichia colimethionine aminopeptidase and its gene structure. J. Bacteriol.169:751–757.
30. Queck SY, Jameson-Lee M, Villaruz AE, Bach TH, Khan BA, Stur-devant DE, Ricklefs SM, Li M, Otto M.2008. RNAIII-independent target gene control by theagrquorum-sensing system: insight into the evolution of virulence regulation inStaphylococcus aureus.Mol. Cell 32:150 –158.
31. Janzon L, Lofdahl S, Arvidson S.1989. Identification and nucleotide sequence of the delta-lysin gene,hld, adjacent to the accessory gene regulator (agr) ofStaphylococcus aureus.Mol. Gen. Genet.219:480 – 485.
32. Rudkin JK, Edwards AM, Bowden MG, Brown EL, Pozzi C, Waters EM, Chan WC, Williams P, O’Gara JP, Massey RC.2012. Methicillin resis-tance reduces the virulence of healthcare-associated methicillin-resistant Staphylococcus aureusby interfering with theagrquorum sensing system. J. Infect. Dis.205:798 – 806.
33. Shaw LN, Aish J, Davenport JE, Brown MC, Lithgow JK, Simmonite K, Crossley H, Travis J, Potempa J, Foster SJ.2006. Investigations into sigmaB-modulated regulatory pathways governing extracellular virulence determinant production inStaphylococcus aureus.J. Bacteriol.188:6070 – 6080.