0022-538X/95/$04.0010
Copyrightq1995, American Society for Microbiology
A Mutant v-rel with Increased Ability To Transform B Lymphocytes
PAOLO ROMERO
1,2*
AND
ERIC H. HUMPHRIES
1,3†
Mary Babb Randolph Cancer Center
1and Departments of Medicine
2and Microbiology and Immunology,
3West Virginia University, Morgantown, West Virginia 26506-9162
Received 17 June 1994/Accepted 14 October 1994
We observed that two strains of REV-T differ in the ability to transform bursal cells in vitro. REV-TW, with
v-
rel
derived from a well-characterized clone and considered the prototype of the wild type, fails to generate
colonies in soft agar. In contrast, REV-S2A3, derived from the S2A3 cell line, readily transforms bursal cells.
With PCR, a 1,591-bp fragment containing v-
rel
from the REV-S2A3 provirus was cloned into plasmid pREV-0.
Except for the absence of v-
rel
, pREV-0 is identical to pREV-TW. Five clones of pREV-PCR, each produced by
an independent amplification, were obtained. The REV-PCR viruses displayed the strong transforming
phenotype of REV-S2A3. Two mutations were identified in the 5
*
region of v-
rel
from REV-PCR1 to REV-PCR5:
a silent mutation and a G-to-T transversion, changing the alanine at position 40 to serine. To confirm the
relevance of this amino acid substitution, a 478-bp fragment containing the mutations was exchanged between
REV-TW and REV-PCR1. Only the mutant viruses were able to form large colonies of bursal cells in liquid
culture and to generate bursal cell colonies in soft agar. When tested on splenocytes, the wild-type viruses
induced predominantly non-B-cell colonies while the mutant viruses gave origin mainly to B-cell colonies. The
above results indicate that the substitution of serine for alanine at position 40 of v-Rel enhances the ability of
REV-T to transform B lymphocytes in vitro. This mutation is close to the DNA-binding region, and the variant
v-Rel oncoprotein shows increased
k
B-binding activity, thus confirming the relevance of this property for
transformation.
Several aspects of transformation by v-rel render this
onco-gene an attractive tool for investigating the pathoonco-genesis of
hematological malignancies. v-rel is the transforming gene of
avian reticuloendotheliosis virus strain T (REV-T). This virus
is one of the most virulent oncogenic retroviruses, causing
death in 7 to 10 days from polyclonal lymphoid neoplasias after
injection into newly hatched chickens (20, 59). The v-Rel
oncoprotein belongs to the NF-
k
B/Rel family of transcription
factors that includes the dorsal morphogen in Drosophila
melanogaster, NF-
k
B1 (p105/p50), NF-
k
B2 (p100/p52), RelA
(p65), RelB, and Rel (4, 19, 34, 46, 48, 52). After binding to
k
B
sites, members of this class of molecules modulate the
tran-scription of genes involved in embryogenesis, immune
re-sponse, inflammation, and viral replication (2, 12, 22, 38).
Homodimerization or heterodimerization is a prerequisite for
transactivation, and the combinatorial possibilities
consider-ably expand the range of target genes (50). The activity of the
members of this family is regulated by a set of inhibitory factors
including I
k
B-
a
, I
k
B-
g
, Bcl-3, and cactus. These proteins
contain five to eight copies of the ankyrin repeat and function
by sequestering NF-
k
B/Rel complexes in the cytoplasm and
inhibiting their binding to DNA in the nucleus (3, 10, 31, 32,
38). The members of the NF-
k
B/Rel family share the Rel
homology region that encompasses 300 N-terminal amino
acids and mediates DNA binding, dimerization, nuclear
tar-geting, and interactions with inhibitor proteins. The
N-termi-nal motif RxxRxRxxC has recently been found to be essential
for DNA binding and is conserved in all NF-
k
B/Rel proteins
(36, 40). In v-Rel, this sequence is located between amino acids
27 and 35 (56, 62).
The v-rel oncogene is a truncated and mutated form of
turkey c-rel. v-Rel is actually an Env-Rel-Env fusion protein
containing 11 Env-derived amino acids in place of the first 2
amino acids of the c-Rel protein, 474 c-Rel-derived amino
acids, and 18 amino acids derived from out-of-frame Env
sequences at its C terminus which replace the terminal 118
amino acids of c-Rel. In addition, v-Rel contains 14 amino acid
substitutions and three small deletions compared with c-Rel
(12, 62). The exact contribution of each of these changes to the
activation of the full oncogenic potential of c-Rel is not known
(20). It is conceivable that each modification brought about a
discrete increment in the transforming activity and that v-rel
represents the result of an evolutionary process under strong
selective pressure favoring a more tumorigenic phenotype
(45). In accordance with this notion, one would expect to find
additional mutations with enhanced transforming ability.
v-rel was transduced once and transforms only avian cells.
Avian v-rel expressed from a murine retrovirus caused a
cytopathic effect in mouse fibroblasts and bone marrow cells
and produced no transformation. The inability to transform
murine cells has been ascribed to this cytopathic effect (55).
However, activation through rearrangement of REL, NFKB2,
and BCL-3 has been implicated in the pathogenesis of human
lymphoid malignancies (31, 39, 46, 49, 63). The full range of
target cells susceptible to transformation by v-rel remains to be
defined. Immature lymphoid cells, B cells, T cells, myeloid
cells, and fibroblasts have been reported to be transformed by
REV-T (5, 7, 8, 20, 37, 42, 43, 65, 66). A number of factors have
been considered to be important in determining the type of cell
rendered neoplastic, including the age of the animal and the
type of helper virus used (6).
Most of the in vitro studies done have analyzed the
gener-ation of colonies in soft agar by splenocytes after infection with
REV-T (24). While carrying out similar experiments with
bursal cells, we became aware that two strains of REV-T differ
in the ability to transform B lymphocytes. The virus derived
from nonproducer cell line S2A3 (6, 37) transforms B cells with
high efficiency in both liquid and soft agar assays, while the
* Corresponding author. Mailing address: Section of Hematology-Oncology, West Virginia University, P.O. Box 9162, Morgantown, WV 26506-9162. Phone: (304) 293-4229. Fax: (304) 293-2519.
† Deceased.
301
on November 9, 2019 by guest
http://jvi.asm.org/
wild type (14, 45, 62) fails to do so. The experiments described
in the present report were aimed at establishing the molecular
basis for this difference. We demonstrate that substitution of
serine for alanine at position 40 of the v-Rel protein accounts
for this enhanced oncogenic potential towards B lymphocytes.
This gain-of-function mutation is located near the
DNA-binding site. In electrophoretic mobility shift assays using in
vitro translation products, the mutant v-Rel oncoprotein binds
a
k
B site more avidly than does the wild type.
MATERIALS AND METHODS
Plasmids and retroviruses used.All recombinant DNA manipulations were performed in accordance with standard procedures (53). Unless otherwise specified, the pTZ-18R phagemid (Bio-Rad Laboratories, Richmond, Calif.) served as the vector for the constructs used. pREV-T3 is a circularly permuted clone of transforming REV-T DNA containing a single long terminal repeat and was cloned into the SalI site of pBR322 (14). pREV-T10 was derived from REV-T3 by adding a 59 long terminal repeat and a leader sequence, thus completing the viral genome (45). pREV-0 is a pREV-T3-derived vector designed for expression of exogenous genes in avian cells. Briefly, the REV-T3 clone was transferred from pBR322 to pTZ-18R, the viral structure was completed by addition of a 59long terminal repeat and leader sequences, XhoI and BssHII sites were created in noncoding regions, and v-rel was replaced by a short, synthetic XhoI-BssHII fragment (45). pREV-TW was constructed by insertion of an XbaI-NruI fragment containing all of v-rel from pREV-T3 into the XhoI-BssHII site of pREV-0 by using intermediate plasmid pTZXN-R contain-ing a synthetic XhoI-XbaI-NruI-BssHII adaptor (45). v-rel from the REV-T provirus present in the S2A3 cell line (REV-S2A3) was amplified five times by PCR and directly inserted into pREV-0 to create five independent clones, PCR1 to PCR5. The plasmids for hybrid retroviruses pREV-pcrTW and pREV-twPCR were obtained by exchanging a 478-bp XhoI-ClaI fragment, coding for the first 148 amino acids of v-Rel, between pREV-TW and pREV-PCR1 (Fig. 1).
For in vitro translation experiments, v-rel genes from pREV-pcrTW and pREV-twPCR were inserted into pTZXN-R (45) as XhoI-BssHII fragments.
pCSV11S3 is a genomic clone of chicken syncytial virus (CSV) isolated in this laboratory from the 11S3 cell line (7). This retrovirus was used as a helper virus in all of the experiments described here. Plasmid pREVA6 was used to obtain the viral stocks of REV-A utilized in immunocytochemical assays (25).
PCR cloning.PCR amplification of all of v-rel from the REV-T provirus present in cell line S2A3 was done with the following oligomers: 59-ACTCTC GAGTCTAGAAAGCTCCTG-39, spanning positions237 to214 with respect
to v-rel and containing a C-to-G change at position229, creating an XhoI site, and 59-CGTTTCCGCGCGCCAAGGTC-39, complementary to positions145 to
126 and including a T-to-G change at position133 and an A-to-C change at position138, converting an NruI site to a BssHII site (Fig. 2) (56, 62). The reaction mixture was subjected to 30 cycles of amplification (948C for 70 s, 558C for 60 s, and 728C for 180 s) followed by a 7-min extension at 728C with a thermal cycler and the GeneAmp kit (Perkin-Elmer, Norwalk, Conn.). The 1,591-bp amplified fragment was isolated by polyacrylamide gel electrophoresis after digestion with XhoI and BssHII and inserted into the XhoI-BssHII site of pREV-0. The products of five independent amplifications were used to generate pREV-PCR1 to pREV-PCR5.
Nucleotide sequence determination was performed by the dideoxy-chain termination method on single-strand phagemids with custom-made primers and T7 DNA polymerase (Sequenase; U.S. Biochemical, Cleveland, Ohio).
Chickens, cell lines, and media.Embryonated SC eggs were purchased from Hyline International Hatcheries (Dallas Center, Iowa) and incubated with humidity at 398C. For transformation experiments, the animals were sacrificed 3 to 5 weeks after hatching. S2A3 is a nonproducer cell line developed in the laboratory of H. R. Bose by in vitro infection of spleen cells with REV-T (6, 37). Chicken embryonic fibroblasts (CEF) were prepared from 10-day-old embryos. All in vitro cell cultures used Dulbecco’s modified Eagle’s medium supplemented with 10% bovine calf serum (HyClone Laboratories, Inc., Logan, Utah), 5% chicken serum (GIBCO Biologicals, Grand Island, N.Y.), 10% tryptose phos-phate broth (Difco Laboratories, Detroit, Mich.), 50mM 2-mercaptoethanol, and antibiotics (penicillin G at 100 U/ml and streptomycin at 50mg/ml).
Generation of virus stocks and virus titrations.Transfection of fibroblasts was performed by using a modified calcium phosphate procedure as described elsewhere (53). Exponentially growing CEF were plated at 53105
cells per 90-mm-diameter tissue culture dish and cotransfected with 20mg of a plasmid containing a replication-defective retrovirus (pREV-TW, pREV-T10, pREV-T3, pREV-PCR1-5, pREV-pcrTW, or pREV-twPCR) and 2mg of plasmid pCSV 11S3, containing replication-competent helper virus CSV. pREV-T3 was di-gested with SalI and ligated before transfection. Adequate expression of genes from both viruses was verified by immunofluorescence. REV-A stocks were obtained by transfection of CEF with 2mg of plasmid pREVA6. Virus stocks were harvested after 6 days of culture and stored frozen.
The infectious titers of the replication-defective viruses and the CSV helper virus were determined by a modification of the quantitative immunocytochemical assay (25, 57). Secondary cultures of CEF were plated at 73105cells per
[image:2.612.59.296.74.223.2]60-mm-diameter tissue culture dish. After 24 h, the cells were infected with 0.2 FIG. 1. Schematic representation of the retroviruses used. The abilities of
the viruses to transform bursal cells in liquid culture assays are summarized. The long terminal repeats and v-rel are represented as boxes. The dark boxes denote v-rel genes or parts thereof derived from the REV-S2A3 provirus. The open boxes indicate v-rel genes originating from plasmid pREV-T3. The XhoI and ClaI sites used in the construction of hybrid retroviruses pcrTW and REV-twPCR are shown. The amino acid at position 40 of the v-Rel oncoprotein is indicated. The immunocytochemical (immunocytoch.) titer refers to the number of foci of CEF reactive with anti-v-Rel antibody per milliliter of viral stock. The titers of the helper virus ranged from 0.43106to 53106/ml. Colonies were
scored as large if single, easily seen by eye, and growing in wells with acidification of the medium. Total colonies are colonies of any size scored with a microscope. The large-colony/total-colony ratio represent the ratio of the respective means. On average, four independent experiments were performed for each virus.
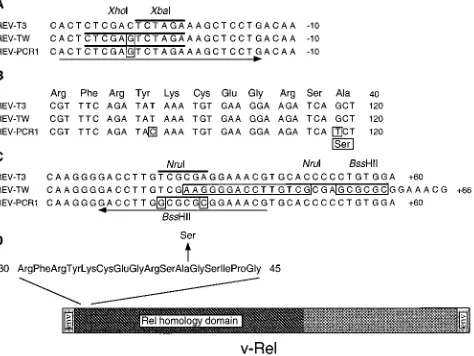
FIG. 2. Results of DNA sequence analysis of REV-PCR1 and REV-TW compared with the published sequence of REV-T3 (56, 62). The nucleotides and amino acid differing from those of REV-T3 are boxed. The arrows underline the stretches identical (59) or complementary (39) to the oligonucleotides used for PCR amplification. The bars overlie the indicated restriction sites. The negative numbers indicate positions relative to the start codon. The numbers preceded by a plus sign denote positions from the stop codon. The numbers without a sign refer to the v-rel open reading frame. (A) Findings 59of v-rel showing the expected transversion from C to G introduced to generate the XhoI site in both REV-TW and REV-PCR1. (B) Comparison of the 59 sequences of v-rel. REV-T3 and REV-TW are identical. REV-PCR1 contains a silent transition from T to C at position 99 and a G-to-T transversion at position 118 converting codon 40 from alanine to serine. (C) Sequences 39of v-rel displaying the extra 21 bp in REV-TW and the expected changes in REV-PCR1 converting the NruI site into a BssHII site. (D) Diagram showing the only difference between the wild-type and mutant v-Rel oncoproteins.
on November 9, 2019 by guest
http://jvi.asm.org/
[image:2.612.320.557.417.595.2]ml of virus dilutions and incubated for 1 h at 378C. For titration of the v-rel-coding viruses, the CEF were then superinfected with 106infectious units of
REV-A for 1 h. After addition of 4 ml of fresh medium, the plates were incubated for 48 h and then washed in phosphate-buffered saline (PBS), fixed in 2% paraformaldehyde in PBS, and stained for the presence of the v-Rel protein. For titration of CSV, the CEF were overlaid with medium containing 0.7% agar and processed in a similar fashion. Monoclonal antibody HY87 was used to detect the v-Rel protein in the titration of the replication-defective viruses, and monoclonal antibody HY83 was utilized for assay of the CSV helper virus. After addition of biotinylated goat anti-mouse immunoglobulin antibody and strepta-vidin-linked alkaline phosphatase, foci of immunoreactive cells were scored with a microscope (anti-v-Rel antibody) or by eye (anti-CSV antibody).
Transformation assays.Bursal or splenic cells were isolated from fresh tissues taken from 3- to 5-week-old SC chickens. Cells (1.23107
) were incubated with the appropriate dilution of the viral stock at 378C in 200 to 300ml for 1 h. After addition of culture medium, the infected cells were transferred to 24 wells in 96-well trays for the liquid assay or to two 60-mm-diameter petri dishes in 0.35% agar for the soft agar assay. The number of wells containing at least one colony of any size (total colonies) was scored after 8 days of incubation at 378C. Colonies were defined as large if single, easily seen by eye, and associated with yellow discoloration of the medium. The macroscopic colonies formed in soft agar were scored after 14 days of incubation at 378C. The results are expressed as the number of colonies per milliliter of viral stock.
Indirect immunofluorescence and antibodies. Cells from single colonies generated in liquid or soft agar transformation assays were subcultured in 1 ml of medium in 24-well tissue culture trays. After 3 days, 105
cells were cytocen-trifuged onto microscope slides, air dried, fixed in methanol-acetone (1:1) for 5 min, washed three times with PBS, and incubated for 30 min in 0.1% bovine serum albumin in PBS. The slides were then incubated for 1 h with hybridoma supernatant containing a monoclonal antibody diluted 1:1, washed with PBS, and incubated for 1 h with a fluorescein isothiocyanate-conjugated goat anti-mouse immunoglobulin antibody. Slides were washed three times in PBS, with the final wash overnight, and mounted with Fluoromount-G (Southern Biotechnology Associates, Birmingham, Ala.). The intensity of staining was analyzed under a fluorescence microscope.
The following monoclonal antibodies were employed: HY19 anti-chicken immunoglobulin M; HY83 CSV, HY87 v-Rel protein, and CT-3 anti-chicken CD3 (13, 25).
In vitro translation and electrophoretic mobility shift assay.v-rel genes derived from pREV-twPCR (wild type) and pREV-pcrTW (mutant) were introduced into pTZ-18R-based plasmid pTZXN-R as described above. The v-Rel proteins were produced by using the Promega T7 TnT wheat germ extract coupled transcription-translation system in accordance with the protocol of the manufacturer. The relative yield of translation products was estimated by Western immunoblotting (53). A 32P-labeled DNA probe was prepared by
annealing the 27-base template 59-CAACGGCAGGGGAATCTCCCTCTCC TT-39to the complementary 10-base primer 59-AAGGAGAGGG-39and filling the overhang with the Klenow fragment of DNA polymerase I essentially as described by Ballard et al. (4). The template used corresponds to thekB enhancer present in the interleukin-2 receptorapromoter (4). By using in vitro translation products, DNA-binding reactions were carried out at room temper-ature for 30 min in 10 mM Tris-HCl (pH 7.5)–50 mM KCl–1 mM EDTA–5% glycerol in the presence of 0.2 ng (30,000 cpm) of radioactive probe and 2mg of poly(dI-dC) (Pharmacia, Piscataway, N.J.). The resulting complexes were re-solved by 5% nondenaturing polyacrylamide gels and visualized by autoradiog-raphy overnight at room temperature.
RESULTS
REV-S2A3 transforms bursal cells more efficiently than
does wild-type REV-T.
REV-T is the only naturally occurring
retroviral isolate that encodes v-rel (20, 59). REV-T has been
independently cloned and characterized by two groups, and the
nucleotide sequence of v-rel was found to be identical (14, 56,
62). pREV-T3 is the clone used by Chen and Temin (14) and
is considered the prototype of the wild type. pREV-TW was
constructed in our laboratory by inserting v-rel from pREV-T3
into pREV-0, a retroviral vector also derived from pREV-T3
(45). S2A3 is one of the several nonproducer cell lines
obtained by Bose et al. by infecting avian hematopoietic cells
with uncloned REV-T (37). Infectious stocks of this virus were
obtained after superinfecting the S2A3 cell line with the CSV
helper virus. When comparing the in vitro properties of
REV-TW and REV-S2A3, we observed a higher efficiency of
bursal cell transformation by the S2A3 strain. Only this virus
was able to generate colonies in soft agar and to form large
clones in liquid assays (Fig. 1 and Table 1).
v-
rel
accounts for enhanced B-cell transformation by
REV-S2A3.
Two mechanisms could explain the above observations.
Differences in the non-v-rel part of the viral genome could
influence the level of v-rel expression. Transformation of B
cells may require a higher concentration of v-Rel protein than
transformation of other hematopoietic cells, and this
require-ment is met only by REV-S2A3. Alternatively, the two viruses
encode structurally distinct forms of v-rel with unique
onco-genic potentials. Northern studies of cell lines infected with the
two viruses failed to show a significant difference in RNA
expression (data not shown). On the basis of these preliminary
results, we tested the hypothesis that REV-S2A3 codes for a
mutant v-rel with enhanced ability to transform B cells. The
strategy followed was to compare the two oncogenes directly
against the same REV-TW background by inserting v-rel from
REV-S2A3 into REV-0 (Fig. 1). Southern blot analysis
dem-onstrated the existence of only one v-rel-containing provirus in
cell line S2A3, indicating that a single molecular species of
REV-T was responsible for the altered phenotype (data not
shown). PCR amplification of v-rel from S2A3 cells was done
with a 5
9
primer containing an XhoI site and a 3
9
primer
containing a BssHII site. A 1,591-bp fragment was isolated and
cloned into pREV-0 by using the XhoI and BssHII sites. This
procedure resulted in three base pair changes outside v-rel
compared with the published sequence of REV-T (56, 62).
Five clones of pREV-PCR, each produced by an independent
amplification, were obtained.
[image:3.612.316.555.83.173.2]The pREV-PCR plasmids were cotransfected with pCSV
11S3 into CEF to obtain REV-PCR(CSV) viral stocks.
Immu-nofluorescence studies confirmed the expression in fibroblasts
of v-Rel encoded by the recombinant plasmids (data not
shown). We then tested the ability of PCR1 to
REV-PCR4 to form bursal cell colonies in liquid culture assays and
compared it with that of REV-S2A3, REV-TW, REV-T10, and
REV-T3. These experiments delineated two distinct
pheno-types (Fig. 1). Only the retroviruses containing v-rel from
REV-S2A3 generated large colonies. These are easily
recog-nizable by the change in medium color from pink to yellow
caused by a drop in pH as the cell number per well approaches
10
6(see below). In calculating the titer of large CFU; we took
into account only the wells with a single colony, since the
yellow discoloration could also be due to multiple colonies with
limited numbers of cells. On the other hand, REV-TW,
REV-T10, and REV-T3, all related genealogically and
encod-ing the same wild-type v-rel, formed only small colonies. These
TABLE 1. Properties of transformed bursal cells
Virus No. of colonies in soft agara
Colonies in liquid assay
Cell no. 105b % Reaching P10c
REV-TW 0 1.9 11
REV-twPCR 0 1 19
REV-S2A3 1,510 NDd ND
REV-pcrTW 900 10 97
REV-PCR1 250 ND ND
REV-PCR2 900 13.6 87
aMean number of macroscopic colonies per milliliter of viral stock. On average, three experiments were performed for each virus.
bMean total cell number per well containing a single colony. On average, 34 colonies, generated in five transformation experiments, were studied for each virus.
cColonies were serially passaged 10 times in 24-well trays at 48-h intervals. Results are expressed as the percentage of colonies showing growth at passage ten (P10). On average, 25 colonies, obtained in four transformation experiments, were studied for each virus.
d
ND, not done.
on November 9, 2019 by guest
http://jvi.asm.org/
findings indicated that efficient B-cell oncogenesis is due to
qualitative changes in v-rel and not to alterations in other parts
of the viral genome.
An alanine-to-serine mutation at position 40 of the v-Rel
protein confers B-cell-transforming activity.
To define the
structural basis for this difference, the entire v-rel genes and
the flanking segments from both REV-TW and REV-PCR1
were sequenced. Two mutations were found in the 5
9
region of
v-rel from REV-PCR1: a silent mutation at codon 33 and a
G-to-T transversion, changing the alanine at position 40 to
serine (Fig. 2B). These two mutations were also present in the
other four REV-PCR viruses and thus do not represent
PCR-induced artifacts. The v-rel sequence from REV-TW was
found to be identical to the published one (56, 62), confirming
the wild-type genotype of this virus. 5
9
of v-rel, both REV-TW
and REV-PCR1 have the expected change from C to G at
position
2
29, creating the XhoI site used for cloning (Fig. 2A).
In REV-PCR1, analysis of the sequences 3
9
of v-rel revealed
the anticipated changes from T to G and from A to C at
positions
1
33 and
1
38, respectively, converting the NruI site
into a BssHII site (Fig. 2C). The sequence of REV-TW 3
9
of
v-rel contains an extra 21 bp, starting at position
1
36. This
stretch represents an unexpected cloning artifact and includes
a 15-bp repeat of the sequence from position
1
21 to position
1
35 and 6 bp introduced to create the BssHII site (Fig. 2C).
The above alterations outside v-rel may not be relevant to the
oncogenic properties of REV-TW, since they are located in
noncoding parts of the viral genome. These changes, however,
could modulate the expression of v-rel. The only structural
modification of v-Rel that can account for the increased
transformation of B cells by REV-S2A3 is replacement of the
wild-type alanine at position 40 with a serine (Fig. 2D).
To establish the pathogenic relevance of this mutation, two
hybrid retroviruses were constructed by exchanging the
XhoI-ClaI fragment (478 bp), coding for the first 148 amino acids of
the v-Rel protein, between REV-TW and REV-PCR1. These
chimeric constructs are designated REV-pcrTW (serine at
position 40) and REV-twPCR (alanine at position 40) (Fig. 1).
The exchange was verified by DNA sequence analysis of the
mutation site and of the flanking sequences 3
9
of v-rel. After
transfection into CEF to rescue the viruses, the transforming
activities of the two viruses and the parental strains were
compared. Only the mutant viruses were able to form large
colonies of B cells in liquid culture (Fig. 1). The 15-bp repeat
present in the 3
9
part of REV-TW does not inhibit
transfor-mation, since REV-pcrTW contains this sequence and is fully
oncogenic. On the other hand, REV-twPCR, which lacks the
two mutations and codes for an alanine at position 40 but is
otherwise identical to transforming REV-PCR1, displays the
weakly oncogenic phenotype. Collectively, these results
indi-cate that the change from alanine to serine at position 40 of the
v-Rel protein correlates with the increased transformation of
bursal cells by REV-S2A3 (Fig. 1).
Comparison of the transforming properties of wild-type and
mutant v-
rel
in vitro.
The test that most clearly was able to
differentiate between the two phenotypes was formation of
macroscopic colonies of bursal cells in soft agar. On repeated
experiments, REV-TW and REV-twPCR failed to generate
colonies while the mutant viruses consistently scored positive
results, although with a variable titer (Table 1). Particularly
revealing was the comparison between REV-twPCR and
REV-pcrTW. These two hybrid viruses were obtained with the
same transfection and displayed similar titers on
immunocyto-chemical assay (Fig. 1), indicating that the two viral stocks
contain comparable numbers of viral particles coding for the
oncogene. However, only REV-pcrTW, coding for v-Rel with
serine at position 40, can form colonies in soft agar. This assay
is considered the best in vitro correlate of in vivo oncogenicity;
however, the colony formation titers were variable and
rela-tively low compared with the immunocytochemical titers.
Higer titers and more consistent results were obtained by using
the liquid transformation assay and scoring the formation of
large colonies 8 days after infection. The large number of
experiments done with this test are summarized in Fig. 1.
Although the sizes of the colonies formed a continuous
spectrum, we found it useful to divide them into large and
small ones. The large ones were defined by yellow
discolora-tion of the medium as the cells accumulated in the wells and
the pH decreased. The small ones only occasionally progressed
to become large and tended to regress after day 8 of culture.
The total numbers of colonies generated by the two groups of
viruses were similar, with titers ranging from 10,000 to
50,000/ml (data not shown), confirming the comparable
immu-nocytochemical titers. At the lower dilutions, several wells
turned yellow, reflecting the presence of multiple colonies.
Accordingly, the exact titer of large-colony production by the
wild-type viruses is not known. On the other hand,
approxi-mately one of every two single-hit colonies generated by the
mutant viruses was scored as large on day 8 (Fig. 1). The
remaining colonies were of considerable size and commonly
progressed to cause a yellow discoloration in 2 to 3 days. The
trays displayed two distinct color patterns that consistently
mirrored the genotypes of the viruses. To have more insight
into the effects of this gain-of-function mutation, we compared
some aspects of the proliferative activity of bursal cell colonies
induced by the two forms of v-rel. Average cell counts of wells
containing single colonies revealed a 10-fold difference
be-tween the two types of virus (Table 1). Interestingly, the clones
induced by the wild-type viruses had a much higher percentage
of dead cells (30 versus 5%; data not shown), indicating that
the small colonies may represent transient proliferation of cells
destined to die and not full-fledged transformation. We
stud-ied the ability of the clones to be passaged 10 times as an
indicator of immortalization by different forms of v-rel. Only 10
to 20% of the clones generated by the wild type passed that
mark, while 85 to 95% of the clones driven by the mutant
oncoprotein could be cultured for 10 passages (Table 1). These
data indicate that the mutant v-rel confers an increased
prolif-erative activity on B cells.
We were interested in determining if the mutant viruses
transform other cell types more efficiently. Parallel
experi-ments were performed with spleen cells as the target (Table 2).
The soft agar assay was not as discriminatory as for bursal cells,
since the wild-type viruses were also able to generate colonies,
although at a titer lower than that of the mutant viruses. The
same trend was apparent in the liquid assay, where an
approx-imately 10-fold difference in the titer of large colony formation
was observed. This is in contrast to the 100-fold difference for
the same assay employing bursal cells. These data are
consis-tent with the notion that the enhanced transforming ability of
the REV-S2A3 type of virus is selective for B cells, while the
two kinds of virus transform non-B cells with similar
efficien-cies. Immunophenotypic analysis of colonies derived from
spleen cells supported this hypothesis. The REV-TW type of
virus gave rise predominantly to T-cell or non-T, non-B-cell
colonies, while the majority of the colonies produced by the
mutant viruses were of B-cell origin (Table 3).
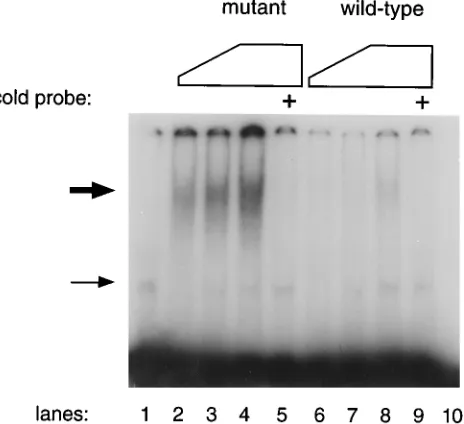
Mutant v-Rel has increased DNA-binding activity.
The
v-Rel oncoprotein is believed to be pathogenic by acting as a
transcription factor either activating or repressing gene
expres-sion after binding to enhancers containing the
k
B motif (4, 16,
18, 27, 41, 54). DNA-binding is a conserved property of
on November 9, 2019 by guest
http://jvi.asm.org/
transforming v-Rel proteins (4, 11, 28, 35, 60, 61). The mutated
residue is located very close to the putative DNA-binding site
encompassing amino acids 27 to 35 (36, 40). The change from
alanine to serine at position 40 could significantly affect the
DNA-protein interaction. We tested this possibility in an
electrophoretic mobility shift assay using in vitro-translated
proteins and an oligomer containing the
k
B element present in
the promoter of the interleukin-2 receptor
a
gene (4). Figure
3 shows the results of a typical experiment with wheat germ
extracts programmed with v-rel derived from REV-pcrTW
(mutant) or REV-twPCR (wild type). Western blotting showed
that the extracts contained equivalent amounts of the
transla-tion product (data not shown). v-Rel with serine at positransla-tion 40
displayed a DNA-binding activity at least fivefold higher than
that of the alanine-containing oncoprotein. In the presence of
an excess of cold probe, the specific band was completely
inhibited while the nonspecific binding activity present in the
extract was not affected.
DISCUSSION
Malignant transformation is considered to be a multistep
process. Many oncogenes transduced by retroviruses require
the cooperation of another oncogene. Synergism between
cytoplasmic and nuclear oncoproteins has been described in
several experimental systems (26). The unusually potent
trans-forming activity of REV-T in vivo implies that v-Rel performs
more than one function during tumorigenesis. In the present
report, we describe a naturally occurring gain-of-function point
mutation of this oncogene that specifically enhances the ability
of the wild type to transform B lymphocytes in vitro. This
variant of v-rel may provide a novel system for elucidating one
function critical to B-cell transformation.
[image:5.612.321.553.74.286.2] [image:5.612.62.298.82.207.2]The structural requirements for v-rel-induced oncogenesis
have been extensively investigated by using a number of
artificial constructs and mutants. Alterations in both the Rel
homology region and the C terminus are required for
trans-formation (9, 17, 29, 43, 45, 54, 58, 60). The data collected so
far do not conclusively favor one of the different pathogenetic
models that have been proposed (20). Acting as a
trans-dominant factor, v-Rel could repress or activate transcription
of critical genes either by occupying
k
B sites directly or by
forming inactive complexes with other Rel proteins or I
k
B
molecules, respectively. The latter indirect models are
consis-tent with the observation that v-Rel is primarily cytoplasmic in
transformed splenocytes (21). Furthermore, mutated forms of
v-Rel can act either in the nucleus or in the cytoplasm to
transform spleen cells and to activate transcription (23).
Sev-eral reports based on transient-expression experiments have
indicated that v-rel-induced transformation is associated with
transcriptional repression of genes regulated by NF-
k
B/Rel
FIG. 3. Comparison of the DNA-binding activities of the mutant and wild-type v-Rel proteins. Increasing amounts of in vitro-translated proteins were incubated with a radiolabeled probe containing akB motif and analyzed by electrophoretic mobility shift assay. Wheat germ extract was programmed with plasmids containing v-rel from REV-pcrTW (lanes 2, 3, 4, and 5) or REV-twPCR (lanes 6, 7, 8, and 9). The control extract (lane 1) was programmed with a plasmid containing a nontranslated form of REV-T. The following amounts of extract were added: lanes 2 and 6, 1ml; lanes 3 and 7, 2.5ml; lanes 1, 4, 5, 8, and 9, 5ml. A 200-fold excess of the cold double-stranded oligonucleotide was added to the samples in lanes 5 and 9. Only the labeled probe (30,000 cpm; 0.2 ng) was loaded on lane 10. The thick arrow indicates the specific band, while the thin arrow corresponds to the nonspecific band.TABLE 2. Transformation of spleen cellsa
Virus
Immuno-cytochemical
titer (106)b
Liquid assay
No. of macroscopic colonies in soft
agar assay Total no. of
colonies (104)
No. of large colonies
(104)
REV-T3 0.5 1.5 ,0.1 NDc
REV-TW 0.3 0.3 ,0.1 20
REV-twPCR 1.5 1.2 ,0.1 65
REV-S2A3 0.5 5 0.6 ND
REV-PCR2 5 3.6 0.6 210
REV-pcrTW 2 3.1 0.3 340
CSV 1.5 0 0 0
None 0 0 0
aThe results are expressed as the mean number of colonies per milliliter of viral stock. For the liquid assay, on average, five experiments were performed for each virus. For the soft agar assay, four experiments were done for each virus. The total colonies were colonies of any size scored with a microscope. Colonies were scored as large if single, easily seen by eye, and growing in wells with yellow discoloration of the medium.
b
Number of foci of CEF reactive with anti-v-Rel antibody per milliliter of viral stock. Antibody HY83 was used for the CSV titer.
c
ND, not done.
TABLE 3. Immunophenotypes of the colonies derived from spleen cells
Assay and virus
No. of expts (no. of colo-nies
ana-lyzed)
No. (%)a with B
cells
No. (%)b with T
cells
No. (%)c with non-B, non-T cells
Liquid
REV-TW 2 (7) 2 (28) 3 (43) 2 (28)
REV-twPCR 3 (20) 4 (20) 12 (60) 4 (20)
REV-pcrTW 3 (21) 16 (76) 3 (14) 2 (9.5)
REV-PCR2 2 (12) 9 (75) 2 (16) 1 (8.3)
Soft agar
REV-twPCR 1 (10) 2 (20) 7 (70) 1 (10)
REV-pcrTW 1 (10) 10 (100) 0 0
a
Colonies with.70% of cells reactive with anti-immunoglobulin M antibody HY19.
b
Colonies with.70% of cells reactive with anti-CD3 antibody CT-3. c
Colonies with,5% of cells staining with antibodies HY19 or CT-3. All of the scored colonies stained strongly with anti-v-Rel antibody.
on November 9, 2019 by guest
http://jvi.asm.org/
[image:5.612.58.297.557.684.2]proteins (4, 27, 41, 44, 51). However, other studies have
suggested that transforming forms of v-Rel activate
transcrip-tion from promoters containing
k
B sites (16, 18, 25, 45, 54, 60).
The data obtained with the REV-PCR viruses indicated that
v-rel derived from the S2A3 cell line confers the ability to
transform bursal cells efficiently. Sequence analysis confirmed
that REV-TW codes for a v-rel identical to the one already
characterized and considered the wild type (14, 56, 62). All of
the PCR viruses, coding for a v-rel derived from
REV-S2A3, contained a mutation changing the alanine at position
40 to serine. The experiments with the hybrid viruses
estab-lished the importance of this change for efficient B-lymphocyte
transformation. The mutant v-rel confers an expanded
prolif-erative potential on infected B cells. This was manifested by
increased colony size in liquid culture and colony formation in
soft agar.
As a biochemical correlate of the enhanced transforming
activity, we have shown that the serine at position 40 increases
the ability of v-Rel to bind a
k
B site. This finding is consistent
with the observation that residues 27 to 35 of v-Rel are
involved in redox regulation of DNA binding (36, 40).
Re-cently, a temperature-sensitive transforming mutant of v-rel
was obtained by changing codon 37 from glycine to glutamic
acid. The resultant v-Rel protein was temperature sensitive for
DNA binding, confirming the notion that the latter function is
critical in transformation (61). The N-terminal 50 residues of
the c-Rel protein have been reported to mediate the
interac-tion with the inhibitor I
k
B and the TATA-binding protein (32,
33). The v-Rel oncoprotein was unable to bind the latter
polypeptide (33). However, these findings are somewhat
con-troversial, since another group has implicated the C-terminal
activation domains of c-Rel and v-Rel in the association with
the TATA-binding protein and transcription factor IIB (64).
Alterations in each of the above interactions can be envisioned
to affect the transforming ability of v-rel through
transcrip-tional modulation of critical genes. It is also possible that some
other function, like dimerization or binding to a novel protein,
could be altered by the mutation. The serine is not located in
a typical recognition motif for the known protein kinases,
although the RXS sequence can be phosphorylated in vitro by
cyclic AMP-dependent protein kinase (30). It remains to be
shown if this posttranslational modification is involved in
regulation of the activity of the mutant v-Rel.
The alanine at position 40 probably plays an important role
since it is conserved in dorsal, RelA, RelB, and c-Rel (48, 52,
62). A histidine replaces alanine in NF-
k
B1 and NF-
k
B2, two
homologous members of the family with distinctive features
and separate evolutionary histories (19, 22, 46). The above
observation and the fact that an alanine is present in the two
clones of v-rel characterized in the past (56, 62) suggest that
REV-S2A3 arose because of a mutation of a REV-T3 type of
virus and not vice versa.
Our data indicate that the main effect of the mutation is to
expand the ability to transform B lymphocytes, rather than
general enhancement of the oncogenic potential toward all
hematopoietic cells. This observation raises the possibility that
a specific cellular component unique to B cells constitutes the
target for the more active v-Rel protein. This target could be
either a regulatory DNA element or a protein present in active
form only in B cells. In retrospect, the present results may
explain in part the efficient transformation of mature B cells
observed in our previous studies (6, 7). In these investigations,
the S2A3 cell line was used to produce the stocks of
REV-T(CSV) that readily induced immunoglobulin M-positive
lym-phomas and cell lines.
Three non-mutually exclusive mechanisms can account for
the 10-fold difference in the sizes of the bursal cell colonies in
liquid culture assays: (i) the cells transformed by mutant v-rel
are prevented from entering G
0and remain in cycle, (ii) the
mutant oncogene enables B lymphocytes to escape the normal
fate of programmed cell death, and (iii) wild-type v-Rel is more
toxic for B cells, and the mutant oncoprotein is deficient in a
function that is detrimental to B lymphocytes. The increased
number of dead cells in the colonies generated by wild-type
v-rel is consistent with the latter two explanations. Under some
conditions, overexpression of c-rel induces apoptosis in avian
bone marrow cells (1). One of the functions of v-Rel could be
prevention of c-Rel-mediated programmed cell death.
A large proportion of oncogenes transduced by acutely
transforming retroviruses are transcriptional stimulators.
Fur-thermore, activation of transcription factors through
chromo-somal translocation is one of the most common pathogenetic
mechanisms implicated in human hematological malignancies
(15, 47). However, the chain of events leading to subversion of
the normal differentiation pattern and to unrestrained
prolif-eration has not been elucidated. Nor has an explanation been
provided for the association between activation of specific
transcription factors and the cell lineage of the resulting
malignancy. Further studies of the molecular consequences of
this variant v-Rel may offer clues to the solution of the above
problems.
ACKNOWLEDGMENTS
We thank Radmila Hrdlicˇkova´ and Jirˇı´ Nehyba for critical reading of the manuscript; for plasmids pREV-TW, pREV-T10, and pREV-0; and for monoclonal antibodies HY83 and HY87. We are also grateful to the late Howard M. Temin for plasmid pREV-T3, Henry R. Bose for the S2A3 cell line, and Max D. Cooper for monoclonal antibody CT3. We acknowledge the excellent technical assistance of Alvin Lim and Yi Chen. We are grateful to Daniel C. Flynn, Kenneth Landreth, and Vinay Pathek for advice during preparation of the manuscript.
REFERENCES
1. Abbadie, C., N. Kabrun, F. Bouali, J. Smardova, D. Ste´helin, B. Vanden-bunder, and P. J. Enrietto. 1993. High levels of c-rel expression are associated with programmed cell death in the developing avian embryo and in bone marrow cells in vitro. Cell 75:899–912.
2. Baeuerle, P. A. 1991. The inducible transcription activator NF-kB: regulation by distinct protein subunits. Biochim. Biophys. Acta 1072:63–80. 3. Baeuerle, P. A., and D. Baltimore. 1988. IkB: a specific inhibitor of the
NF-kB transcription factor. Science 242:540–546.
4. Ballard, D. W., W. H. Walker, S. Doerre, P. Sista, J. A. Molitor, E. P. Dixon, N. J. Peffer, M. Hannink, and W. C. Greene.1990. The v-rel oncogene encodes akB enhancer binding protein that inhibits NF-kB function. Cell 63: 803–814.
5. Barth, C. F., D. L. Ewert, W. C. Olson, and E. H. Humphries. 1990. Reticuloendotheliosis virus REV-T(REV-A)-induced neoplasia: develop-ment of tumors within the T-lymphoid and myeloid lineages. J. Virol. 64: 6054–6062.
6. Barth, C. F., and E. H. Humphries. 1988. A nonimmunosuppressive helper virus allows high efficiency induction of B cell lymphomas by reticuloendo-theliosis virus strain T. J. Exp. Med. 167:89–108.
7. Barth, C. F., and E. H. Humphries. 1988. Expression of v-rel induces mature B-cell lines that reflect the diversity of avian immunoglobulin heavy- and light-chain rearrangements. Mol. Cell. Biol. 8:5358–5368.
8. Beug, H., H. Mu¨ller, S. Grieser, G. Doederlein, and T. Graf.1981. Hema-topoietic cells transformed in vitro by REVTavian reticuloendotheliosis virus
express characteristics of very immature lymphoid cells. Virology 115: 295–309.
9. Bhat, G. V., and H. M. Temin. 1990. Mutational analysis of v-rel, the oncogene of reticuloendotheliosis virus strain T. Oncogene 5:625–634. 10. Blank, V., P. Kourilsky, and A. Israe¨l. 1992. NF-kB and related proteins:
Rel/dorsal homologies meet ankyrin-like repeats. Trends Biochem. Sci. 17: 135–140.
11. Boehmelt, G., A. Walker, N. Kabrun, G. Mellitzer, H. Beug, M. Zenke, and P. J. Enrietto. 1992. Hormone-regulated v-rel estrogen receptor fusion protein: reversible induction of cell transformation and cellular gene expres-sion. EMBO J. 11:4641–4652.
12. Bose, H. R., Jr. 1992. The Rel family: models for transcriptional regulation
on November 9, 2019 by guest
http://jvi.asm.org/
and oncogenic transformation. Biochim. Biophys. Acta 1114:1–17. 13. Chen, C.-L. H., L. L. Ager, G. L. Gartland, and M. D. Cooper. 1986.
Identification of a T3/T cell receptor complex in chickens. J. Exp. Med. 164: 375–380.
14. Chen, I. S. Y., and H. M. Temin. 1982. Substitution of 59 helper virus sequences into non-rel portion of reticuloendotheliosis virus strain T sup-presses transformation of chicken spleen cells. Cell 31:111–120.
15. Cleary, M. L. 1991. Oncogenic conversion of transcription factors by chromosomal translocations. Cell 66:619–622.
16. Diehl, J. A., and M. Hannink. 1993. Heterologous C-terminal sequences disrupt transcriptional activation and oncogenesis by p59v-rel. J. Virol. 67:
7161–7171.
17. Diehl, J. A., T. A. McKinsey, and M. Hannink. 1993. Differential pp40IkB-b inhibition of DNA binding by rel proteins. Mol. Cell. Biol. 13:1769–1778. 18. Ge´linas, C., and H. M. Temin. 1988. The v-rel oncogene encodes a cell
specific transcriptional activator of certain promoters. Oncogene 3:349–355. 19. Ghosh, S., A. M. Gifford, L. R. Riviere, P. Tempst, G. P. Nolan, and D. Baltimore. 1990. Cloning of the p50 DNA binding subunit of NF-kB: homology to rel and dorsal. Cell 62:1019–1029.
20. Gilmore, T. D. 1991. Malignant transformation by mutant Rel proteins. Trends Genet. 7:318–322.
21. Gilmore, T. D., and H. M. Temin. 1986. Different localization of the product of the v-rel oncogene in chicken fibroblasts and spleen cells correlates with transformation by REV-T. Cell 44:791–800.
22. Grilli, M., J.-J.-S. Chiu, and M. J. Lenardo. 1993. NF-kB and Rel: participants in a multiform transcriptional regulatory system. Int. Rev. Cytol. 143:1–62.
23. Hannink, M., and H. M. Temin. 1989. Transactivation of gene expression by nuclear and cytoplasmic rel proteins. Mol. Cell. Biol. 9:4323–4336. 24. Hoelzer, J. D., R. B. Lewis, C. R. Wasmuth, and H. R. Bose, Jr. 1980.
Hematopoietic cell transformation by reticuloendotheliosis virus: character-ization of the genetic defect. Virology 100:462–474.
25. Hrdlicˇkova´, R., J. Nehyba, and E. H. Humphries. 1994. v-rel induces expression of three avian immunoregulatory surface receptors more effi-ciently than c-rel. J. Virol. 68:308–319.
26. Hunter, T. 1991. Cooperation between oncogenes. Cell 64:249–270. 27. Inoue, J.-I., L. D. Kerr, L. J. Ransone, E. Bengal, T. Hunter, and I. M.
Verma.1991. c-rel activates but v-rel suppresses transcription fromkB sites. Proc. Natl. Acad. Sci. USA 88:3715–3719.
28. Kabrun, N., J. W. Hodgson, M. Doemer, G. Mak, B. R. Franza, Jr., and P. J. Enrietto.1991. Interaction of the v-rel protein with an NF-kB DNA binding site. Proc. Natl. Acad. Sci. USA 88:1783–1787.
29. Kamens, J., P. Richardson, G. Mosialos, R. Brent, and T. Gilmore. 1990. Oncogenic transformation by vRel requires an amino-terminal activation domain. Mol. Cell. Biol. 10:2840–2847.
30. Kemp, B. E., and R. B. Pearson. 1990. Protein kinase recognition sequence motifs. Trends Biochem. Sci. 15:342–346.
31. Kerr, L. D., C. S. Duckett, P. Wamsley, Q. Zhang, P. Chiao, G. Nabel, T. W. McKeithan, P. A. Baeuerle, and I. M. Verma.1992. The proto-oncogene BCL-3 encodes an IkB protein. Genes Dev. 6:2352–2363.
32. Kerr, L. D., J.-I. Inoue, N. Davis, E. Link, P. A. Baeuerle, H. R. Bose, Jr., and I. M. Verma.1991. The Rel-associated pp40 protein prevents DNA binding of Rel and NF-kB: relationship with IkBband regulation by phosphoryla-tion. Genes Dev. 5:1464–1476.
33. Kerr, L. D., L. J. Ransone, P. Wamsley, M. J. Schmitt, T. G. Boyer, Q. Zhou, A. J. Berk, and I. M. Verma.1993. Association between proto-oncoprotein Rel and TATA-binding protein mediates transcriptional activation by
NF-kB. Nature (London) 365:412–419.
34. Kieran, M., V. Blank, F. Logeat, J. Vandekerckhove, F. Lottspeich, O. Le Bail, M. B. Urban, P. Kourilsky, P. A. Baeuerle, and A. Israe¨l.1990. The DNA binding subunit of NF-kB is identical to factor KBF1 and homologous to the rel oncogene product. Cell 62:1007–1018.
35. Kochel, T., and N. R. Rice. 1992. v-rel- and c-rel-protein complexes bind to the NF-kB site in vitro. Oncogene 7:567–572.
36. Kumar, S., A. B. Rabson, and C. Ge´linas. 1992. The RxxRxRxxC motif conserved in all Rel/kB proteins is essential for the DNA-binding activity and redox regulation of the v-Rel oncoprotein. Mol. Cell. Biol. 12:3094– 3106.
37. Lewis, R. B., J. McClure, B. Rup, D. W. Niesel, R. F. Garry, J. D. Hoelzer, K. Nazerian, and H. R. Bose, Jr.1981. Avian reticuloendotheliosis virus: identification of the hematopoietic target cell for transformation. Cell 25: 421–431.
38. Liou, H.-C., and D. Baltimore. 1993. Regulation of the NF-kB/rel transcrip-tion factor and IkB inhibitor system. Curr. Opin. Cell Biol. 5:477–487. 39. Lu, D., J. D. Thompson, K. G. Gorski, N. R. Rice, M. G. Mayer, and J. J.
Yunis.1991. Alterations at the rel locus in human lymphoma. Oncogene 6: 1235–1241.
40. Matthews, J. R., W. Kaszubska, G. Turcatti, T. N. C. Wells, and R. T. Hay.
1993. Role of cysteine62in DNA recognition by the P50 subunit of NF-kB.
Nucleic Acids Res. 21:1727–1734.
41. McDonnell, P. C., S. Kumar, A. B. Rabson, and C. Ge´linas. 1992. Transcrip-tional activity of rel family proteins. Oncogene 7:163–170.
42. Moore, B. E., and H. R. Bose, Jr. 1988. Expression of the v-rel oncogene in reticuloendotheliosis virus-transformed fibroblasts. Virology 162:377–387. 43. Morrison, L. E., G. Boehmelt, and P. J. Enrietto. 1992. Mutations in the
rel-homology domain alter the biochemical properties of v-rel and render it transformation defective in chicken embryo fibroblasts. Oncogene 7:1137– 1147.
44. Mosialos, G., P. Hamer, A. J. Capobianco, R. A. Laursen, and T. D. Gilmore. 1991. A protein kinase-A recognition sequence is structurally linked to transformation by p59v-reland cytoplasmic retention of p68c-rel. Mol. Cell.
Biol. 11:5867–5877.
45. Nehyba, J., R. Hrdlicˇkova´, and E. H. Humphries.1994. Evolution of the oncogenic potential of v-rel: rel-induced expression of immunoregulatory receptors correlates with tumor development and in vitro transformation. J. Virol. 68:2039–2050.
46. Neri, A., C.-C. Chang, L. Lombardi, M. Salina, P. Corradini, A. T. Maiolo, R. S. K. Chaganti, and R. Dalla-Favera.1991. B cell lymphoma-associated chromosomal translocation involves candidate oncogene lyt-10, homologous to NF-kB p50. Cell 67:1075–1087.
47. Nichols, J., and S. D. Nimer. 1992. Transcription factors, translocations, and leukemia. Blood 80:2953–2963.
48. Nolan, G. P., S. Ghosh, H.-C. Liou, P. Tempst, and D. Baltimore. 1991. DNA binding and IkB inhibition of the cloned p65 subunit of NFkB, a rel-related polypeptide. Cell 64:961–969.
49. Ohno, H., G. Takimoto, and T. W. McKeithan. 1990. The candidate proto-oncogene bcl-3 is related to genes implicated in cell lineage determi-nation and cell cycle control. Cell 60:991–997.
50. Perkins, N. D., R. M. Schmid, C. S. Duckett, K. Leung, N. R. Rice, and G. J. Nabel.1992. Distinct combinations of NF-kB subunits determine the spec-ificity of transcriptional activation. Proc. Natl. Acad. Sci. USA 89:1529–1533. 51. Richardson, P. M., and T. D. Gilmore. 1991. vRel is an inactive member of the Rel family of transcriptional activating proteins. J. Virol. 65:3122–3130. 52. Ryseck, R.-P., P. Bull, M. Takamiya, V. Bours, U. Siebenlist, P. Dobrzanski, and R. Bravo.1992. RelB, a new Rel family transcription activator that can interact with p50-NF-kB. Mol. Cell. Biol. 12:674–684.
53. Sambrook, J., E. F. Fritsch, and T. Maniatis. 1989. Molecular cloning: a laboratory manual, 2nd ed. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, N.Y.
54. Sarkar, S., and T. D. Gilmore. 1993. Transformation by the vRel oncoprotein requires sequences carboxy-terminal to the Rel homology domain. Onco-gene 8:2245–2252.
55. Schwartz, R. C., and O. N. Witte. 1988. A recombinant murine retrovirus expressing v-rel is cytopathic. Virology 165:182–190.
56. Stephens, R. M., N. R. Rice, R. R. Hiebsch, H. R. Bose, Jr., and R. V. Gilden. 1983. Nucleotide sequence of v-rel: the oncogene of reticuloendotheliosis virus. Proc. Natl. Acad. Sci. USA 80:6229–6233.
57. Stoker, A. W., and M. J. Bissell. 1987. Quantitative immunocytochemical assay for infectious avian retroviruses. J. Gen. Virol. 68:2481–2485. 58. Sylla, B. S., and H. M. Temin. 1986. Activation of oncogenicity of the c-rel
proto-oncogene. Mol. Cell. Biol. 6:4709–4716.
59. Theilen, G., R. Zeigel, and M. Tweihaus. 1966. Biological studies with RE virus (strain T) that induces reticuloendotheliosis in turkeys, chickens, and Japanese quails. J. Natl. Cancer Inst. 37:747–749.
60. Walker, W. H., B. Stein, P. A. Ganchi, J. A. Hoffman, P. A. Kaufman, D. W. Ballard, M. Hannink, and W. C. Greene.1992. The v-rel oncogene: insights into the mechanism of transcriptional activation, repression, and transfor-mation. J. Virol. 66:5018–5029.
61. White, D. W., and T. D. Gilmore. 1993. Temperature-sensitive transforming mutants of the v-rel oncogene. J. Virol. 67:6876–6881.
62. Wilhelmsen, K. C., K. Eggleton, and H. M. Temin. 1984. Nucleic acid sequences of the oncogene v-rel in reticuloendotheliosis virus strain T and its cellular homolog, the proto-oncogene c-rel. J. Virol. 52:172–182. 63. Wulczyn, F. G., M. Naumann, and C. Scheidereit. 1992. Candidate
proto-oncogene bcl-3 encodes a subunit-specific inhibitor of transcription factor NF-kB. Nature (London) 358:597–599.
64. Xu, X., C. Prorock, H. Ishikawa, E. Maldonado, Y. Ito, and C. Ge´linas. Functional interaction of the v-Rel and c-Rel oncoproteins with the TATA-binding protein and association with transcription factor IIB. Mol. Cell. Biol. 13:6733–6741.
65. Zhang, J., W. Bargmann, and H. R. Bose, Jr. 1989. Rearrangement and diversification of immunoglobulin light-chain genes in lymphoid cells trans-formed by reticuloendotheliosis virus. Mol. Cell. Biol. 9:4970–4976. 66. Zhang, J., W. Olson, D. Ewert, W. Bargmann, and H. R. Bose, Jr. 1991. The
v-rel oncogene of avian reticuloendotheliosis virus transforms immature and mature lymphoid cells of the B cell lineage in vitro. Virology 183:457–466.