Interleukin-12 inhibits hepatitis B virus replication in transgenic mice.
Full text
Figure
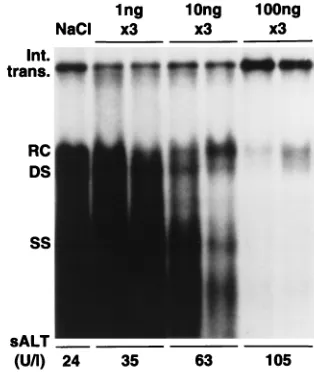
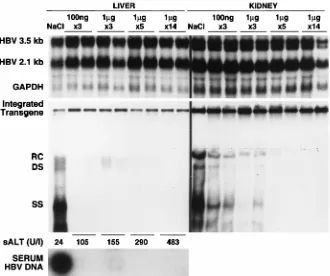
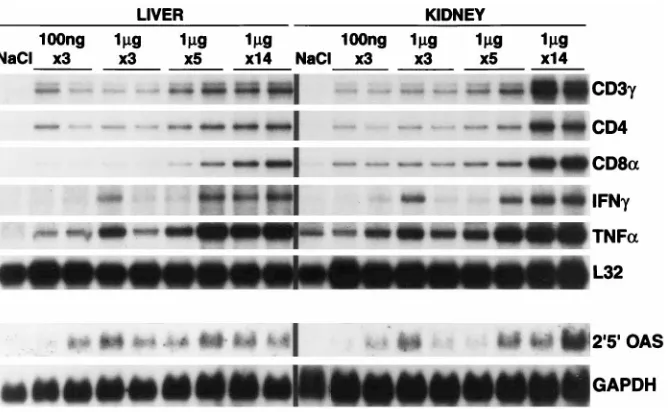
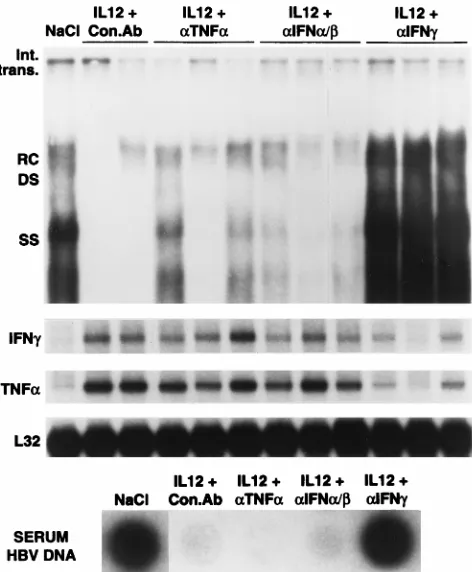
Related documents
Importantly, transfer of splenic and intrahepatic lymphocytes from HBVTg mice treated with IL-15 plus IFN- ␣ , but not from those receiving the LUC control, to C57BL/6 mice
The HBV transgenic mouse experiments demonstrated that proteasome inhibitor MLN-273 enhanced viral replication markedly in the HBVX ⫺ mice but only slightly in the HBVWT
The mean serum glucose levels plus standard deviations are shown for the following groups of HBV transgenic mice: five control male FoxA3 ⫹/⫺ , six fasted male FoxA3 ⫹/⫺ ,
The ratio of viral replication intermediates to HBV 3.5-kb transcripts in individ- ual mice indicates that in rat HNF3  -expressing HBV trans- genic mice, four- to sevenfold less
3, hepatic HBV replicative DNA forms were significantly reduced by the first day after MCMV infec- tion in the absence of histological (not shown) and biochemical evidence of
Collectively, these results suggest that IL-2 alters the steady-state content of hepatic HBV mRNA by a posttranscriptional mechanism in vivo, that this effect is mediated by TNF-oi,
The significant Table 1, Table 2 linear stepwise regres- sion quantifies a DCP Effects Model (DCP-EM) for the HBV mice showing a significant relationship between the inhibition
We stained peritoneal cells, spleen cells, and bone marrow cells from cadmium- injected 116 transgenic mice or cadmium-injected control mice with anti-Ly-1 mAb together with
