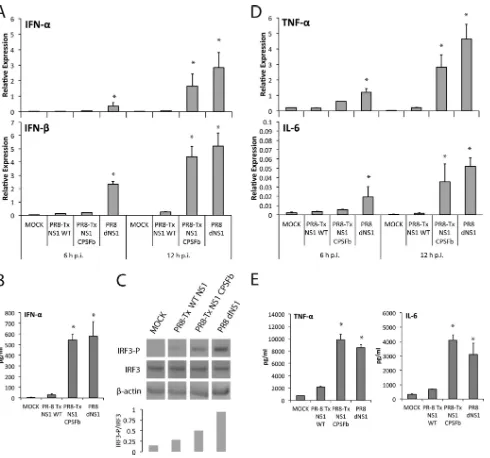
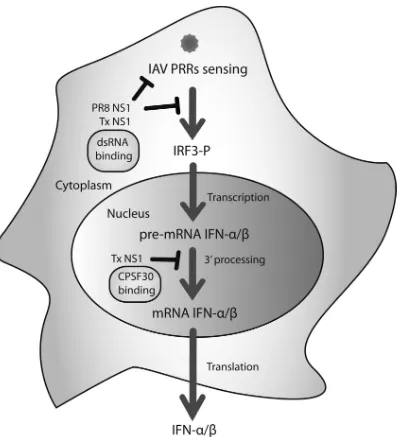
Contribution of Double-Stranded RNA and CPSF30 Binding Domains of Influenza Virus NS1 to the Inhibition of Type I Interferon Production and Activation of Human Dendritic Cells
Full text
Figure
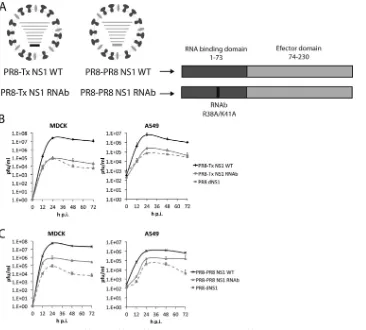

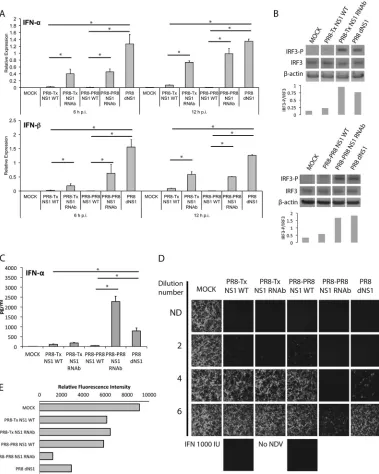
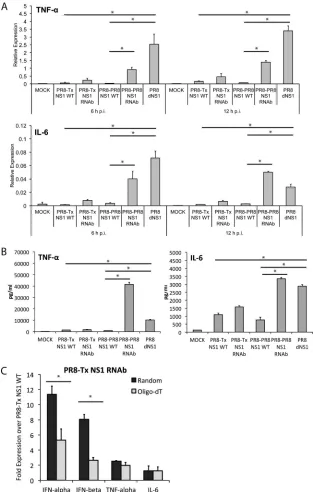
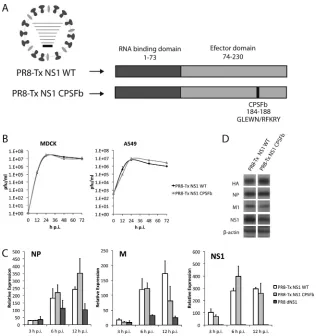
Related documents
This type of equations arises from a variety of mathematical models in engineering and physical sciences; for example, inverse scattering problems in quantum physics, an inverse
In this paper, we establish the weighted sharp maximal function inequalities for a multilinear operator associated to a singular integral operator with non-smooth kernel.. As
To verify the regions of diversity (L 2 -D hy- brids), single amplicons of these regions (the tarp gene, incA , hctB , and toxin locus genes; the IGR upstream of ftsK ) that
Our study successfully implemented a referral program for maternal contraceptive services within pediatric care by having pediatric residents assess postpartum women ’ s needs
Evidence published by the Scottish Government demon- strates that the social housing sector has a much stronger association with child poverty than other types of housing tenure: 63
Moreover, there is also a consensus that contributions should be based on the risk profile of each bank – as an essential precondition to providing individual banks with the proper
European Union Foreign Affairs Ministers agreed that they should continue to follow thE roadmap for EU negotiations when they discussed EU enlargement at the
To overcome this issue, various feeding techniques have proposed .there are many aspects that affect the performance of the antenna like dimensions, selection of the
