Terminating Nucleotides by Human Immunodeficiency Virus Type 1
Reverse Transcriptase Containing the M184V Mutation
Antonio J. Acosta-Hoyos, Suzanne E. Matsuura, Peter R. Meyer,* and Walter A. Scott
Department of Biochemistry and Molecular Biology, University of Miami Miller School of Medicine, Miami, Florida, USA
Resistance to nucleoside reverse transcriptase (RT) inhibitors is conferred on human immunodeficiency virus type 1 through
thymidine analogue resistance mutations (TAMs) that increase the ability of RT to excise chain-terminating nucleotides after
they have been incorporated. The RT mutation M184V is a potent suppressor of TAMs. In RT containing TAMs, the addition of
M184V suppressed the excision of 3
=
-deoxy-3
=
-azidothymidine monophosphate (AZTMP) to a greater extent on an RNA
tem-plate than on a DNA temtem-plate with the same sequence. The catalytically inactive RNase H mutation E478Q abolished this
differ-ence. The reduction in excision activity was similar with either ATP or pyrophosphate as the acceptor substrate. Decreased
exci-sion of AZTMP was associated with increased cleavage of the RNA template at position
ⴚ
7 relative to the primer terminus,
which led to increased primer-template dissociation. Whether M184V was present or not, RT did not initially bind at the
ⴚ
7
cleavage site. Cleavage at the initial site was followed by RT dissociation and rebinding at the
ⴚ
7 cleavage site, and the
dissocia-tion and rebinding were enhanced when the M184V mutadissocia-tion was present. In contrast to the effect of M184V, the K65R
muta-tion suppressed the excision activity of RT to the same extent on either an RNA or a DNA template and did not alter the RNase H
cleavage pattern. Based on these results, we propose that enhanced RNase H cleavage near the primer terminus plays a role in
M184V suppression of AZT resistance, while K65R suppression occurs through a different mechanism.
R
everse transcriptase (RT) of human immunodeficiency virus
type 1 (HIV-1) is the key enzyme responsible for the synthesis
of a double-stranded copy of the HIV genome that is subsequently
integrated into the host chromosome during HIV infection.
Treatment of HIV-1-infected patients with RT inhibitors such as
3
=
-deoxy-3
=
-azidothymidine (zidovudine, AZT) leads to selection
of mutations in RT known as thymidine analogue resistance
mu-tations (TAMs), which include M41L, D67N, K70R, L210W,
T215Y or F, and K219Q or E. RTs containing various
combina-tions of these mutacombina-tions have an elevated excision activity that
allows them to remove AZT monophosphate (AZTMP) and other
chain-terminating nucleotides after they have been incorporated
(1, 2, 10, 33). Treatment of HIV-1-infected patients with
(
⫺
)2
=
,3
=
-dideoxy-3
=
-thiacytidine (lamivudine, 3TC) or (
⫺
)2
=
,
3
=
-dideoxy-5-fluoro-3
=
-thiacytidine (emtricitabine, FTC) leads to
selection of the M184I mutation in RT, which is rapidly replaced
with M184V (7, 25, 52, 57). The M184V mutation is a potent
suppressor of AZT resistance conferred by TAMs (7, 30, 40, 57),
and this suppressor activity is thought to contribute to the
bene-ficial effects of therapies that include 3TC or FTC in combination
with other nucleoside RT inhibitors (19, 30, 40, 44, 56).
Methionine 184 is part of the YMDD signature motif that
makes up the polymerase active site of HIV-1 RT and lies near the
binding sites for the primer terminus and the incoming
deoxy-nucleoside triphosphate (dNTP) (28, 31). M184I is usually
se-lected first during therapy with 3TC or FTC, and structural studies
to investigate the molecular mechanism of drug resistance have
focused on RT containing this mutation. A cocrystal structure of
M184I mutant RT with a DNA-DNA primer-template (P/T)
shows changes in the positioning of the primer terminus and the
dNTP binding site due to the mutation, leading to a model that
explains 3TC and FTC resistance through an increased ability of
the mutant enzyme to exclude the analogs in favor of the natural
substrate, dCTP (50). M184I RT is defective in binding to natural
dNTPs and has defects in primer extension and processivity, as
well as increased strand-switching activity (26, 29). These defects
result in impaired fitness
in vivo
, and the M184I mutation is
rap-idly replaced with M184V during prolonged therapy (52, 53).
M184V RT also has a mild defect in processivity (3–5, 8, 23, 39, 54)
and ternary complex (RT-dNTP-P/T) stability (18, 24)
accompa-nied by modest reductions in affinity for natural dNTPs (15, 20,
60), but this mutant is selected because of a fitness advantage
in
vivo
. Selection experiments in a TAM background resulted in
M184V with no evidence of an M184I intermediate (30), and
M184V is usually the only mutation detected at RT position 184 in
HIV-1-infected patients undergoing prolonged 3TC therapy (53).
The present study focused on M184V since it is the mutation most
relevant to ongoing clinical suppression of AZT resistance.
The biochemical basis of M184V suppression of TAMs has
been the subject of several studies (9, 21, 22, 27, 32, 38, 39, 49, 58,
62). Typically, the M184V mutation confers a reduction in
exci-sion activity that can be observed on either RNA or DNA
tem-plates. Here we compared M184V-dependent reductions in
exci-sion activity on both RNA and DNA templates with the same
sequence. Suppression of excision was consistently greater on an
RNA template than on a DNA template. This increased
suppres-sion depended on the RNase H activity of RT and was associated
Received22 July 2011 Accepted21 February 2012
Published ahead of print29 February 2012
Address correspondence to Walter A. Scott, [email protected].
* Present address: Shaw Science Partners, Atlanta, Georgia, USA.
Copyright © 2012, American Society for Microbiology. All Rights Reserved.
doi:10.1128/JVI.05767-11
on November 7, 2019 by guest
http://jvi.asm.org/
with enhanced RNase H cleavage at sites in the template near the
primer terminus that result in dissociation of the P/T duplex. The
M184V mutation favored dissociation of the mutant RT from
the site of initial cleavage, resulting in enhanced binding and
cleavage at a downstream site near the primer terminus, leading to
the dissociation of P/T, which disfavors excision.
MATERIALS AND METHODS
Expression and purification of wild-type (WT) RT (RTWT) and mutant
HIV-1 RTs.His-tagged HIV-1 RT was prepared as previously described, using expression vector pKRT2, which contains RT coding sequence from the HIV-1 BH10 background (12), or pTRc99 (Pharmacia Biotech), which contains RT coding sequence from the HIV-1 LAI background (55). pKRT2 was obtained from the AIDS Research and Reference Re-agent Program (contributed by Richard D’Aquila and William C. Sum-mers). Proviral clones of HIV-1 WT-LAI and RT mutants in the LAI background containing restriction sites XbaI and XmaI bracketing most of the RT coding sequence were provided by John Mellors. The XbaI-XmaI fragment was transferred into a pTRc99 expression vector, also provided by John Mellors. The expression vectors were further modified by adding an N-terminal polyhistidine tail as previously described (34, 35). RTK65R, RTD67N/K70R/T215Y/K219Q, and RTK65R/D67N/K70R/T215Y/K219Q were expressed in pTRc99 constructs and compared with RTWT-LAI. The other mutant RTs used in this study (RTM41L/T215Y, RTM41L/M184V/T215Y, RTM41L/T215Y/E478Q, RTM41L/M184V/T215Y/E478Q, RTD67N/K70R/T215F/K219Q, and RTD67N/K70R/M184V/T215F/K219Q) were expressed in pKRT2 constructs and compared with RTWT-BH10. Mutations were introduced by the megaprimer method (51) or by site-directed mutagenesis using the QuikChange II kit (Agilent Technologies) and confirmed by sequencing. WT and mutant RTs were purified as previously described (34) and were predominantly homodimeric. For all assays, RT was added in 20- to 40-fold excess over the P/T concentration based on the RT protein concen-tration with no adjustment made for differences in specific activity.
Labeled and unlabeled oligonucleotides.Oligodeoxynucleotides L33 (5=-CTACTAGTTTTCTCCATCTAGACGATACCAGAA-3=), D19 (5=-C TACTAGTTTTCTCCATC-3=), and D50 (5=-GAGTGCTGAGGTCTTCA
TTCTGGTATCGTCTAGATGGAGAAAACTAGTAG-3=) were
pur-chased from Sigma Genosys and gel purified prior to use. Where indicated, L33 was 5=labeled with [␥-32P]ATP (Perkin-Elmer Life Sci-ences) and T4 polynucleotide kinase (Promega Corp.). Labeled DNA was separated from unincorporated nucleotide using Micro Bio-Spin Col-umns (P-30 colCol-umns, RNase-Free; Bio-Rad Laboratories), followed by phenol-chloroform extraction and ethanol precipitation. The 5= -biotin-ylated L33 oligodeoxynucleotide was purchased from Eurofins MWG Operon and gel purified before use. RNA template R52 (5=-GGGAGUG CUGAGGUCUUCAUUCUGGUAUCGUCUAGAUGGAGAAAACUAG UAG-3=) was synthesized byin vitrotranscription by following the man-ufacturer’s protocol (T7-MEGA-shortscript kit; Ambion, Inc.). In brief, 500 nM DNA duplex formed by the oligonucleotide 5=-AATTTAATACG ACTCACTATAGGGAGTGCTGAGGTCTTCATTCTGGTATCGTCTAG ATGGAGAAAACTAGTAG-3=annealed to its complement was incubated for 4 h with T7 RNA polymerase and NTPs at 37°C, treated with RNase-free DNase I (Ambion, Inc.) for 20 min at 37°C, and then heated at 95°C for 5 min. Unincorporated nucleotides were removed with P-30 columns, followed by treatment with 1 U shrimp alkaline phosphatase (Promega Corp.) in the presence of RNase inhibitor (20 U of RNasin-Plus; Promega Corp.) for 30 min at 37°C. The phosphatase was inactivated by heating for 5 min at 95°C, and the unlabeled RNA was gel purified, phenol-chloro-form extracted, and ethanol precipitated. Alternatively, after gel purifica-tion, the RNA was 5=labeled with [␥-32P]ATP and T4 polynucleotide kinase in a reaction mixture containing 20 U of RNasin-Plus, and labeled nucleotide was removed by centrifugation through a P-30 column, fol-lowed by phenol-chloroform extraction and ethanol precipitation.
Excision rescue of AZTMP-terminated P/Ts.Sixteen picomoles of 5=-32P-labeled L33 was annealed with 32 pmol of unlabeled R52 or D50
and incubated with 10M AZTTP and 190 nM RTM41L/T215Y/E478Qin RB-10 buffer (final concentrations, 10 mM MgCl2, 40 mM HEPES [pH, 7.5], 60 mM KCl, 1 mM dithiothreitol, 2.5% glycerol, and 80 g/ml bovine serum albumin) and 20 U of RNasin-Plus in a total volume of 300l for 1 h at 37°C. The32P-labeled, chain-terminated P/T was purified by centrifugation through a P-30 column, phenol-chloroform extraction, and ethanol precipitation and resuspended in buffer containing 10 mM Na-HEPES (pH 7.4) and 60 mM KCl. Chain termination was 90 to 94% complete based on primer extension ex-periments. Excision rescue experiments were initiated by adding WT or mutant RT (110 to 250 nM) to a mixture containing 5 nM 5=-32 P-L33-AZT primer annealed to unlabeled R52 or D50 template in RB-10 buffer with the four dNTPs at 10M (each) and 3.2 mM ATP or 15 or 50M pyrophosphate (PPi) in a final volume of 10l. The ATP and dNTPs were purchased from GE Healthcare and treated with thermo-stable pyrophosphatase (Roche Applied Science) as previously de-scribed (37). The reaction mixtures were incubated at 37°C for various times (1 to 30 min) and terminated by heating at 90°C for 4 min. An equal volume of 2⫻urea-TTE loading buffer (16 M urea, 180 mM Tris, 58 mM taurine, 1 mM EDTA, 0.5% bromphenol blue, 0.5% xylene cyanol) was added, and the primer extension products were separated from unextended primer by electrophoresis on 10% polyacrylamide denaturing gels containing 8 M urea. The amount of radioactivity in extended primer was determined on a dried gel with a PhosphorI-mager (Molecular Dynamics Storm 840) and expressed as a percentage of the total labeled primer detected in the gel. Results are presented without correction for primer extension that occurred in the absence of added ATP or PPi(6 to 10%). Results of repeated experiments were averaged after normalization to the percent extended primer formed in the same experiment by RTWTafter 15 min of incubation.
Detection and quantification of RNase H cleavage products.5=-32 P-labeled R52 template (12 pmol) was annealed with 24 pmol of unla-beled L33 or D19 primer and incubated with 10M AZTTP, 125 nM RTM41L/T215Y/E478Q, and 20 U of RNasin-Plus in RB-10 buffer in a total volume of 150l for 1 h at 37°C. Unincorporated AZTTP was removed by passage through a P-30 spin column. AZTMP-terminated P/T was isolated by phenol-chloroform extraction and ethanol precipitation and resuspended in buffer containing 10 mM Na-HEPES (pH 7.4) and 60 mM KCl. Aliquots of labeled template-primer were incubated with WT or mutant RTs in RB-10 buffer containing 20 U of RNasin-Plus (final volume, 10l) for various times (5 to 30 min), and the reactions were terminated by heating for 4 min at 90°C and the addition of an equal volume of 2⫻urea-TTE loading buffer. The RNase H cleavage products were fractionated by electrophoresis on 10% polyacryl-amide denaturing gels, and degradation products were quantified by PhosphorImager and expressed as a percentage of the total RNA tem-plate.
Detection of primer-bound and released RNase H cleavage frag-ments.5=-32P-labeled R52 template (12 pmol) was annealed with 24 pmol of unlabeled 5=-biotinylated L33 oligonucleotide and terminated with AZTMP as described above. After purification by P-30 spin col-umns, phenol-chloroform extraction, and ethanol precipitation, the AZT-terminated, biotinylated P/T was resuspended at a concentration of 100 nM and incubated with streptavidin (SA)-conjugated magnetic beads (Dynabeads M-280 SA; Invitrogen) (30g of SA-beads, 0.1 pmol of biotinylated primer) for 30 min at room temperature. The indicated mutant RT (110 to 250 nM) was added, and the suspension was incubated as described for RNase H cleavage assays. At the time points indicated, free and SA-bound fractions were separated using a magnet. Samples were diluted with an equal volume of 2⫻urea-TTE loading buffer and heated for 4 min at 90°C, and free and SA-bound RNase H cleavage products were fractionated by gel electrophoresis and detected as described above.
on November 7, 2019 by guest
http://jvi.asm.org/
RESULTS
Effect of the M184V mutation on ATP-mediated excision of
AZTMP from a chain-terminated primer.
Excision rescue of a
5
=
-
32P-labeled AZTMP-terminated DNA primer was evaluated by
primer extension assays carried out in the presence of WT or
mu-tant RT, ATP as the excision acceptor substrate, and all four
dNTPs. Transfer of the chain-terminating AZTMP to ATP by the
excision activity of RT produces an unblocked primer that is
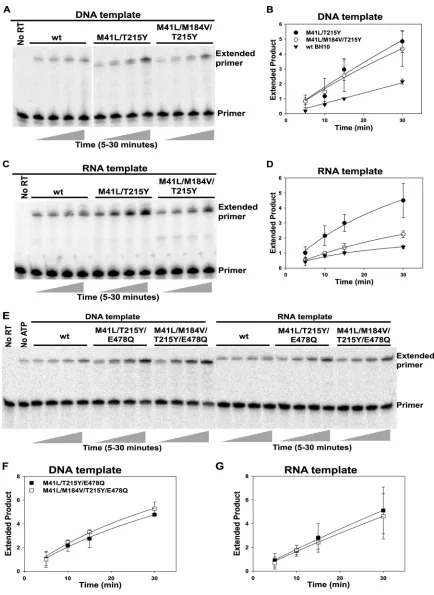
ex-tended by the DNA-polymerizing activity of RT. Figure 1 shows
the rescue of AZTMP-terminated primer by WT and mutant RTs
carried out with DNA template (Fig. 1A, B, E, and F) or with RNA
template (Fig. 1C, D, E, and G). As previously reported (35),
ex-cision activity was substantially increased in RT containing the
M41L and T215Y mutations (RT
M41L/T215Y) in comparison with
RT
WT. The increases were similar for RNA and DNA templates.
Addition of the M184V mutation to an M41L/T215Y background
(RT
M41L/M184V/T215Y) had a variable effect on excision when tested
on a DNA template (Fig. 1A and B). In some experiments, the
M184V mutation suppressed primer rescue by as much as 25%,
and in other experiments, the reduction was less than 10% (the
error bars reflect this variability). On an RNA template, the
addi-tion of M184V to an M41L/T215Y background consistently
re-duced primer rescue by 50 to 60% (Fig. 1C and D).
To determine whether the RNase H activity of RT was
re-sponsible for the difference seen with RNA or DNA templates,
the E478Q mutation was introduced into these enzymes
(RT
M41L/T215Y/E478Qand RT
M41L/M184V/T215Y/E478Q). The E478Q
mutation knocks out RNase H catalytic activity in HIV-1 RT.
When E478Q was present, the M184V mutation failed to
sig-nificantly reduce excision rescue on either a DNA or an RNA
template (Fig. 1E, F, and G). For a quantitative summary of
multiple experiments, see Fig. 6A. These results suggest that a
major portion of the effect of M184V on excision is dependent
on the RNase H activity of RT.
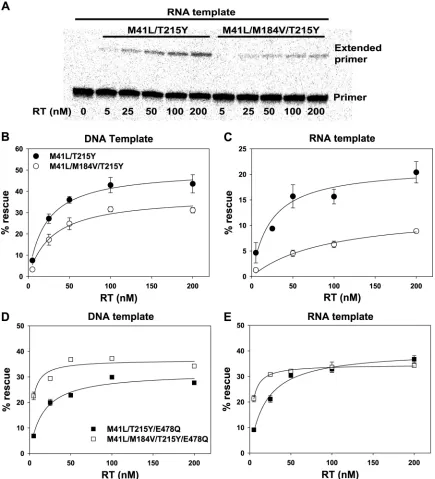
Dependence of AZTMP excision on RT concentration.
Rates
of excision with different concentrations of the mutant RTs were
compared (Fig. 2). With all four mutant RTs, the excision reaction
approached saturation at 200 nM RT. On an RNA template,
exci-sion rescue was reduced 60 to 70% when M184V was present, with
little dependence on the RT concentration (Fig. 2A and C). On a
DNA template, excision rescue was reduced 25 to 30% when
M184V was present over the same range of RT concentrations
(Fig. 2B). In contrast, when E478Q was present, M184V enhanced
the rate of excision on both RNA and DNA templates at low RT
concentrations. At higher RT concentrations, M184V stimulated
excision on a DNA template but not on an RNA template (Fig. 2D
and E). Since our assays measure excision as a single-turnover
reaction, which is approximately linear for 15 min, the initial rates
of excision were estimated from the percentage of primer rescue
observed after 15 min of incubation. The excision rate data as a
function of the RT concentration were fitted to a hyperbolic
equa-tion to determine the apparent
K
Dand maximum rate of excision
(
k
excis) at saturating RT and physiological ATP concentrations
(Table 1). Addition of M184V to RT
M41L/T215Yincreased the
ap-parent
K
Dfrom 23 to 93 nM on an RNA template. The effect was
much smaller on a DNA template (21 nM without M184V
com-pared with 29 nM when the M184V mutation was present). A
reduction of
k
exciswas observed on both DNA and RNA templates
when M184V was present. When M184V was added to RT lacking
RNase H activity (RT
M41L/T215Y/E478Q), the
K
D
was decreased to a
similar extent on either an RNA or a DNA template and there was
no significant effect of M184V on
k
excison either template.
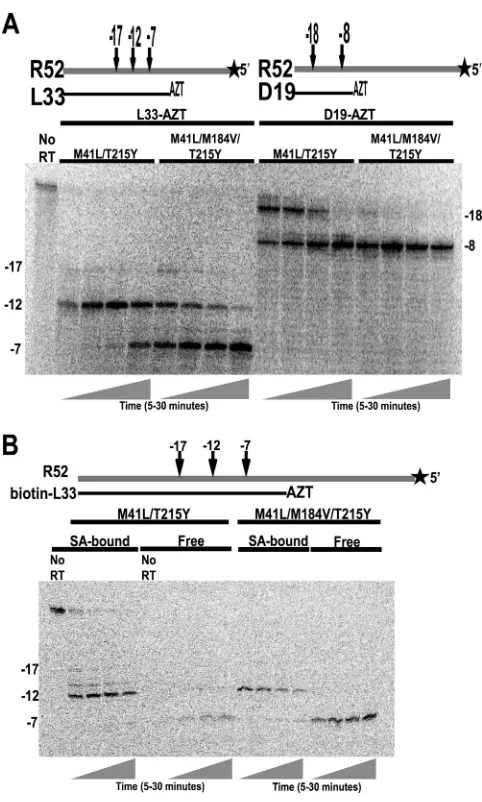
RNase H cleavage and P/T dissociation after cleavage.
RNA
template (R52) was labeled with
32P and annealed to primer
L33-AZT or D19-L33-AZT (shown at the top of Fig. 3A), and RNase H
cleavage products were monitored by gel electrophoresis (Fig.
3A). We observed major cleavage sites at
⫺
12 and
⫺
7 for P/T
L33-AZT/R52 and at
⫺
18 and
⫺
8 for P/T D19-AZT/R52.
Nucle-otide numbering is given relative to the primer terminus, which is
designated
⫺
1. In both sequence contexts, RT that contained the
M184V mutation showed enhanced secondary-site cleavage. The
product of cleavage at
⫺
7 predominated after 5 min of
incuba-tion. For quantification of the
⫺
7 cleavage products formed after
15 min of incubation with the L33-AZT primer and the R52
tem-plate, see Fig. 6B.
To determine whether the RNase H cleavages observed in these
experiments resulted in P/T dissociation, a biotin-labeled,
AZT-terminated primer (biotin-L33) was annealed to 5
=
-
32P-labeled
R52 template (shown at the top of Fig. 3B), and SA-conjugated
magnetic beads were used to separate RNase H cleavage products
that remained associated with the primer from those that were
released during incubation (Fig. 3B). The results show that
⫺
12
cleavage products remained associated with the primer but
⫺
7
cleavage products were quantitatively released. DNA-RNA
du-plexes with less than 10 bp have a reduced ability to support either
polymerization or ATP-dependent excision, presumably due to
the instability of the duplex structure (16, 46). Addition of M184V
to the RT
M41L/T215Ybackground enhanced the release of
⫺
7
cleav-age products, indicating that the remaining DNA-RNA P/T
du-plex was unstable. Similar effects of the M184V mutation were
observed for ddAMP-terminated primer annealed to R52
tem-plate and for ddAMP-terminated P/T with an unrelated sequence
(data not shown). These results suggest that cleavages within 7 bp
of the primer terminus are enhanced when the M184V mutation is
present, giving rise to increased P/T dissociation.
RNase H cleavage patterns in the presence of a trap.
To
eval-uate the role of the initial RT binding sites in determining the
RNase H cleavage events, mutant RTs with or without the M184V
mutation were preincubated with a 5
=
-labeled RNA template
an-nealed to an unlabeled, AZTMP-terminated DNA primer in the
absence of MgCl
2. After 5 min of preincubation, 15 mM MgCl
2and an excess of poly(rA)-oligo(dT) were added to the mixture
and the incubation was continued. Samples were taken after
var-ious times of additional incubation, and RNase H cleavage
prod-ucts were fractionated by electrophoresis (Fig. 4A). For both
RT
M41L/T215Yand RT
M41L/M184V/T215Y, the predominant cleavage
products corresponded to RNase H cleavage at positions
⫺
17 and
⫺
12, but cleavage at
⫺
7 was not observed, even after 30 min of
incubation. Cleavage at
⫺
17 occurred more rapidly than that at
⫺
12. After 30 min, 70 to 90% of the uncleaved template was
de-pleted, indicating that most of the P/T was saturated with RT
during the preincubation and that binding of RT usually led to
cleavage prior to dissociation.
These results indicate that RT binds predominantly at two
al-ternative positions on this P/T. Binding in the upstream position
placed the RNase H active-site residues at the primary RNA
cleav-age site (leading to
⫺
17 cleavage). Binding at the downstream
position placed the RNase H active-site residues at a secondary
cleavage site (leading to
⫺
12 cleavage). RT
M41L/T215Yfavored
⫺
17
on November 7, 2019 by guest
http://jvi.asm.org/
FIG 1Effects of RT mutations on rescue of AZTMP-terminated primer annealed to RNA or DNA templates. Excess DNA (D50) or RNA (R52) template was annealed to 5=-32P-labeled L33 and terminated with AZTMP. P/T at 5 nM was incubated with different RTs as indicated in panels A, C, and E for 5, 10, 15, or 30
min in the presence of 3.2 mM ATP and 10M dNTPs. Controls without RT (No RT) or with RT and dNTPs but no ATP (No ATP) were incubated for 30 min. Reaction products were fractionated by electrophoresis on 10% denaturing polyacrylamide gels. The positions of the unextended primer (Primer) and primer extension products (Extended primer) are indicated on the right. (B, D, F, and G) The full-length extended primer was quantified using a PhosphorImager and expressed as a ratio to extended product formed in 15 min by RTWT-BH10. Values shown are the mean and standard deviation of two or more experiments. wt,
wild type.
on November 7, 2019 by guest
http://jvi.asm.org/
[image:4.585.74.508.63.658.2]cleavage, whereas RT
M41L/M184V/T215Yslightly favored the
⫺
12
cleavage site. Figure 4B indicates that about 23% of the initial
binding by RT
M41L/T215Yoccurs at the
⫺
12 cleavage site, whereas
greater than 40% of the initial binding by RT
M41L/M184V/T215Yoc-curs at this site. Neither enzyme was initially bound in a position
that allowed cleavage at
⫺
7 to occur, despite the fact that
incuba-tion in the absence of a trap resulted in most of the template being
cleaved at
⫺
7 after 30 min. This agrees with results previously
reported by Brehm et al. (11) and suggests that RT dissociates after
cleavage at the initial binding site and rebinds at the downstream
site on the P/T, leading to cleavage near the primer terminus and
P/T dissociation.
FIG 2Effect of RT concentration on rescue of AZTMP-terminated primer annealed to RNA or DNA template. Experiments were carried out as described in the legend to Fig. 1, except that the RT concentrations were 5, 25, 50, 100, and 200 nM and incubation was for 15 min. (A) Representative autoradiogram of rescue of AZTMP-terminated primer annealed to RNA template by mutant RTs as indicated. Reaction mixtures were fractionated by electrophoresis on 10% denaturing polyacrylamide gels. The positions of the unextended primer (Primer) and primer extension products (Extended primer) are indicated to the right. Excess DNA template (B and D) or RNA template (C and E) was annealed to 5=-32P-labeled L33 and terminated with AZTMP. P/T at 5 nM was incubated with different RTs
as indicated for 15 min in the presence of 3.2 mM ATP and 10M dNTPs. The percentage of the primer that was extended (percent rescue) was determined using a PhosphorImager. Values are the mean of two or more experiments⫾the standard deviation. Where error bars are not visible, they are hidden by the data symbols. The apparentKDfor the RT-P/T interaction and the apparentkexcisfor the maximum rate of excision at a saturating RT concentration and a
physiological ATP concentration are given in Table 1.
on November 7, 2019 by guest
http://jvi.asm.org/
[image:5.585.75.510.67.546.2]Additional experiments were carried out to evaluate the effect
of M184V on the dissociation of RT from the initial binding sites
(Fig. 4C). RT
M41L/T215Yand RT
M41L/M184V/T215Ywere
preincu-bated with labeled, AZT-terminated P/T in the absence of MgCl
2,
and various concentrations of unlabeled, AZT-terminated P/T
(trap) were added to the reaction mixture together with 15 mM
MgCl
2to initiate RNase H activity. Unlabeled P/T competed with
labeled P/T for binding of RT after its dissociation from the initial
binding site, leading to reduced
⫺
7 cleavage and protection of the
⫺
17 product from secondary-site cleavage. When the M184V
mutation was present, higher concentrations of the trap were
re-quired to inhibit
⫺
7 cleavage and stabilize the
⫺
17 cleavage
prod-uct, suggesting that dissociation of the initially bound RT from the
labeled P/T was enhanced by the M184V mutation.
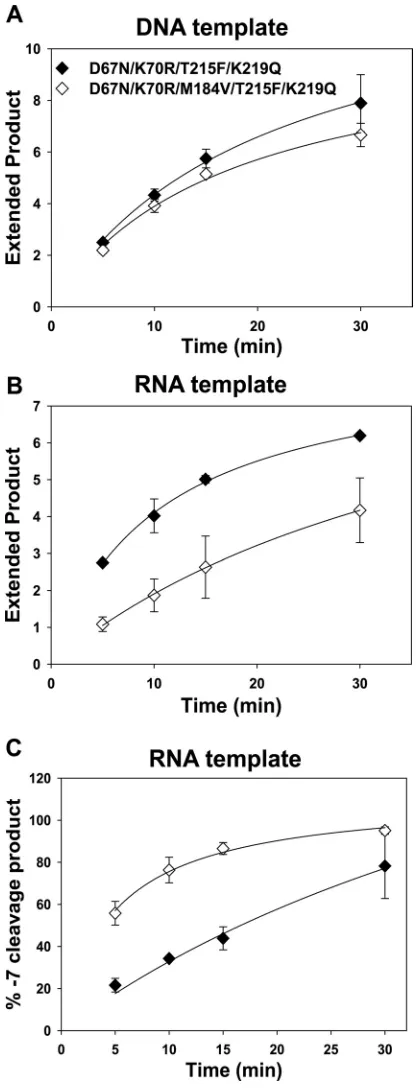
Effect of the M184V mutation on ATP-mediated excision
rescue and RNase H cleavage in a D67N/K70R/T215F/K219Q
background.
RT
D67N/K70R/T215F/K219Qsupports a high level of
ex-cision. The addition of M184V to these mutations reduced primer
rescue by 30 to 60% when tested on an RNA template (Fig. 5B) but
did not reach the level of significance when tested on a DNA
tem-plate (Fig. 5A). Quantitative comparisons after 15 min of
incuba-tion are shown in Fig. 6A (expressed as the amount of extended
primer relative to RT
WT). A similar effect of M184V was observed
in either the RT
M41L/T215Yor the RT
D67N/K70R/T215F/K219Qback-ground.
Figure 5C shows the formation of
⫺
7 cleavage products on
L33-AZTMP/R52 P/T as a function of time of incubation, and a
quanti-tative comparison after 15 min of incubation is summarized in Fig.
6B. The addition of M184V to the RT
D67N/K70R/T215F/K219Qback-ground resulted in an about 2-fold increase in the formation of the
⫺
7 cleavage product, which is less than the 5-fold increase seen after
the addition of M184V to the RT
M41L/T215Ybackground. Both results
are consistent with a major role for increased cleavage at
⫺
7 in the
RNA template in M184V suppression of AZT resistance.
Effect of M184V on excision mediated by PP
i.
The results
pre-sented above suggest an indirect mechanism for M184V
suppres-sion of TAMs leading to the prediction that the reduced primer
rescue may be independent of excision acceptor substrate. Figure
7 shows that the addition of M184V to an RT
D67N/K70R/T215F/K219Qbackground reduced excision rescue mediated by 15
M PP
iby 36
to 43% on an RNA template (Fig. 7B) and had little effect when a
DNA template was used (Fig. 7A). The rate of excision was
greater when 50
M PP
iwas provided as the acceptor substrate,
but the effect of M184V was similar (51 to 54% reduced rescue
after 5 to 10 min) (Fig. 7C). The effect of M184V on primer
rescue was quantitatively similar with ATP as the acceptor
sub-strate in this TAM background (46% reduction, Fig. 6A). PP
i-TABLE 1Kinetic parameters of mutant RT and template/primera
RT mutations
DNA template RNA template
KD(nM) kexcis(min⫺1) KD(nM) kexcis(min⫺1)
M41L/T215Y 21.4⫾5.7 0.17⫾0.02 23.0⫾12.9 0.07⫾0.01
M41L/M184V/T215Y 29.0⫾9.1 0.13⫾0.02 93.3⫾35.2 0.04⫾0.01
M41L/T215Y/E478Q 16.0⫾4.1 0.12⫾0.01 19.2⫾3.3 0.13⫾0.01
M41L/M184V/T215Y/E478Q 3.3⫾1.0 0.12⫾0.01 3.2⫾0.4 0.12⫾0.00
aApparent binding constants (K
D) were determined by fitting the observed percent rescue plots (Fig. 2) to the following equation: product⫽kexcis⫻[RT]/(KD⫹[RT]). The
values shown are means⫾standard deviations of two or three experiments.
FIG 3Effect of the M184V mutation in the M41L/T215Y background on the formation and release of RNase H cleavage products. (A) Excess unla-beled L33 or D19 was annealed to 5=-32P-labeled R52, terminated with
AZTMP, and incubated with the indicated mutant RTs for 5, 10, 15 or 30 min. Major RNase H cleavage positions (measured from the primer termi-nal AZT) are shown at the top and on the left (for L33-AZT P/T) and right (for D19-AZT P/T) of the gel. (B) Biotinylated L33 was annealed to 5=-32
P-labeled R52, terminated with AZTMP, and incubated with the indicated mutant RTs in the presence of SA-tagged magnetic beads. At 5, 10, 15, or 30 min, SA-bound and unbound (free) fractions were separated with a mag-net. Samples were fractionated by electrophoresis on 10% denaturing poly-acrylamide gels.
on November 7, 2019 by guest
http://jvi.asm.org/
[image:6.585.42.544.77.149.2] [image:6.585.42.283.215.616.2]mediated excision was also reduced by the addition of M184V
to an RT
M41L/T215Ybackground (data not shown). No
reduc-tion was seen when the E478Q RNase H-negative mutareduc-tion was
also present.
FIG 4Effect of the M184V mutation on initial binding and RNase H cleavages in the presence of a trap. The 5=-32P-labeled R52 template was annealed with the L33
primer and terminated with AZTMP. Mutant RTs (200 nM) were preincubated for 5 min with 5 nM labeled, AZTMP-terminated P/T in the presence of 5 mM EDTA. (A) MgCl2at 15 mM and excess poly(rA)-oligo(dT) were added, and
incubation was continued for 5, 10, 15, or 30 min. Reaction products were frac-tionated by electrophoresis on 10% polyacrylamide gels. Major RNase H cleavage products are indicated at the right. Controls without MgCl2addition (No Mg2⫹),
without poly(rA)-oligo(dT) addition (No trap), or with poly(rA)-oligo(dT) added during preincubation for 5 min in the absence of P/T (Pre trap) are indi-cated at the top. Controls were incubated for 30 min. (B) The⫺12 cleavage prod-uct in panel A was quantified as a percentage of the total radioactivity in each lane and plotted against the time of incubation. The mean⫾standard deviation of two independent experiments is shown. (C) 5=-32P-labeled R52 template annealed to
unlabeled L33 primer, terminated with AZTMP, and preincubated as described above. Unlabeled L33-AZTMP/R52 P/T trap (1, 5, 50, 100, 200, and 600 nM) was added together with15 mM MgCl2. After 15 min of further incubation, the
reac-tion products were fracreac-tionated by electrophoresis as described above. Controls without L33-AZTMP/R52 ((⫺) Trap) are indicated.
FIG 5Effect of the M184V mutation in a D67N/K70R/T215F/K219Q back-ground on rescue of AZTMP-terminated primer and formation of⫺7 RNase H cleavage product. (A) DNA (D50) or (B) RNA (R52) template was annealed to 5=-32P-labeled L33 primer, terminated with AZTMP, and incubated with
mutant RTs as indicated. The extended product is expressed as a ratio of the extended products formed in 15 min by RTWT-BH10as described in the legend
to Fig. 1. (C) Unlabeled L33 primer was annealed with 5=-32P-labeled R52,
terminated with AZTMP, and incubated with mutant RTs. Products of tem-plate cleavage at position⫺7 were detected as described in the legend to Fig. 3 and expressed as a percentage of the total radioactivity in the lane.
on November 7, 2019 by guest
http://jvi.asm.org/
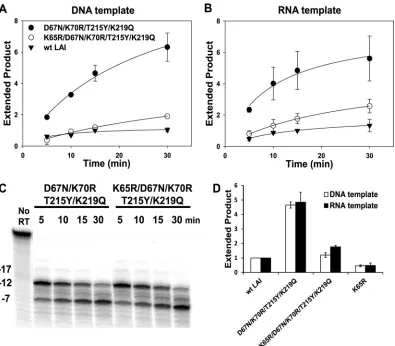
[image:7.585.44.285.61.498.2] [image:7.585.317.525.66.610.2]Effect of K65R on excision rescue and formation of RNase
H cleavage products.
K65R is also a suppressor of AZT
resis-tance by TAMs (6, 36, 43, 59), and AZTMP excision activity is
reduced in RTs containing this mutation (21, 36, 59). In
con-trast to the results obtained with M184V, the addition of K65R
to the RT
D67N/K70R/T215Y/K219Qbackground reduced AZTMP
excision activity to similar extents on both DNA (Fig. 8A) and
RNA templates (Fig. 8B). Quantitative comparison of excision
after incubation for 15 min is summarized in Fig. 8D. The
RNase H cleavage pattern was not significantly different for
RT
D67N/K70R/T215Y/K219Qwith or without K65R (Fig. 8C). These
results suggest that the mechanisms of suppression of excision
rescue are different for K65R and M184V.
FIG 6Rescue of AZTMP-terminated primers and RNase H cleavage at posi-tion⫺7 by WT or mutant RT. (A) Primer rescue determined after 15 min of incubation with 3.2 mM ATP for WT and mutant RTs on DNA or RNA templates as described in the legend to Fig. 1. (B) Formation of⫺7 cleavage product after 15 min of incubation with labeled RNA template for WT and mutant RTs as described in the legend to Fig. 3. All values are expressed relative to that obtained with RTWT-BH10. The mean for two or three experiments⫾
the standard deviation is shown.
FIG 7Effect of M184V on rescue of AZTMP-terminated primer using PPias
the excision acceptor substrate. (A) A DNA (D50) or (B) RNA (R52) template was annealed to 5=-32P-labeled L33, terminated with AZTMP, and incubated
with WT or mutant RTs as indicated for 5, 10, 15, and 30 min in the presence of 15M PPiand 10M dNTPs. Radioactivity was quantified using a
Phos-phorImager and expressed as a percentage of that of the primer that was ex-tended (percent rescue). (C) Same as panel B, except that incubation was for 1, 3, 5, or 10 min in the presence of 50M PPiand 10M dNTPs.
on November 7, 2019 by guest
http://jvi.asm.org/
[image:8.585.60.271.61.475.2] [image:8.585.307.530.63.635.2]DISCUSSION
Enhanced AZTMP excision by RTs containing two to four TAMs
was suppressed by the M184V mutation as previously reported.
We show that M184V suppression was greater when DNA
synthe-sis occurred on an RNA template than when it occurred on a DNA
template. The excision activity was reduced to a similar extent
when measured with either ATP or PP
ias the acceptor substrate.
While total RNase H activity, as measured by disappearance of the
full-length template, was not significantly altered by the M184V
mutation (in agreement with reference 62, we show that in the
presence of TAMs, M184V selectively enhanced cleavage at a
downstream RNase H cleavage site near the primer terminus
(po-sition
⫺
7 on the template). This cleavage leads to P/T dissociation.
Trap experiments that detect only cleavage at the initial binding
sites indicated that RT first bound and cleaved at
⫺
17 or
⫺
12 and
then dissociated and rebound at the downstream site to produce
⫺
7 cleavage products. Our results suggest that the M184V
muta-tion enhances dissociamuta-tion from the initial binding sites, resulting
in increased downstream binding.
Previous studies have shown that M184V added to a TAM
background reduced excision on RNA templates (32, 48, 58, 62)
and DNA templates (9, 21, 22, 27, 38, 49, 62) by 16 to 58%, though
no decrease was detected in one study (39). We found the effect on
a DNA template to be variable, ranging from no effect to a 34%
decrease. We observed a consistently greater reduction in excision
with an RNA template than with a DNA template with the same
sequence. This difference was dependent on the RNase H activity
of RT. Enhanced cleavage at position
⫺
7 on the template due to
the presence of the M184V mutation leads to P/T dissociation and
would account for reduced excision.
Delviks-Frankenberry et al. (13) have shown that several
con-nection domain mutations, including G335C/D, N348I, A360I/N,
V365I, and A376S, obtained from treatment-experienced
HIV-1-infected patients increased AZT resistance in the context of TAMs.
These connection domain mutations also produced significantly
lower RNase H activity and greater AZTMP excision on an RNA
template than on a DNA template. These authors proposed that a
balance exists between the RNase H and excision activities of
HIV-1 RT that is disrupted by these mutations (13, 14, 41, 42).
Reduced RNase H cleavage indirectly enhances AZTMP excision
by increasing the time during which excision can occur before
RNase H cleavage reduces the length of the P/T duplex to the point
FIG 8Effect of the K65R mutation in a D67N/K70R/T215Y/K219Q background on rescue of AZTMP-terminated primer and formation of RNase H cleavage products. (A) A DNA (D50) or (B) RNA (R52) template was annealed to 5=-32P-labeled L33, terminated with AZTMP, and incubated with mutant RTs asindicated. The extended product is expressed as a ratio of the extended products formed in 15 min by RTWT-LAI. (C) Unlabeled L33 was annealed with
5=-32P-labeled R52, terminated with AZTMP, and incubated with indicated mutant RTs, and reaction products were analyzed as described in the legend to Fig.
3. Major cleavage products are indicated at the left. (D) Ratios of the extended products formed in 15 min of incubation by mutant RTs to the products formed by RTWT-LAI. Bars indicate the average of two experiments⫾the standard deviation.
on November 7, 2019 by guest
http://jvi.asm.org/
[image:9.585.96.491.65.411.2]where it dissociates. The stimulation of excision activity was
ob-served for either ATP or PP
ias the acceptor substrate (13, 16, 58),
which is consistent with the indirect mechanism proposed.
Con-siderable experimental support for this mechanism has been
re-ported (11, 13, 16, 17, 42, 46–48, 61).
The model also predicts that mutations that enhance RNA
cleavage will decrease excision activity. This prediction is
sup-ported by our results showing that enhanced
⫺
7 cleavage by RT
due to M184V is correlated with decreased excision activity. One
additional prediction of the model is that connection domain
mu-tations such as N348I, which reduce secondary-site RNase H
cleavage, may compensate for the reduced excision caused by
M184V. Compensation was not observed in two studies (41, 61),
but partial compensation was observed in two other studies (48,
58). We show that M184V strongly enhances
⫺
7 cleavage when
present in a background of TAMs; however, the effect is much less
in the absence of TAMs, as has also been reported by other
labo-ratories (24, 26, 58, 62).
The binding affinity of RT for P/T is driven primarily by
resi-dues near the primer terminus causing RT to bind preferentially in
the polymerase mode (16). RTs containing the M184V or M184I
mutation are known to have reduced processivity for DNA
syn-thesis (3–5, 8, 23, 54), and the RT-dNTP-P/T ternary complexes
formed by these mutant RTs have decreased stability (18, 24;
Acosta-Hoyos, unpublished data), suggesting that RTs containing
M184V or M184I are bound more weakly and dissociate more
readily from the P/T. Increased dissociation of the mutant RT
from its initial binding sites would increase the likelihood of
re-binding at secondary cleavage sites, thereby increasing the rate of
cleavage at these sites. We show that initial binding of
TAM-con-taining RTs with or without M184V occurs at the primer
termi-nus, leading to
⫺
17 cleavage, and at a secondary site five
nucleo-tides downstream, leading to
⫺
12 cleavage. Initial binding is
shifted to favor the
⫺
12 cleavage position by addition of M184V,
which is consistent with reduced affinity at the primer terminus
due to this mutation. Our results (Fig. 4A) and those of Brehm et
al. (11) show that cleavage within 10 nt of the primer terminus
occurs only after RT has first bound and cleaved at an initial
up-stream binding site. The mechanism of this sequential pattern of
cleavage is not clear, but it may help RT avoid cleavage events that
would lead to P/T dissociation during ongoing DNA synthesis
(45). A model of reduced excision due to the M184V mutation is
shown in Fig. 9.
Other mutations in HIV-1 RT are known to reduce excision
activity and suppress the AZT-resistant phenotype of TAMs
(re-viewed in reference 1). One of these is K65R, which affects
inter-actions with the phosphate chain of the incoming dNTP. In this
report, we show that addition of K65R to a TAM background
reduced excision activity to similar extents on DNA and RNA
templates. We found no evidence of an RNase H-dependent
com-ponent for K65R suppression and conclude that the mechanism of
suppression by this mutation is distinct from that of M184V.
Other known TAM suppressors include the L74V and Y181C
mu-tations. Radzio et al. (48) have shown an increase in
⫺
10 site
RNase H cleavage when Y181C is added to an N348I background.
Parallel experiments did not show a similar effect of L74V on
⫺
10
cleavage.
The ability of the M184V mutation to increase the sensitivity of
HIV-1 to nucleoside RT inhibitors has played an important role in
antiretroviral therapy for at least 15 years due to the extensive
therapeutic use of 3TC or FTC together with thymidine analogue
nucleoside RT inhibitors. Our studies contribute to the
under-standing of the biochemical mechanisms that underlie the ability
of this mutation to suppress resistance to AZT and other
nucleo-sides. These insights may lead to the improvement of familiar
antiviral therapies and the development of new therapeutic
strat-egies.
ACKNOWLEDGMENTS
This work was supported by NIH grant AI-39973 (W.A.S.), the University of Miami Developmental Center for AIDS Research (5P30-AI-073961), and fellowships from the American Heart Association (0615079B to A.J.A.-H.) and amfAR (70567-31-RF to P.R.M.).
REFERENCES
1.Acosta-Hoyos AJ, Scott WA.2010. The role of nucleotide excision by reverse transcriptase in HIV drug resistance. Viruses2:372–394. 2.Arion D, Kaushik N, McCormick S, Borkow G, Parniak MA. 1998.
Phenotypic mechanism of HIV-1 resistance to 3=-azido-3= -deoxythymidine (AZT): increased polymerization processivity and en-hanced sensitivity to pyrophosphate of the mutant viral reverse transcrip-tase. Biochemistry37:15908 –15917.
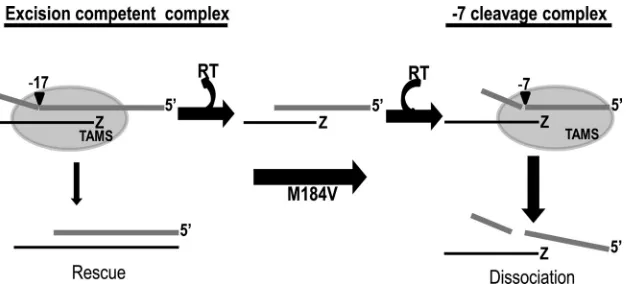
3.Avidan O, Hizi A.1998. The processivity of DNA synthesis exhibited by FIG 9Model of increased RNase H secondary cleavage by M184V and inhibition of excision. Addition of M184V to a TAM background reduces the binding affinity of RT at the AZTMP termination site (Z) on the P/T and stimulates RT release after cleavage of the template at a site (⫺17) directed by the initial binding position. This leads to increased rebinding and RNase H cleavage at secondary positions. RNase H cleavage near the primer terminus (⫺7) results in P/T dissociation, which reduces the opportunity for excision to occur.
on November 7, 2019 by guest
http://jvi.asm.org/
[image:10.585.137.450.65.207.2]drug-resistant variants of human immunodeficiency virus type-1 reverse transcriptase. Nucleic Acids Res.26:1713–1717.
4.Back NKT, Berkhout B.1997. Limiting deoxynucleoside triphosphate concentrations emphasize the processivity defect of lamivudine-resistant variants of human immunodeficiency virus type 1 reverse transcriptase. Antimicrob. Agents Chemother.41:2484 –2491.
5.Back NKT, et al.1996. Reduced replication of 3TC-resistant HIV-1 vari-ants in primary cells due to a processivity defect of the reverse transcrip-tase enzyme. EMBO J.15:4040 – 4049.
6.Bazmi HZ, et al.2000. In vitro selection of mutations in the human immunodeficiency virus type 1 reverse transcriptase that decrease suscep-tibility to (⫺)--D-dioxolane-guanosine and suppress resistance to 3= -azido-3=-deoxythymidine. Antimicrob. Agents Chemother.44:1783– 1788.
7.Boucher CAB, et al.1993. High-level resistance to (⫺) enantiomeric 2=-deoxy-3=-thiacytidine in vitro is due to one amino acid substitution in the catalytic site of human immunodeficiency virus type 1 reverse trans-criptase. Antimicrob. Agents Chemother.37:2231–2234.
8.Boyer PL, Hughes SH.1995. Analysis of mutations at position 184 in reverse transcriptase of human immunodeficiency virus type 1. Antimi-crob. Agents Chemother.39:1624 –1628.
9.Boyer PL, Sarafianos SG, Arnold E, Hughes SH.2001. Selective excision of AZTMP by drug-resistant human immunodeficiency virus reverse transcriptase. J. Virol.75:4832– 4842.
10. Boyer PL, Sarafianos SG, Arnold E, Hughes SH. 2002. The M184V mutation reduces the selective excision of zidovudine 5=-monophosphate (AZTMP) by the reverse transcriptase of human immunodeficiency virus type 1. J. Virol.76:3248 –3256.
11. Brehm JH, Mellors JW, Sluis-Cremer N.2008. Mechanism by which a glutamine to leucine substitution at residue 509 in the ribonuclease H domain of HIV-1 reverse transcriptase confers zidovudine resistance. Bio-chemistry47:14020 –14027.
12. D’Aquila RT, Summers WC. 1989. HIV-1 reverse transcriptase/ ribonuclease H: high level expression inEscherichia colifrom a plasmid constructed using the polymerase chain reaction. J. Acquir. Immune Defic. Syndr.2:579 –587.
13. Delviks-Frankenberry KA, et al.2008. HIV-1 reverse transcriptase con-nection subdomain mutations reduce template RNA degradation and en-hance AZT excision. Proc. Natl. Acad. Sci. U. S. A.105:10943–10948. 14. Delviks-Frankenberry KA, Nikolenko GN, Pathak VK.2010. The
“con-nection” between HIV drug resistance and RNase H. Viruses2:1476 – 1503.
15. Deval J, et al.2004. Mechanistic basis for reduced viral and enzymatic fitness of HIV-1 reverse transcriptase containing both K65R and M184V mutations. J. Biol. Chem.279:509 –516.
16. Ehteshami M, et al.2008. Connection domain mutations N348I and A360V in HIV-1 reverse transcriptase enhance resistance to 3=-azido-3= -deoxythymidine through both RNase H-dependent and -independent mechanisms. J. Biol. Chem.283:22222–22232.
17. Ehteshami M, Götte M.2008. Effects of mutations in the connection and RNase H domains of HIV-1 reverse transcriptase on drug susceptibility. AIDS Rev.10:224 –235.
18. Ehteshami M, et al.2008. Mutations M184V and Y115F in HIV-1 reverse transcriptase discriminate against “nucleotide-competing reverse trans-criptase inhibitors.” J. Biol. Chem.283:29904 –29911.
19. Eron JJ, et al.1995. Treatment with lamivudine, zidovudine, or both in HIV-positive patients with 200 to 500 CD4⫹cells per cubic millimeter. N. Engl. J. Med.333:1662–1669.
20. Feng JY, Anderson KS.1999. Mechanistic studies examining the effi-ciency and fidelity of DNA synthesis by the 3TC-resistant mutant (184V) of HIV-1 reverse transcriptase. Biochemistry38:9440 –9448.
21. Frankel FA, Invernizzi CF, Oliveira M, Wainberg MA.2007. Dimin-ished efficiency of HIV-1 reverse transcriptase containing the K65R and M184V drug resistance mutations. AIDS21:665– 675.
22. Frankel FA, Marchand B, Turner D, Götte M, Wainberg MA.2005. Impaired rescue of chain-terminated DNA synthesis associated with the L74V mutation in human immunodeficiency virus type 1 reverse trans-criptase. Antimicrob. Agents Chemother.49:2657–2664.
23. Gao H-Q, Boyer PL, Arnold E, Hughes SH.1998. Effects of mutations in the polymerase domain on the polymerase, RNase H and strand transfer activities of human immunodeficiency virus type 1 reverse transcriptase. J. Mol. Biol.277:559 –572.
24. Gao H-Q, Boyer PL, Sarafianos SG, Arnold E, Hughes SH.2000. The
role of steric hindrance in 3TC resistance of human immunodeficiency virus type-1 reverse transcriptase. J. Mol. Biol.300:403– 418.
25. Gao Q, et al.1993. The same mutation that encodes low-level human immunodeficiency virus type 1 resistance to 2=,3=-dideoxyinosine and 2=,3=-dideoxycytidine confers high-level resistance to the (⫺) enantiomer of 2=,3=-dideoxy-3=-thiacytidine. Antimicrob. Agents Chemother.37: 1390 –1392.
26. Gao L, et al.2008. Apparent defects in processive DNA synthesis, strand transfer, and primer elongation of Met-184 mutants of HIV-1 reverse transcriptase derive solely from a dNTP utilization defect. J. Biol. Chem. 283:9196 –9205.
27. Götte M, Arion D, Parniak MA, Wainberg MA. 2000. The M184V mutation in the reverse transcriptase of human immunodeficiency virus type 1 impairs rescue of chain-terminated DNA synthesis. J. Virol.74: 3579 –3585.
28. Huang H, Chopra R, Verdine GL, Harrison SC.1998. Structure of a covalently trapped catalytic complex of HIV-1 reverse transcriptase: im-plications for drug resistance. Science282:1669 –1675.
29. Jamburuthugoda VK, et al.2008. Reduced dNTP binding affinity of 3TC-resistant M184I HIV-1 reverse transcriptase variants responsible for viral infection failure in macrophage. J. Biol. Chem.283:9206 –9216. 30. Larder BA, Kemp SD, Harrigan PR. 1995. Potential mechanism for
sustained antiretroviral efficacy of AZT-3TC combination therapy. Sci-ence269:696 – 699.
31. Larder BA, Purifoy DJM, Powell KL, Darby G.1987. Site-specific mu-tagenesis of AIDS virus reverse transcriptase. Nature327:716 –717. 32. Lennerstrand J, Hertogs K, Stammers DK, Larder BA.2001. Correlation
between viral resistance to zidovudine and resistance at the reverse trans-criptase level for a panel of human immunodeficiency virus type 1 mu-tants. J. Virol.75:7202–7205.
33. Meyer PR, Matsuura SE, Mian AM, So AG, Scott WA.1999. A mech-anism of AZT resistance: an increase in nucleotide-dependent primer un-blocking by mutant HIV-1 reverse transcriptase. Mol. Cell4:35– 43. 34. Meyer PR, Matsuura SE, So AG, Scott WA.1998. Unblocking of
chain-terminated primer by HIV-1 reverse transcriptase through a nucleotide-dependent mechanism. Proc. Natl. Acad. Sci. U. S. A.95:13471–13476. 35. Meyer PR, et al.2002. Effects of specific zidovudine resistance mutations
and substrate structure on nucleotide-dependent primer unblocking by human immunodeficiency virus type 1 reverse transcriptase. Antimicrob. Agents Chemother.46:1540 –1545.
36. Meyer PR, et al.2003. Relationship between 3=-azido-3=-deoxythymidine resistance and primer unblocking activity in foscarnet-resistant mutants of human immunodeficiency virus type 1 reverse transcriptase. J. Virol. 77:6127– 6137.
37. Meyer PR, Smith AJ, Matsuura SE, Scott WA.2004. Effects of primer-template sequence on ATP-dependent removal of chain-terminating nu-cleotide analogues by HIV-1 reverse transcriptase. J. Biol. Chem.279: 45389 – 45398.
38. Miranda LR, Götte M, Liang F, Kuritzkes DR.2005. The L74V mutation in human immunodeficiency virus type 1 reverse transcriptase counter-acts enhanced excision of zidovudine monophosphate associated with thymidine analog resistance mutations. Antimicrob. Agents Chemother. 49:2648 –2656.
39. Naeger LK, Margot NA, Miller MD.2001. Increased drug susceptibility of HIV-1 reverse transcriptase mutants containing M184V and zidovu-dine-associated mutations: analysis of enzyme processivity, chain-terminator removal and viral replication. Antivir. Ther.6:115–126. 40. Nijhuis M, et al.1997. Lamivudine-resistant human immunodeficiency
virus type 1 variants (184V) require multiple amino acid changes to be-come co-resistant to zidovudine in vivo. J. Infect. Dis.176:398 – 405. 41. Nikolenko GN, et al.2007. Mutations in the connection domain of HIV-1
reverse transcriptase increase 3=-azido-3=-deoxythymidine resistance. Proc. Natl. Acad. Sci. U. S. A.104:317–322.
42. Nikolenko GN, et al.2005. Mechanism for nucleoside analog-mediated abrogation of HIV-1 replication: balance between RNase H activity and nucleotide excision. Proc. Natl. Acad. Sci. U. S. A.102:2093–2098. 43. Parikh UM, Bacheler L, Koontz D, Mellors JW.2006. The K65R
muta-tion in human immunodeficiency virus type 1 reverse transcriptase exhib-its bidirectional phenotypic antagonism with thymidine analog muta-tions. J. Virol.80:4971– 4977.
44. Petrella M, Wainberg MA.2002. Might the M184V substitution in HIV-1 RT confer clinical benefit? AIDS Rev.4:224 –232.
45. Purohit V, Roques BP, Kim B, Bambara RA.2007. Mechanisms that
on November 7, 2019 by guest
http://jvi.asm.org/
prevent template inactivation by HIV-1 reverse transcriptase RNase H cleavages. J. Biol. Chem.282:12598 –12609.
46. Radzio J, Sluis-Cremer N. 2008. Efavirenz accelerates HIV-1 reverse transcriptase ribonuclease H cleavage, leading to diminished zidovudine excision. Mol. Pharmacol.73:601– 606.
47. Radzio J, Sluis-Cremer N.2011. Subunit-specific mutational analysis of residue N348 in HIV-1 reverse transcriptase. Retrovirology8:69. 48. Radzio J, Yap S-H, Tachedjian G, Sluis-Cremer N. 2010. N348I in
reverse transcriptase provides a genetic pathway for HIV-1 to select thy-midine analogue mutations and mutations antagonistic to thythy-midine an-alogue mutations. AIDS24:659 – 667.
49. Ray AS, Basavapathruni A, Anderson KS.2002. Mechanistic studies to understand the progressive development of resistance in human immu-nodeficiency virus type 1 reverse transcriptase to abacavir. J. Biol. Chem. 277:40479 – 40490.
50. Sarafianos SG, et al.1999. Lamivudine (3TC) resistance in HIV-1 reverse transcriptase involves steric hindrance with-branched amino acids. Proc. Natl. Acad. Sci. U. S. A.96:10027–10032.
51. Sarkar G, Sommer SS.1990. The “megaprimer” method of site-directed mutagenesis. Biotechniques8:404 – 407.
52. Schinazi RF, et al.1993. Characterization of human immunodeficiency viruses resistant to oxathiolane-cytosine nucleosides. Antimicrob. Agents Chemother.37:875– 881.
53. Schuurman R, et al.1995. Rapid changes in human immunodeficiency virus type 1 RNA load and appearance of drug-resistant virus populations in persons treated with lamivudine (3TC). J. Infect. Dis.171:1411–1419. 54. Sharma PL, Crumpacker CS.1999. Decreased processivity of human immunodeficiency virus type 1 reverse transcriptase (RT) containing di-danosine-selected mutation Leu74Val: a comparative analysis of RT
vari-ants Leu74Val and lamivudine-selected Met184Val. J. Virol.73:8448 – 8456.
55. Shi C, Mellors JW.1997. A recombinant retroviral system for rapid in vivo analysis of human immunodeficiency virus type 1 susceptibility to reverse transcriptase inhibitors. Antimicrob. Agents Chemother.41: 2781–2785.
56. Staszewski S, et al.1997. Reductions in HIV-1 disease progression for zidovudine/lamivudine relative to control treatments: a meta-analysis of controlled trials. AIDS11:477– 483.
57. Tisdale M, Kemp SD, Parry NR, Larder BA.1993. Rapidin vitroselection of human immunodeficiency virus type 1 resistant to 3=-thiacytidine in-hibitors due to a mutation in the YMDD region of reverse transcriptase. Proc. Natl. Acad. Sci. U. S. A.90:5653–5656.
58. von Wyl V, et al.2010. Epidemiological and biological evidence for a compensatory effect of connection domain mutation N348I on M184V in HIV-1 reverse transcriptase. J. Infect. Dis.201:1054 –1062.
59. White KL, et al.2006. The K65R reverse transcriptase mutation in HIV-1 reverses the excision phenotype of zidovudine resistance mutations. An-tivir. Ther.11:155–163.
60. Wilson JE, et al.1996. Human immunodeficiency virus type-1 reverse transcriptase. Contribution of Met-184 to binding of nucleoside 5= -triphosphate. J. Biol. Chem.271:13656 –13662.
61. Yap S-H, et al.2007. N348I in the connection domain of HIV-1 reverse transcriptase confers zidovudine and nevirapine resistance. PLoS Med. 4:e335.
62. Zelina S, Sheen C-W, Radzio J, Mellors JW, Sluis-Cremer N. 2008. Mechanisms by which the G333D mutation in human immunodeficiency virus type 1 reverse transcriptase facilitates dual resistance to zidovudine and lamivudine. Antimicrob. Agents Chemother.52:157–163.
on November 7, 2019 by guest
http://jvi.asm.org/