Copyright © 2000, American Society for Microbiology. All Rights Reserved.
Differences in Determinants Required for Complex Formation
and Transactivation in Related VP16 Proteins
MATTHEW GRAPES†ANDPETER O’HARE*
Marie Curie Research Institute, Oxted, Surrey RH8 OTL, United Kingdom
Received 1 June 2000/Accepted 26 July 2000
VP16-H is an essential structural protein of herpes simplex virus type 1 (HSV-1) and is also a potent activator of virus immediate-early (IE) gene expression. Current models of functional determinants within VP16-H indicate that it consists of two domains, an N-terminal domain involved in recruiting VP16-H to a multicomponent DNA binding complex with two host proteins, Oct-1 and host cell factor (HCF), and an acidic C-terminal domain exclusively involved in transactivation. VP16-E, from equine herpesvirus 1 (EHV-1), exhibits strong conservation with the N-terminal domain of VP16-H but, with the exception of a short segment at the extreme C terminus, lacks almost the entire acidic C-terminal domain. Studies of key activation determinants within the C terminus of VP16-H would predict that VP16-E may activate poorly, if at all. However, VP16-E is a potent activator of both EHV-1 and HSV-1 IE gene transcription. We show that VP16-E does not follow the simple two-domain model of VP16-H. Thus, despite the conservation in the N-terminal domains, this region in VP16-E is not sufficient for assembly into the DNA binding complex with Oct-1 and HCF. The short conserved determinant close to the C terminus is completely dispensable in VP16-H but is absolutely required in VP16-E. In activation studies, the potency of intact VP16-E was not recapitulated in chimeric proteins in which it was fused with a GAL4 DNA binding domain. Furthermore, a chimeric protein consisting of the C-terminal region of VP16-E fused to the N-terminal domain of VP16-H, while able to promote complex formation, nevertheless exhibited very weak activation. These results indicate that the mode of recruitment of the activation domain, i.e., through complex formation with Oct-1 and HCF, may be crucial for activation and that key determinants required for activation in VP16-E, and possibly VP16-H, may involve interactions between regions of the C terminus and the N terminus rather than discrete domains with independent functions.
VP16 is encoded by the UL48 gene of herpes simplex virus type 1 (HSV-1) and is an essential structural protein, assem-bled into the tegument of the virion at approximately 1,200 to 1,500 molecules per particle (14). It is also a potent activator of the transcription of viral immediate-early (IE) genes (2, 3, 32). Transcriptional activation is initiated by the recruitment of VP16, together with two cellular proteins, Oct-1 (9, 30, 33, 38) and host cell factor (HCF) (17, 50, 51), into a multicomponent complex formed on regulatory sites (TAATGARAT motifs) present within each of the IE gene promoters (for reviews, see references 29 and 49). Previous analyses from several labora-tories are consistent with a model of VP16 whereby the func-tions of recruitment to the DNA binding complex and tran-scriptional activation are separate activities located in two discrete domains (4, 10, 43). An amino-terminal domain, re-fined to within residues 49 to 390, is involved in the binding of VP16 to HCF and the recruitment of this binary complex to the Oct-1–DNA complex (11). Analysis by limited proteolysis demonstrated that the region at about residue 370 within this domain is present in a surface-exposed loop (13); results from site-directed mutagenesis showed that, while complex forma-tion is sensitive to alteraforma-tions in other regions, key residues involved in interactions with Oct-1 and HCF are located within residues 360 to 390 (1, 11, 21, 48).
VP16 extends for another 100 residues beyond the
C-termi-nal boundary of the domain required for complex formation, and this C-terminal extension is highly enriched in acidic amino acids (6, 31). This region encompasses determinants required for transcriptional activation, since C-terminal trun-cations or insertions within the intact protein abolished acti-vation without having any detectable effect on complex forma-tion (1, 10, 35, 48). However, most studies of the determinants involved in transcriptional activation per se have been per-formed in the context of fusion proteins in which the C-termi-nal region has been fused to a heterologous DNA binding domain, e.g., that from the yeast GAL4 protein, and activation has been studied with target promoters containing GAL4 rec-ognition sites (4, 37). From such studies, the C-terminal region has been generally recognized as a physical and functional domain which can be split broadly into two subdomains, the H1 or N region (residues 410 to 452) and the H2 or C region (residues 453 to 490) (35, 43, 46). Although the net negative charge in the C-terminal region contributes to activation, the pattern of hydrophobic and aromatic residues appears to be more critical, with particularly important residues being phe-nylalanines at position 442 in H1 (N) and at positions 473, 475, and 479 in H2 (C) (5, 35, 46). The number of targets proposed to bind to the VP16 activation domain is confusingly large and includes members of the basal transcription initiation complex (18, 23, 39), mediator proteins and RNA polymerase II ho-loenzyme (15, 19), histone acetylases (44), and many other members of the transcriptional apparatus, and it is presently difficult to reconcile a role for all of these factors in a physio-logically relevant way (for a review, see reference 41). More-over, comparison with the homologues of VP16 from other alphaherpesviruses has emphasized some of the difficulties in understanding the detailed mechanism of activation.
* Corresponding author. Mailing address: Marie Curie Research Institute, The Chart, Oxted, Surrey RH8 OTL, United Kingdom. Phone: 44 1883 715028. Fax: 44 1883 714375. E-mail: P.OHare@mcri .ac.uk.
† Present address: Montreal Neurological Institute, Montreal, Que-bec, Canada H3A 2B4.
10112
on November 9, 2019 by guest
http://jvi.asm.org/
For example, within the VP16 homologues from bovine her-pesvirus 1 (BHV-1), equine herher-pesvirus 1 (EHV-1), and vari-cella-zoster virus (VZV), the N terminus is well conserved, but there is significant divergence in the C terminus among these proteins. Indeed, based on the known requirements within VP16-H and the differences found within the C termini, it was predicted that, e.g., the EHV-1 homologue might not activate IE gene expression (40). However, each of the VP16 species, including that of EHV-1, has been reported to activate IE gene expression (8, 22, 25, 28, 34). The C terminus of the EHV-1 protein (termed VP16-E for ease of reference in this work) exhibits some homology with that of VP16-H but appears to be a truncated version and does not have the same preponderance of negatively charged residues. However, this region of VP16-E has been shown to be required for its transcriptional activity (8).
As part of a program to clarify the mechanisms involved in IE gene activation, we sought to directly compare VP16-H and VP16-E, particularly with respect to the involvement of C-terminal regions in complex formation and transactivation. Our results indicate some significant differences between the two proteins, since a determinant within the C terminus of VP16-E is required for the assembly of the complex with Oct-1 and HCF. Paradoxically, this region, while representing a se-lectively conserved segment within the otherwise diverged C termini, is dispensable in VP16-H. Moreover, we show that the activities of GAL4 fusion proteins do not reflect the activities of the intact parental proteins, indicating that the mode of recruitment of the activation domain may be crucial for acti-vation. Key determinants required for activation in VP16-E, and possibly VP16-H, may involve interactions between re-gions of the C terminus and the N terminus rather than dis-crete domains with independent functions.
MATERIALS AND METHODS
Cells, transfections, and CAT assays.COS-1 and Vero cells were grown in Dulbecco’s modified minimal essential medium containing 10% newborn calf serum. Transfections were performed by the calcium phosphate method with various amounts of expression plasmids made up to 2g with pUC19 DNA as described previously (11). Cells were harvested approximately 40 h after trans-fection and assayed for chloramphenicol acetyltransferase (CAT) activity exactly as described previously (11).
Plasmids.VP16-H cloned in pcDNA1 (Invitrogen) was transferred from the parental construct (26) into pcDNA1/amp as aHindIII-EcoRI fragment. VP16-E was initially produced by PCR amplification of gene 12 from EHV-1 strain Ab1 in vector GE126 (8). Alignment of the coding sequences of various VP16 species indicated that in this original construct, translation of VP16-E may have initiated from an upstream in-frame methionine, yielding a protein with an extra 30 residues. VP16-E was therefore recloned by PCR amplification with primers containingEcoRI andXbaI sites at the 5⬘and 3⬘ends, respectively. The 5⬘primer was positioned to amplify from the next available methionine, yielding a protein about 30 residues shorter than the original one. The fragment was inserted between theEcoRI andXbaI sites of pcDNA1/amp to yield the expression vector for wild-type VP16-E, pcDNA1/amp.VP16-E. (The lack of 30 residues from the original construct had no deleterious effect on activity but appeared to increase the efficiency of expression.) Mutations in VP16-E were introduced by subclon-ing from plasmids GE130, GE134, GE135, GE136, and GE137, which contain mutant VP16-E species (8), by swapping the C-terminalBglII-HpaI fragment of the parental vector with the corresponding fragment from each of the mutants in the GE series of plasmids to yield E.(1-425), E.(1-445), VP16-E.(⌬444), VP16-E.(⌬442-444), and VP16-E.(⌬442-445).
A further truncation was produced to yield VP16-E.(1-393) by digesting pcDNA1/amp.VP16-E withPshAI andXbaI and then using an annealed oligo-nucleotide to replace the coding frame downstream of residue 393 with a ter-mination codon and an XbaI site. All of the VP16-H and VP16-E variants examined were therefore expressed in identical backgrounds. GAL–VP16-E was produced by digesting pcDNA1/amp.VP16-E withEcoRI andXbaI and cloning the appropriate fragment into the GAL fusion vector pM3 (36) to yield an in-frame fusion between the GAL4 DNA binding domain and the complete VP16-E protein. This vector was subcloned back into pcDNA1/amp.VP16-E usingBglII to create pcDNA1/amp.GAL-VP16-E. The GAL4–VP16-H acidic domain fusion, pPO64 (46), contains just the C-terminal 80 residues of VP16-H,
while pGal4-VP16-H contains full-length VP16-H and was produced by inserting theBamHI-PstI fragment of MK6 (13) into the GAL vector pM2 (36).
The chimera of VP16-H and VP16-E was produced by PCR amplification of residues 393 to 448 from the C-terminal end of VP16-E. The PCR fragment introduced aSalI site at the 5⬘end and aBglII site at the 3⬘end, and the fragment was cloned intoSalI-BglII-digested pGE138 (8). This procedure created a fusion protein linking the N-terminal region up to position 411 of VP16-H in frame to the C-terminal region beginning at position 393 of VP16-E. This chimera, des-ignated pMG3, was subcloned into the pcDNA1/amp backbone by digestion of pMG3 with AgeI and HpaI and insertion of the appropriate fragment into pcDNA1/amp.VP16-H to form VP16-H/E.
A version of VP16-E (pSV5-VP16-E) containing an epitope tag (from the paramyxovirus SV5 matrix protein) at its N terminus was constructed by first inserting the SV5 epitope tag into pcDNA1/amp and then cloning into this vector theBamHI-HpaI fragment of pcDNA1/amp.VP16-E. This construction intro-duced an extra 36 bp between the SV5 tag and the start of the VP16-E reading frame which, together with the SV5 tag, added 24 residues to the N terminus of VP16-E. SV5-VP16-E.(1-393) was produced by digesting pcDNA1/amp.VP16-E(1-393) withSalI andHpaI and ligating the resulting fragment into pSV5-VP16-E.
Target reporter vectors used in CAT assays were as follows. For analysis of VP16-E and VP16-H, the constructs were pCAT_TAAT (8), which contains the CAT gene driven by the natural EHV-1 IE gene promoter region (⫺360 to⫹78) encompassing four octamer binding sites, and pAB5, which contains the CAT gene driven by the HSV-1 IE110 gene promoter-regulatory region (⫺165 to ⫹150). For analysis of the GAL4 fusion proteins, the target plasmid was pUAS10CAT (4), which contains two strong and two weaker GAL4 binding sites.
Gel retardation assays.Binding reactions were carried out as previously de-scribed (16) using 0.02l (0.2 ng) of the purified POU domain, in vitro-trans-lated HCF, and 1 ng of the end-labeled probe. VP16 species were supplied by in vitro translation (TnT; Promega) or from soluble extracts of transfected cells prepared as described previously. The probes encompassed the E1 octamer site of the EHV-1 IE gene promoter (AGCTGAGGAGACGCATGCAGATGAG ATGTGCATCGAGG) (8) or the octamer motif at position⫺160 of the HSV-1 IE110 gene promoter (TAAT24) as previously described (CCATGGAGATCT CGTGCATGCTAATGATATTCTTCCATGG) (underlined sequences indicate the octamer site within the probe) (47). Poly(dI-dC) was routinely added to the mixture at 1g per reaction to reduce nonspecific binding. The mixture was incubated with the labeled probe for 15 min, and the complexes were resolved in nondenaturing 6% polyacrylamide (acrylamide-bisacrylamide, 37:5) gels in 0.5⫻ Tris-borate-EDTA (TBE). Electrophoresis was performed at a constant voltage of 200 V for 90 min. The gels were dried, and complexes were detected by autoradiography.
Immunofluorescence.COS-1 cells to be processed for immunofluorescence were seeded at 1.25⫻105cells per well in six-well cluster plates (Costar) on
25-mm glass coverslips. Approximately 40 h after transfection, cells were washed with phosphate-buffered saline (PBS), fixed for 15 min with ice-cold methanol, and blocked in PBS containing 10% calf serum (blocking solution) for 20 min. Monoclonal antibody against the SV5 tag was added in the same solution (1: 2,000) for 20 min. VP16-H was detected by using the monoclonal antibody LP1 as previously described (20). For secondary antibodies, fluorescein isothiocya-nate-conjugated anti-mouse immunoglobulin G (Vector Laboratories) was used at a dilution of 1:100 and tetramethyl rhodamine isothiocyanate-conjugated anti-rabbit immunoglobulin G (Sigma) was used at a dilution of 1:200. These antibodies were added in blocking solution and incubated for 20 min. After three 5-min washes in PBS, the coverslips were mounted in Vector Shield (Vector Laboratories) and visualized using either a Bio-Rad MRC600 confocal micro-scope or a Zeiss LSM 410 confocal micromicro-scope. Images were processed with Adobe Photoshop software.
RESULTS
Requirements for a conserved determinant in the extreme C
terminus. A schematic summary of the relationship between
the VP16-H and VP16-E proteins is shown in Fig. 1a. A more detailed alignment of the C-terminal region and an indication of some of the variants used in this work are illustrated in Fig. 1b. The N-terminal regions of the proteins, corresponding ap-proximately up to residue 390 of VP16-H, are well conserved. However, after these regions, there is a notable divergence in the sequences, indicated by a transition from yellow to green shading, where VP16-B, VP16-E, and VZV VP16 exhibit con-tinued conservation but VP16-H does not (Fig. 1b). After this section, the C-terminal region of VP16-E is much shorter than that of VP16-H, is not noticeably acidic, and completely lacks the section around the critical phenylalanine residue 442. The exception to the general lack of homology between the C-terminal regions is found at the extreme C terminus, where a
VOL. 74, 2000 COMPARISON OF VP16 FROM EHV AND HSV 10113
on November 9, 2019 by guest
http://jvi.asm.org/
short section of approximately 15 residues is well conserved. This section is within the region of VP16-H which we previ-ously termed H2 (46) and, for the sake of clarity, has been labeled in this work (Fig. 1b) as the H2 core conserved region (H2.CCR).
Transactivation by VP16-E and its variants was examined in
[image:3.612.61.542.72.564.2]transfection assays using a CAT reporter construct, pCAT_TAAT, which contains the native EHV-1 IE gene pro-moter (⫺360 to⫹78) and encompasses four octamer elements (8). Direct comparison was made with VP16-H and additional VP16-E variants. The target vector (200 ng) was cotransfected with VP16-H, VP16-H.(1-453), VP16-H.(1-411), VP16-E, and
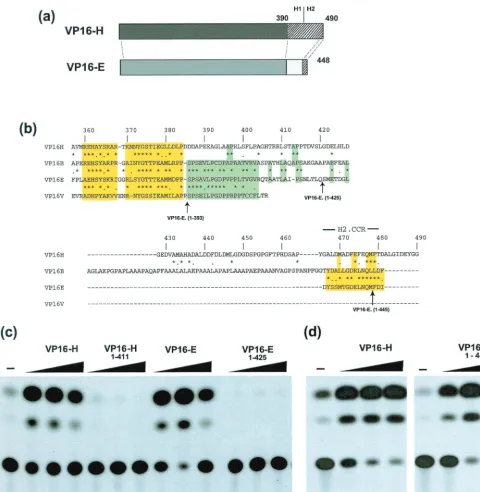
FIG. 1. Comparison of activation by VP16-H and VP16-E. (a) Schematic representation of the similarities between VP16-H and VP16-E. The N-terminal shaded regions are well conserved, while the C-terminal regions are not, with the exception of a short section retained in both species, as discussed in the text. (b) Primary sequence alignment of the VP16 homologues extending from residue 357 in VP16-H. Yellow shading indicates the region well conserved in all species, while green shading indicates the region which appears to be selectively conserved in VP16-E, VP16-B, and VZV VP16. The H2.CCR at the extreme C termini of all species is highlighted, while the C-terminal boundaries of variants of VP16-E analyzed in this work are indicated. (c) Comparison of activation by VP16 variants. The CAT reporter construct pCAT_TAAT (8), which contains the EHV-1 IE gene promoter region (⫺360 to⫹78), was transfected (200 ng) into Vero cells either alone (⫺) or together with the indicated VP16 constructs (at doses of 1, 10, and 100 ng). Cells were harvested 40 h later, and aliquots were assayed for CAT activity. (d) As for panel c but with the VP16-H constructs indicated and the target vector pAB5, which contains the HSV-1 IE110 gene promoter from⫺165 to⫹150 (30).
on November 9, 2019 by guest
http://jvi.asm.org/
a deletion mutant which lacks the H2.CCR, VP16-E.(1-425), at doses of 1, 10, or 100 ng. The results demonstrate that VP16-E was as potent an activator as VP16-H (Fig. 1c, cf. lanes 2 to 4 and 8 to 10). Deletion of the C-terminal 23 residues encom-passing the H2.CCR completely abrogated transactivation by VP16-E (Fig. 1c, lanes 11 to 13). However, in contrast, deletion of all H2 C-terminal 37 residues from VP16-H, encompassing the conserved section, had comparatively little effect on acti-vation (Fig. 1d). Similar results were obtained irrespective of the target IE gene promoter and are consistent with earlier observations (11, 42). VP16-H.(1-411), which lacks the entire acidic domain, was inactive, as expected (Fig. 1b, lanes 5 to 7). While the results for VP16-E.(1-425) are consistent with pre-vious findings (8), the direct comparison shown here illustrates two points. First, wild-type VP16-E which, compared to VP16-H, contains a truncated, nonacidic C-terminal region, is as potent an activator as VP16-H; second, a short extreme C-terminal determinant which has been specifically conserved despite the overall lack of conservation within the C terminus is paradoxically critical for VP16-E activity but dispensable in VP16-H.
Differences in requirements for recruitment to the DNA
binding complex.Although the C-terminal region of VP16-H
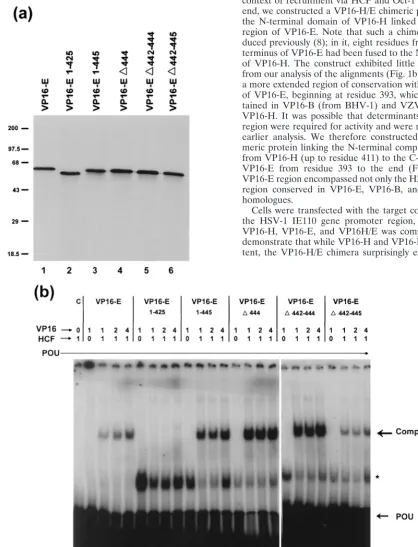
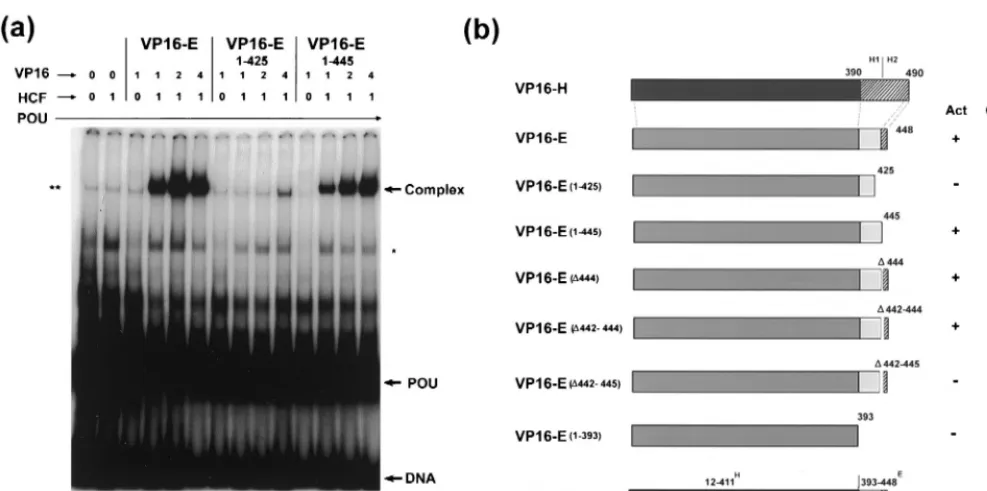
is not required (10) for the assembly of the octamer binding complex (TRF-C) with Oct-1 and HCF, it was nonetheless possible that the failure of VP16-E.(1-425) to activate expres-sion was due to a failure to be recruited into the corresponding complex. To examine this possibility, wild-type E, VP16-E.(1-425), and several additional C-terminal deletion variants were expressed in vitro and assayed for the ability to promote complex formation in gel retardation assays. Each of the pro-teins was expressed in vitro at approximately the same level (Fig. 2a), and equal amounts of the products were incubated with in vitro-translated HCF, the purified POU domain (0.25 ng), and the E1 octamer probe from the EHV-1 IE gene promoter (Fig. 2b). Independent binding of the POU domain to the E1 probe was observed (Fig. 2b, POU), together with a complex (asterisk) which originated from the TnT control ly-sate and was observed in all experiments to various degrees. The formation of TRF-C (Fig. 2b, Complex) was observed dependent upon the addition of both VP16-E and HCF (lanes 3 to 5). Either component alone failed to promote complex formation (Fig. 2b, lanes 1 and 2). Surprisingly, deletion of the C-terminal 23 residues [VP16-E.(1-425)] almost completely eliminated complex formation (Fig. 2b, lanes 7 to 9). A variant containing a further deletion to residue 393 similarly failed to promote complex formation (data not shown). Smaller dele-tions within and around the extreme C terminus had little effect on complex formation (Fig. 2b, lanes 10 to 25). Note that the relatively poorer complex formation observed in this ex-periment for the wild-type and VP16-E.(⌬442-445) species was not a reproducible effect (see, e.g., Fig. 3; also, data not shown).
To provide additional evidence for a defect in complex for-mation of the VP16-E.(1-425) variant, we expressed the VP16-E species in vivo and examined complex formation using soluble extracts. The results were identical to those obtained using the proteins expressed in vitro. Thus, VP16-E.(1-425) was almost completely defective, while VP16-E.(1-445) was similar to the parental species (Fig. 3a). A summary of the results for complex formation, together with previous results on the transcriptional activity of these mutants (8), is shown in Fig. 3b. Two points are of note. First, unlike that of VP16-H and notwithstanding the homology in the region, the N-termi-nal region of VP16-E is not an independent domain, sufficient for complex formation with Oct-1 and HCF. In some way,
whether directly or indirectly by influencing the presentation of the N-terminal region, the C-terminal 20 residues mapping between positions 425 and 445 and encompassing the H2.CCR are required for complex formation. Second, certain residues within this same C-terminal region are also specifically re-quired for transcriptional activity, since we show that the mu-tant VP16-E.(⌬442-445) is able to promote complex forma-tion, while earlier results showed that this mutant was virtually inactive in the induction of IE gene expression (reference 8 and data not shown).
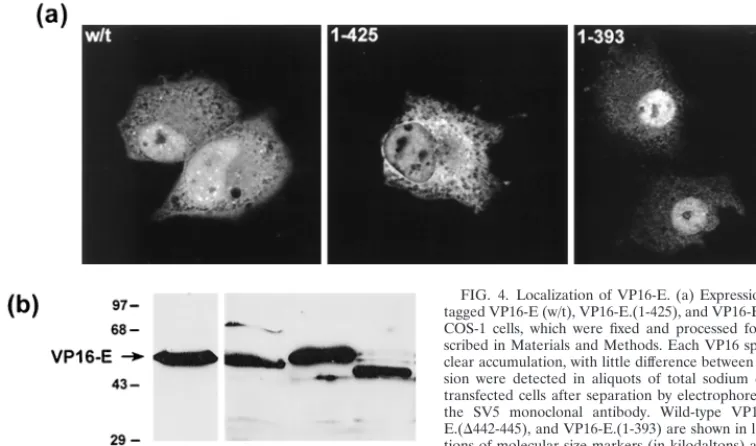
Cellular compartmentalization of VP16-E.To examine the
compartmentalization of VP16-E in comparison to that of VP16-H, VP16-E and variants were tagged at their N termini with an epitope tag (from the SV5 matrix protein), and local-ization in transfected COS-1 cells was assessed by immunoflu-orescence. Cells were fixed in cold methanol, and VP16-E was detected with the anti-SV5 monoclonal antibody. VP16-E typ-ically showed a significant degree of nuclear accumulation, with lower but detectable amounts in the cytoplasm, particu-larly in cells with a higher level of expression (Fig. 4a). Nuclear accumulation of VP16-E was more pronounced than that of VP16-H which, in a parallel analysis, was found in a diffuse, predominantly cytoplasmic pattern, as previously reported (20). Patterns for VP16-E.(1-425) and VP16-E.(1-393) exhib-ited no significant differences in compartmentalization relative to wild-type VP16-E (Fig. 4a), and overall levels of expression were similar (Fig. 4b). The results help support the proposal that there was no gross disruption of VP16-E by virtue of the C-terminal deletions and that the failure to promote activation in vivo was due to a failure to promote complex formation for variants VP16-E.(1-425) and VP16-E.(1-393).
Activation by GAL–VP16-E. The method usually used to
uncouple requirements for DNA binding (whether through protein-protein interactions, as for VP16, or via direct DNA binding) from those for transcriptional activation per se is to link the candidate activation domain to an independent DNA binding domain, for example, that of the GAL4 protein. How-ever, since deletion of the H2.CCR had little effect on VP16-H but had a profound effect on VP16-E, it seemed likely that other determinants, possibly within the N-terminal region it-self, were involved in activation by VP16-E, acting as the equiv-alent of the H1 region of the VP16-H C-terminal activation domain. Indeed, a region close to the N terminus of VP16-E, between residues 23 and 46 (our numbering system; previously labeled as residues 53 to 77), has been indicated to share some homology with the H1 region of VP16-H (35).
Therefore, in order to examine activation by VP16-E sepa-rately from TRF-C formation, we fused the entire VP16-E open reading frame to the GAL4 DNA binding domain and tested the activity of the fusion protein on an upstream acti-vation sequence-containing target reporter gene. Activity was compared with that of GAL4 fusion proteins containing the entire VP16-H open reading frame or just the VP16-H activa-tion domain (Fig. 5b). For the sake of comparison of the two assay systems, we also included a direct parallel examination of the activity of native VP16-E and VP16-H on the native EHV-1 IE gene promoter (Fig. 5a). While, as expected from the above results, VP16-E was as potent as VP16-H (Fig. 5a), GAL4.VP16-E surprisingly was significantly weaker than GAL4. VP16-H and in fact exhibited an activity barely above back-ground (Fig. 5b). The expression of GAL4 fusion proteins was examined by Western blot analysis of transfected-cell extracts with an anti-GAL4 antibody, and expression levels were found to be similar, indicating that the lack of activity was not due to a deficiency in expression levels (data not shown). It is also noteworthy that while GAL4.VP16-H, containing intact
VOL. 74, 2000 COMPARISON OF VP16 FROM EHV AND HSV 10115
on November 9, 2019 by guest
http://jvi.asm.org/
than the fusion protein containing the isolated activation domain Gal4.Acid. This result is considered together with the lack of activity of the GAL4.VP16-E protein in the Discussion.
not recapitulated in the context of GAL4 fusion proteins. One explanation for this result is that the activation function of VP16-E was dependent on or linked to the mode of DNA binding and therefore that a comparison of transcriptional activation by VP16-E and VP16-H required analysis in the context of recruitment via HCF and Oct-1 in TRF-C. To this end, we constructed a VP16-H/E chimeric protein comprising the N-terminal domain of VP16-H linked to the C-terminal region of VP16-E. Note that such a chimera had been pro-duced previously (8); in it, eight residues from the extreme C terminus of VP16-E had been fused to the N-terminal domain of VP16-H. The construct exhibited little activity. However, from our analysis of the alignments (Fig. 1b), there was clearly a more extended region of conservation within the C terminus of VP16-E, beginning at residue 393, which is selectively re-tained in VP16-B (from BHV-1) and VZV VP16 but not in VP16-H. It was possible that determinants within this latter region were required for activity and were not included in the earlier analysis. We therefore constructed a VP16-H/E chi-meric protein linking the N-terminal complex-forming region from VP16-H (up to residue 411) to the C-terminal region of VP16-E from residue 393 to the end (Fig. 6c). Thus, the VP16-E region encompassed not only the H2.CCR but also the region conserved in VP16-E, VP16-B, and the VZV VP16 homologues.
[image:5.612.83.501.116.663.2]Cells were transfected with the target construct containing the HSV-1 IE110 gene promoter region, and activation by VP16-H, VP16-E, and VP16H/E was compared. The results demonstrate that while VP16-H and VP16-E were equally po-tent, the VP16-H/E chimera surprisingly exhibited extremely
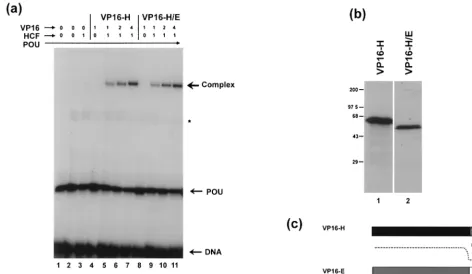
FIG. 2. Complex formation by VP16-E and variants. (a) VP16-E variants were translated in vitro in a rabbit reticulocyte system (50l) in the presence of [35S]methionine, and equal amounts (1l) were separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis and detected by autoradiography. Positions
of molecular size markers (in kilodaltons) are indicated on the left. (b) Equal amounts of in vitro translation reactions for the species indicated (1, 2, or 4l) were incubated with the E1 octamer probe, the purified POU domain, and HCF, supplied from a separate in vitro translation reaction. The position of the complex dependent on all components is indicated. Independent binding of the POU domain is indicated by POU, while a nonspecific complex formed by a component of the reticulocyte extract is indicated by an asterisk. The position of unbound probe is marked as DNA. The results for each VP16 species are shown in four lanes, the first lane of which lacks HCF. Thus, complex formation by competent variants is seen to be dependent upon HCF. The control lanes (lanes 1) show that HCF is not sufficient for complex formation, which requires the appropriate VP16 species.
10116
on November 9, 2019 by guest
http://jvi.asm.org/
weak activity (Fig. 6a). In control experiments with monoclonal antibody LP1, which reacts against an N-terminal determinant of VP16 and could be used to detect both species, the VP16-H/E chimera and VP16-H were expressed in vivo at very sim-ilar levels (Fig. 6b). Although the C terminus of VP16-H can be deleted without a significant effect on the assembly of TRF-C, it was possible that linking the C terminus of VP16-E to VP16-H may have had some dominant negative effect and that the failure of VP16-H/E to activate expression was due to a failure to promote complex formation. We therefore com-pared complex formation by VP16-H and VP16-H/E in gel retardation assays (Fig. 7). The two species were translated in vitro and incubated in similar increasing doses with the puri-fied POU domain, HCF, and the IE110 octamer-GARAT probe. The results demonstrate that VP16-H and VP16-H/E were equally proficient in the assay (Fig. 7a, lanes 5 to 11); thus, the exchange of the acidic domain of VP16-H for the C-terminal domain of VP16-E seems to have little effect on complex assembly.
DISCUSSION
VP16 is a potent activator of HSV-1 IE gene expression, the study of which has yielded significant insights into eukaryotic transcriptional regulation. From such studies, the view has emerged that VP16 comprises two independent domains, a large N-terminal domain involved in binding Oct-1, HCF, and DNA to form the DNA binding complex and a C-terminal domain involved in activation per se. To examine this view further, we have taken the approach of comparing
require-ments within the C-terminal regions of two related VP16 spe-cies, VP16-H and VP16-E, from HSV-1 and EHV-1, respec-tively, for recruitment into the DNA binding complex containing Oct-1 and HCF and for activation. The results in-dicate that VP16-E does not appear to conform to the previ-ously described view of VP16-H of segregation into two inde-pendent domains. In VP16-E, a determinant within the extreme C terminus is directly involved in complex formation or somehow ensures correct presentation of the determinants in the N terminus.
The specific requirement in VP16-E for this C-terminal re-gion in complex formation is something of a paradox. On the one hand, the extreme C termini encompass a short determi-nant (the H2.CCR) which is conserved, despite the overall lack of similarity in the C termini. Deletion of this determinant may therefore have been expected to have a consequence on some activity. This is the case for VP16-E, in that such deletion abolishes transactivation by affecting complex formation. On the other hand, in VP16-H, the removal of the acidic domain, including the H2.CCR determinant, has no effect on complex formation (10). Thus, for VP16-E, the view of the C terminus as being exclusively involved in some aspect of transactivation is not accurate. Notwithstanding that VP16-E retains a high degree of homology with determinants required for complex formation in the N-terminal domain of VP16-H, it appears that features within the C terminus are required for complex as-sembly. It is noteworthy, though, that certain residues within the H2.CCR may be specifically involved in activation, since we show that the mutant VP16-E.(⌬442-445) retained complex-forming activity while being inactive in transactivation (8).
FIG. 3. (a) Requirement for a determinant located between positions 425 and 445 for VP16-E complex formation. COS-1 cells were transfected (2g of DNA) with an expression vector for wild-type VP16-E, VP16-E.(1-445), or VP16-E.(1-425), and soluble extracts were made 40 h after transfection as described in Materials and Methods. Extracts in the amounts indicated (microliters) were incubated with the purified Oct-1 POU domain, in vitro-translated HCF, and radiolabeled EHV-1 E1 probe. Complexes were then separated on a nondenaturing TBE–6% polyacrylamide gel. The nonspecific complex formed by a component in the reticulocyte extract is indicated by a single asterisk, while a component likely representing endogenous Oct-1 in the soluble extract is indicated by a double asterisk. Free probe (DNA), POU binding, and the POU-HCF-VP16 complex are indicated by arrows. Controls in lanes 1 and 2 and the first lane of each series show that the complex was dependent upon HCF and competent VP16-E. Deletion of the 20 residues from positions 445 to 425 abolished complex formation. (b) Summary of each of the VP16-E constructs and their function in complex formation (Comp.) (this work) and IE gene transactivation (Act) (8; this work). Salient features include the lack of complex formation by VP16-E.(1-425) and the competence for complex formation of VP16-E.(⌬442-445), despite its lack of transactivation. In this summary, activation was indicated as negative when it measured less than 10% of control values.
VOL. 74, 2000 COMPARISON OF VP16 FROM EHV AND HSV 10117
on November 9, 2019 by guest
http://jvi.asm.org/
[image:6.612.57.551.71.316.2]The overlap between determinants involved in complex for-mation and those required for transactivation makes analysis of structure-function relationships difficult. To examine activa-tion distinct from complex formaactiva-tion, we fused the entire VP16-E protein to the DNA binding domain of GAL4 and compared activation by VP16-E and VP16-H in the native context to that in the GAL4 fusion setting. While the native proteins exhibited approximately equal activities, in the con-text of the GAL4 fusions, VP16-E was significantly weaker than VP16-H, exhibiting little activity. These results indicate two main points. First, the use of artificial GAL fusion proteins may not reflect the true nature of activation domains, both in qualitative and in quantitative terms. Results from this work and previous work (26) indicate that the GAL4-acidic domain fusion protein is unusually potent and is in fact considerably more potent than GAL4 fused to the entire VP16-H protein. A
positive interpretation of this result could be that the extremely potent activity of the GAL4-acidic domain protein does rep-resent the activity of the acidic domain of native VP16-H when, for example, it becomes released or exposed for full activity through its normal assembly pathway. On the other hand, it is conceivable that the mechanism involved in activation by the GAL4-acidic domain fusion protein does not truly reflect that of the native protein. An alteration in the function of VP16 in the context of a GAL4 chimera has been suggested previously (12). That work showed that VP16-H acting on its native pro-moter could enhance activation only when in a propro-moter- promoter-proximal position, while GAL–VP16-H.(411-490) could addi-tionally activate from a distal position. A related phenomenon was observed with certain HOX proteins, where identification of the activation domains of the HOX proteins varied dramat-ically depending on the context of the proteins (45). Analysis of the binding of HOXD9 as a monomer to a HOX binding site located the activation domain to within residues 76 to 264. However, when the same analysis was carried out using a GAL4 DNA binding domain fused to the HOX protein, only the N-terminal 75 amino acids contained a potential activation domain; this same region, however, could be deleted in a native setting with virtually no effect (45). These and other results from similar types of analysis of other regulatory pro-teins indicate that caution in the interpretation of the results of studies in the context of GAL4 fusion proteins seems justifi-able. It is possible that the mode of DNA binding has direct consequences on the mechanism of activation, for example, in the presentation of the activation domain. With that in mind, we proceeded to construct a VP16 chimera, but here, too, we obtained surprising results.
Our rationale was to explore similarities and differences in VP16-H and VP16-E by constructing a chimera, informed by a previous analysis of the sufficiency of the N-terminal domain of VP16-H for complex formation and our alignment of the ho-mologues to indicate where transitions or boundaries in func-tional sections may occur. Furthermore, recent determination of the crystal structure of VP16-H indicates that there is little ordered structure beyond residue 345, presumably indicative of a disordered unfolded region which appears to have little
[image:7.612.103.481.76.299.2]in-FIG. 4. Localization of VP16-E. (a) Expression vectors (1g) for epitope-tagged VP16-E (w/t), VP16-E.(1-425), and VP16-E.(1-393) were transfected into COS-1 cells, which were fixed and processed for immunofluorescence as de-scribed in Materials and Methods. Each VP16 species exhibited significant nu-clear accumulation, with little difference between variants. (b) Levels of expres-sion were detected in aliquots of total sodium dodecyl sulfate lysates of the transfected cells after separation by electrophoresis and Western blotting with the SV5 monoclonal antibody. Wild-type E, E.(1-425), VP16-E.(⌬442-445), and VP16-E.(1-393) are shown in lanes 1 to 4, respectively. Posi-tions of molecular size markers (in kilodaltons) are indicated on the left.
FIG. 5. Comparison of activation by intact VP16 species versus GAL fusions. (a) Vero cells were transfected with the EHV-1 IE gene target construct pCAT_TAAT (200 ng) alone (⫺) or together with 1, 10, or 100 ng of the VP16-H and VP16-E expression vectors as indicated, and CAT activity was assayed. (b) COS-1 cells were transfected with the upstream activation sequence target con-struct pUAS10CAT (475 ng) alone (⫺) or together with 10, 100, or 1,000 ng of the GAL4 fusion constructs as indicated. The relative strength of VP16-E when the intact proteins were assayed is not reflected in the context of the GAL4 fusion proteins.
on November 9, 2019 by guest
http://jvi.asm.org/
[image:7.612.55.289.510.651.2]fluence on the folding of the N-terminal core domain (24). Thus, a reasonable expectation would be that linking the com-plete N-terminal region to the C-terminal region of a related protein would have little effect on folding. We selected the region of VP16-E to be linked on the basis of the alignments and the transition at residue 393 (of VP16-E) to a C-terminal section showing good conservation in the VP16-B, VP16-E, and VZV VP16 species (Fig. 1b). The chimera was expressed normally; the N terminus folded normally, as indicated by the ability of the chimeric protein to promote complex formation, yet it activated transcription extremely poorly. The salient comparison is between native VP16-E and VP16-H/E. Both retain the C-terminal region, both promote complex forma-tion, yet VP16-E is a potent activator and VP16H/E exhibits little activity. One explanation for this result, illustrated sche-matically in Fig. 7c, could be that for VP16, there exist two C-terminal determinants, H1 and H2, which are redundant for transactivation, with the result that deletion of H2 has little effect. This notion would be consistent with previous results (10, 35, 42). It is reasonable to suggest that VP16-E has only one C-terminal determinant, equivalent to H2, and that dele-tion of this region renders the protein inactive. However, since the VP16-H/E chimera is also inactive, the explanation re-quires a difference between the N termini of VP16-H and VP16-E, with the latter possessing an extra determinant, N1, not present in VP16-H and required together with H2 for activation.
An N-terminal region in VP16-E (residues 23 to 46) exhib-iting some similarity to H1 has been previously reported (27), and this may be the difference between the two proteins;
how-ever, we note that this region in VP16-E does exhibit conser-vation with the extreme N terminus of VP16-H. The determi-nant would also need to be qualitatively distinct from the VP16-H H1 determinant, which appears to suffice in the ab-sence of H2, while the N terminus of VP16-E does not. An alternative explanation is that in both proteins, the N- and C-terminal determinants always function in conjunction. Thus, in VP16-H, activation could be mediated by an N1-H1 or an N1-H2 interaction, while in VP16-E, there would be only an N1-H2 determinant. This explanation, however, is further complicated by the requirement in VP16-E for H2 in complex formation itself; it is possible that in VP16-E an interaction between the N and C termini is required for complex forma-tion, while in VP16-H the N-terminal region is sufficient. An additional chimera in which additional N-terminal regions of VP16-E are spliced into the VP16-H/E chimera may exhibit restored function and identify differences between the two proteins.
[image:8.612.138.474.76.357.2]Finally, however, the basis for the clear selective conserva-tion of the H2.CCR in each of the homologous remains to be established. In this respect, we have previously shown that VP16-H interacts with another tegument protein, VP22, that the C-terminal domain is required, and furthermore that res-idues in the H2.CCR are involved in this interaction (7). It is possible that the other VP16 homologues interact with the corresponding VP22 species and that the conservation within the H2.CCR is related to this activity. Moreover, it is possible that any interaction between the VP16 H2.CCR and VP22 in the virion is relevant to presentation of the activation domain early after entry, i.e., in activation, or late in infection, when
FIG. 6. Lack of activation by VP16-H/E. (a) COS-1 cells were transfected with the HSV-1 IE gene reporter construct pAB5 (100 ng) alone (⫺) or together with expression vectors for VP16-E, VP16-H, and the VP16-H/E chimera [VP16-H.(1-411) plus VP16-E.(393-448)] at doses of 1, 10, and 100 ng, and CAT activity was measured. (b) Levels of expression of VP16-H and VP16-H/E from cells transfected with 100 ng of DNA were compared. Whole-cell extracts were separated on a sodium dodecyl sulfate–10% polyacrylamide gel, and the products were transferred to nitrocellulose and probed with the anti-VP16 monoclonal antibody LP1. Positions of molecular size markers (in kilodaltons) are indicated on the left.
VOL. 74, 2000 COMPARISON OF VP16 FROM EHV AND HSV 10119
on November 9, 2019 by guest
http://jvi.asm.org/
activation may be suppressed. We are currently examining this proposition with cotransfection assays.
In summary, our results indicate that the simplified model of discrete domains within VP16-H for complex formation and activation may not apply to other VP16 homologues and that GAL4 fusion proteins may not reflect determinants or activi-ties of the parental proteins. The result is that at least in certain VP16 species, the separation of determinants involved in re-cruitment to DNA binding complexes from those involved in activation may be more complicated than previously appreci-ated.
REFERENCES
1.Ace, C. I., M. A. Dalrymple, F. H. Ramsay, V. G. Preston, and C. M. Preston.
1988. Mutational analysis of the herpes simplex virus type 1 trans-inducing factor Vmw65. J. Gen. Virol.69:2595–2605.
2.Batterson, W., and B. Roizman.1983. Characterization of the herpes simplex virion-associated factor responsible for the induction of alpha genes. J. Virol.
46:371–377.
3.Campbell, M. E., J. W. Palfreyman, and C. M. Preston.1984. Identification of herpes simplex virus DNA sequences which encode a trans-acting polypeptide responsible for stimulation of immediate early transcription. J. Mol. Biol.180:1–19.
4.Cousens, D. J., R. Greaves, C. R. Goding, and P. O’Hare.1989. The C-terminal 79 amino acids of the herpes simplex virus regulatory protein, Vmw65, efficiently activate transcription in yeast and mammalian cells in chimeric DNA-binding proteins. EMBO J.8:2337–2342.
5.Cress, W. D., and S. J. Triezenberg.1991. Critical structural elements of the VP16 transcriptional activation domain. Science251:87–90.
6.Dalrymple, M. A., D. J. McGeoch, A. J. Davison, and C. M. Preston.1985. DNA sequence of the herpes simplex virus type 1 gene whose product is responsible for transcriptional activation of immediate early promoters. Nu-cleic Acids Res.13:7865–7869.
7.Elliott, G., G. Mouzakitis, and P. O’Hare.1995. VP16 interacts via its activation domain with VP22, a tegument protein of herpes simplex virus, and is relocated to a novel macromolecular assembly in coexpressing cells. J. Virol.69:7932–7941.
8.Elliott, G. D.1994. The extreme carboxyl terminus of the equine herpesvirus 1 homolog of herpes simplex virus VP16 is essential for immediate-early gene activation. J. Virol.68:4890–4897.
9.Gerster, T., and R. G. Roeder.1988. A herpesvirus trans-activating protein interacts with transcription factor OTF-1 and other cellular proteins. Proc. Natl. Acad. Sci. USA85:6347–6351.
10. Greaves, R., and P. O’Hare.1989. Separation of requirements for protein-DNA complex assembly from those for functional activity in the herpes simplex virus regulatory protein Vmw65. J. Virol.63:1641–1650. 11. Greaves, R. F., and P. O’Hare.1990. Structural requirements in the herpes
simplex virus type 1 transactivator Vmw65 for interaction with the cellular octamer-binding protein and target TAATGARAT sequences. J. Virol.64:
2716–2724.
12. Hagmann, M., O. Georgiev, and W. Schaffner.1997. The VP16 paradox: herpes simplex virus VP16 contains a long-range activation domain but within the natural multiprotein complex activates only from promoter-prox-imal positions. J. Virol.71:5952–5962.
13. Hayes, S., and P. O’Hare.1993. Mapping of a major surface-exposed site in the herpes simplex virus protein Vmw65 to a region of direct interaction in a transcription complex assembly. J. Virol.67:852–862.
14. Heine, J. W., R. W. Honess, E. Cassai, and B. Roizman.1974. Proteins specified by herpes simplex virus. XII. The virion polypeptides of type 1 strains. J. Virol.14:640–651.
15. Hengartner, C. J., C. M. Thompson, J. Zhang, D. M. Chao, S.-M. Liao, A. J. Koleske, S. Okamura, and R. A. Young.1995. Association of an activator with an RNA polymerase II holoenzyme. Genes Dev.9:897–910. 16. Hughes, T. A., S. LaBoissiere, and P. O’Hare.1999. Analysis of functional
domains of the host cell factor involved in VP16 complex formation. J. Biol. Chem.274:16437–16443.
17. Katan, M., A. Haigh, C. P. Verrijzer, P. C. van der Vliet, and P. O’Hare.
[image:9.612.67.539.76.349.2]1990. Characterization of a cellular factor which interacts functionally with FIG. 7. Complex formation by VP16-H/E. (a) VP16-H and VP16-H/E were translated in vitro in reticulocyte lysates, and equal amounts of the lysates (in microliters) were incubated with purified POU in the presence or absence of in vitro-expressed HCF exactly as described in the legend to Fig. 2, with the exception that poly(dI-dC) (1g) was included as a nonspecific competitor. The samples were analyzed on a nondenaturing TBE–6% polyacrylamide gel. Free probe (DNA), POU binding, and the POU-HCF-VP16 complex are indicated by arrows. Low amounts of the nonspecific band from the TnT lysate are marked by an asterisk. (b) Sodium dodecyl sulfate-polyacrylamide gel electrophoresis of in vitro-translated VP16-H and VP16-H/E fusion proteins. Positions of molecular size markers (in kilodaltons) are indicated on the left. (c) Schematic summary of determinants in VP16-H and VP16-E. H1 and H2 represent redundant determinants in the C terminus of VP16-H, and H2 is a conserved determinant in VP16-E. N1 represents an N-terminal determinant in VP16-E to account for activation by native VP16-E but not by the VP16-H/E chimera, despite the ability of the latter to promote complex formation.
on November 9, 2019 by guest
http://jvi.asm.org/
Oct-1 in the assembly of a multicomponent transcription complex. Nucleic Acids Res.18:6871–6880.
18.Klemm, R. D., J. A. Goodrich, S. Zhou, and R. Tjian.1995. Molecular cloning and expression of the 32-kDa subunit of human TFIID reveals interactions with VP16 and TFIIB that mediate transcriptional activation. Proc. Natl. Acad. Sci. USA92:5788–5792.
19. Koleske, A. J., and R. A. Young.1994. An RNA polymerase II holoenzyme responsive to activators. Nature368:466–469.
20. LaBoissiere, S., T. Hughes, and P. O’Hare.1999. HCF-dependent nuclear import of VP16. EMBO J.18:480–490.
21. Lai, J. S., and W. Herr.1997. Interdigitated residues within a small region of VP16 interact with Oct-1, HCF, and DNA. Mol. Cell. Biol.17:3937–3946. 22. Lewis, J. B., Y. G. Thompson, and G. B. Caughman.1994. Transcriptional
control of the equine herpesvirus 1 immediate early gene. Virology197:788– 792.
23. Lin, Y.-S., I. Ha, E. Maldonado, D. Reinberg, and M. Green.1991. Binding of general transcription factor TFIIB to an acidic activating region. Nature
353:569–571.
24. Liu, Y., W. Gong, C. C. Huang, W. Herr, and X. Cheng.1999. Crystal structure of the conserved core of the herpes simplex virus transcriptional regulatory protein VP16. Genes Dev.13:1692–1703.
25. Misra, V., A. C. Bratanich, D. Carpenter, and P. O’Hare.1994. Protein and DNA elements involved in transactivation of the promoter of the bovine herpesvirus (BHV) 1 IE-1 transcription unit by the BHV alpha genetrans -inducing factor. J. Virol.68:4898–4909.
26. Misra, V., S. Walker, S. Hayes, and P. O’Hare.1995. The bovine herpesvirus alpha genetrans-inducing factor activates transcription by mechanisms dif-ferent from those of its herpes simplex virus type 1 counterpart VP16. J. Virol.69:5209–5216.
27. Moriuchi, H., M. Moriuchi, R. Pichyangkura, S. J. Triezenberg, S. E. Straus, and J. I. Cohen. 1995. Hydrophobic cluster analysis predicts an amino-terminal domain of varicella-zoster virus open reading frame 10 required for transcriptional activation. Proc. Natl. Acad. Sci. USA92:9333–9337. 28. Moriuchi, H., M. Moriuchi, S. E. Straus, and J. I. Cohen.1993.
Varicella-zoster virus open reading frame 10 protein, the herpes simplex virus VP16 homolog, transactivates herpesvirus immediate-early gene promoters. J. Vi-rol.67:2739–2746.
29. O’Hare, P.1993. The virion transactivator of herpes simplex virus. Semin. Virol.4:145–155.
30. O’Hare, P., and C. R. Goding.1988. Herpes simplex virus regulatory ele-ments and the immunoglobulin octamer domain bind a common factor and are both targets for virion transactivation. Cell52:435–445.
31. Pellett, P. E., J. L. McKnight, F. J. Jenkins, and B. Roizman.1985. Nucle-otide sequence and predicted amino acid sequence of a protein encoded in a small herpes simplex virus DNA fragment capable of trans-inducing alpha genes. Proc. Natl. Acad. Sci. USA82:5870–5874.
32. Post, L. E., S. Mackem, and B. Roizman.1981. Regulation of alpha genes of herpes simplex virus: expression of chimeric genes produced by fusion of thymidine kinase with alpha gene promoters. Cell24:555–565.
33. Preston, C. M., M. C. Frame, and M. E. Campbell.1988. A complex formed between cell components and an HSV structural polypeptide binds to a viral immediate early gene regulatory DNA sequence. Cell52:425–434.
34. Purewal, A. S., R. Allsopp, M. Riggio, E. A. Telford, S. Azam, A. J. Davison, and N. Edington. 1994. Equid herpesviruses 1 and 4 encode functional homologs of the herpes simplex virus type 1 virion transactivator protein, VP16. Virology198:385–389.
35. Regier, J. L., F. Shen, and S. J. Triezenberg.1993. Pattern of aromatic and hydrophobic amino acids critical for one of two subdomains of the VP16 transcriptional activator. Proc. Natl. Acad. Sci. USA90:883–887. 36. Sadowski, I., B. Bell, P. Broad, and M. Hollis.1992. GAL4 fusion vectors for
expression in yeast or mammalian cells. Gene118:137–141.
37. Sadowski, I., J. Ma, S. Triezenberg, and M. Ptashne.1988. GAL4-VP16 is an unusually potent transcriptional activator. Nature335:563–564.
38. Stern, S., M. Tanaka, and W. Herr.1989. The Oct-1 homoeodomain directs formation of a multiprotein-DNA complex with the HSV transactivator VP16. Nature341:624–630.
39. Stringer, K. F., C. J. Ingles, and J. Greenblatt.1990. Direct and selective binding of an acidic transcriptional activation domain to the TATA-box factor TFIID. Nature345:783–786.
40. Telford, E. A., M. S. Watson, K. McBride, and A. J. Davison.1992. The DNA sequence of equine herpesvirus-1. Virology189:304–316.
41. Triezenberg, S. J.1995. Structure and function of transcriptional activation domains. Curr. Opin. Genet. Dev.5:190–196.
42. Triezenberg, S. J., R. C. Kingsbury, and S. L. McKnight.1988. Functional dissection of VP16, the trans-activator of herpes simplex virus immediate early gene expression. Genes Dev.2:718–729.
43. Triezenberg, S. J., K. L. LaMarco, and S. L. McKnight.1988. Evidence of DNA:protein interactions that mediate HSV-1 immediate early gene activa-tion by VP16. Genes Dev.2:730–742.
44. Utley, R. T., K. Ikeda, P. A. Grant, J. Cotes, D. J. Steger, A. Eberharter, S. John, and J. L. Workman.1998. Transcriptional activators direct histone acetyltransferase complexes to nucleosomes. Nature394:498–502. 45. Vigano, M. A., G. Di Rocco, V. Zappavigna, and F. Mavilio.1998. Definition
of the transcriptional activation domains of three human HOX proteins depends on the DNA-binding context. Mol. Cell. Biol.18:6201–6212. 46. Walker, S., R. Greaves, and P. O’Hare.1993. Transcriptional activation by
the acidic domain of Vmw65 requires the integrity of the domain and in-volves additional determinants distinct from those necessary for TFIIB bind-ing. Mol. Cell. Biol.13:5233–5244.
47. Walker, S., S. Hayes, and P. O’Hare.1994. Site-specific conformational alteration of the Oct-1 POU domain-DNA complex as the basis for the differential recognition by Vmw65 (VP16). Cell79:841–852.
48. Werstuck, G., and J. P. Capone.1989. Mutational analysis of the herpes simplex virus trans-inducing factor Vmw65. Gene75:213–224.
49. Wilson, A. C., M. A. Cleary, J. S. Lai, K. LaMarco, M. G. Peterson, and W. Herr.1993. Combinatorial control of transcription: the herpes simplex virus VP16-induced complex. Cold Spring Harbor Symp. Quant. Biol.58:167–178. 50. Wilson, A. C., K. LaMarco, M. G. Peterson, and W. Herr.1993. The VP16 accessory protein HCF is a family of polypeptides processed from a large precursor protein. Cell74:115–125.
51. Xiao, P., and J. P. Capone.1990. A cellular factor binds to the herpes simplex virus type 1 transactivator Vmw65 and is required for Vmw65-dependent protein-DNA complex assembly with Oct-1. Mol. Cell. Biol.10:4974–4977.
VOL. 74, 2000 COMPARISON OF VP16 FROM EHV AND HSV 10121
![FIG. 6. Lack of activation by VP16-H/E. (a) COS-1 cells were transfected with the HSV-1 IE gene reporter construct pAB5 (100 ng) alone (�expression vectors for VP16-E, VP16-H, and the VP16-H/E chimera [VP16-H.(1-411) plus VP16-E.(393-448)] at doses of 1, 1](https://thumb-us.123doks.com/thumbv2/123dok_us/513033.70352/8.612.138.474.76.357/activation-transfected-reporter-construct-expression-vectors-chimera-doses.webp)