JOURNAL OFVIROLOGY, Mar. 1994.p. 1993-1997 Vol.68, No.3 0022-538X/94/$04.00+0
Copyright ©) 1994,American Society for Microbiology
Analysis
of Tat Function in Human
Immunodeficiency Virus
Type
1-Infected Low-Level-Expression
Cell
Lines
Ul and ACH-2
PAULA
CANNON,'
SEON-HEE KIM,2CATHERINEULICH,3 AND SUNYOUNG KIM2*Departmentof Biochemistry, Universityof Oxford, OxfordOX]3QU, United
Kingdom';
Institute for MolecularBiology and Genetics andDepartmentof Microbiology, Seoul National University, Seoul 151-742, Korea-; andDivision of Molecular Virology, UniversityofTexasSoulthwestern Graduate School
of BiomedicalSciences, Dallas, Texas
752353
Received 28 December 1992/Accepted 24November 1993
The Ul and ACH-2 cell lines are subclones of human monocytic and T-lymphoid cells, respectively, persistently infected with human immunodeficiency virus type 1. These cell lines harbor the viral genome but produce only very low levels of viral progeny, which can be increased by stimulation with agents such as phorbol ester andcytokines. As such, they provide an in vitro model for human immunodeficiency virus type 1 latency. Inorder toexamine the basis for their latent state, we have analyzed the activity of endogenous Tat protein in these cells and investigated theeffect on viral replication of the addition of exogenous Tat protein. We find that
Ulcells seem tohave levels of Tatprotein that are suboptimal for long terminal repeat (LTR) transcription, becausetranscriptionfrom atransfectedLTR-chloramphenicol acetyltransferaseplasmidcanbeenhanced by cotransfection of a Tatexpression plasmid. Furthermore, viral replication can be stimulated in this cell line byincubation with purified Tat protein. In contrast, ACH-2 cells are not limited for LTR-chloramphenicol acetyltransferase transcription by endogenous levels of Tat, and virus production is not increased by the additionof exogenous Tatprotein.By semiquantitative PCR analysis of viral RNA, we have demonstrated that Tatprotein caused an increase in human immunodeficiency virus RNA expression inUlcellsbut had no effect inACH-2 cells. This suggests that adifferent mechanism underlies the latent state in Ul and ACH-2 cells.
Theability ofhumanimmunodeficiencyvirus type I (HIV-1) toestablishalatent phase ofinfectionin vivo is characteristic of the pathogenesis of AIDS (19, 20). Accordingly, much interest hasfocused onpossible mechanismsfor the establish-mentandmaintenance oflatentinfectionsby HIV-1(reviewed
in references 1 and 11). Latency can arise at apreintegration
stage ifconditionsin the host cell are unfavorable forreverse
transcription and integration of the provirus, as could occur upon infection ofaquiescenthostcell (28, 29). Alternatively, latency can be established when the provirus is already inte-gratedinthe genomeof thehost cell but is unabletoproduce high levels ofviraltranscripts orprogenyvirions.
TheUl and ACH-2celllinesareHIV-1-infected monocytic
and T-lymphocytic cells, respectively (3, 8). Ul and ACH-2
cells contain one and twocopies ofthe HIV genome,
respec-tively, but they express viral RNA and proteins at very low
levels. For thisreason, these cell lines have been used
exten-sively asin vitro models of HIVlatency.Because the parental
cell linesofUl andACH-2representthe twomajorhost cell types for HIV-1 infection in vivo, they are often studied in
parallel. Viralproduction from these cells can beincreasedby
avariety ofprocesses, including treatmentwith
cytokines
and mitogens andsuperinfection byotherviruses (3, 4, 7, 8, 12, 14, 23, 24). Such studies may provide an indication ofprocesses that could lead to activation of latent virus in the body and causeprogression todisease.RNA transcription in U1 and ACH-2 cells is at a low level and shows an aberrant pattern, with the 2-kbmultiply spliced
*Corresponding author. Mailing address: Institute for Molecular
BiologyandGenetics,Building 105, Seoul NationalUniversity, Kwan-Ak-Gu, Seoul 151-742, Korea. Phone: 82-2-880-7529. Fax: 82-2-875-0907.
transcripts predominating (21, 24). Following stimulation, there is an initial increase in the levels of the 2-kb message, followed bya later increase in the singly spliced (4.3-kb) and unspliced (9.2-kb) species (12, 21, 24). This pattern of
tran-scription is reminiscent of the early stages of a synchronous
one-stepproductive infection (16)and has led to the hypoth-esis that latency could arise because of the inability of an
infection to progress beyond an initial transcriptional phase (16, 24). In HIV-1-infected butasymptomatic patients, it has been demonstrated thatatrulylatent stateofviralexpression
does not exist but that viral transcription and replication proceedat a low level throughout this period (22, 26).
Inter-estingly, it has also been noted that there is a
qualitative
changeinthepatternof viral RNAexpression duringprogres-siontothe
symptomatic
stageof thedisease,withasimilar shifttowardsthe production of
unspliced
messages (22).While the establishment and maintenance of a low-level
infection probably depend on a combination of cellular and
viral factors, the pattern of transcription in these latent cell lines suggests in particular the involvementof the viral
regu-latory factors Tat and Rev
(11).
Transcription from the viral long terminal repeat (LTR) is strongly dependent on the presence of Tat(reviewed
in reference10),
solimiting
amountsof this
key
viralregulatory
protein
couldlead tolowoveralllevelsoftranscription. Rev hasalso been
proposed
as a candidate latency gene, because theproduction
of the un-spliced genomic RNA isdependenton Rev and also because the pattern of viral RNAexpression
resulting
from mutationsin Rev is similar to that observed
during
theearly
transcrip-tional phase
(6, 27).
Although many studies have
investigated
the conditionsunder which virus
production
can be increased in Ul andACH-2cells, little is known about
why
thesecellsare latent in 1993on November 9, 2019 by guest
http://jvi.asm.org/
thefirst place or indeed whether they are latent for the same reasons. As aninitialanalysisof the molecular basis of latency, we have examined the role of Tat in the maintenance of low-level infections in these cells.
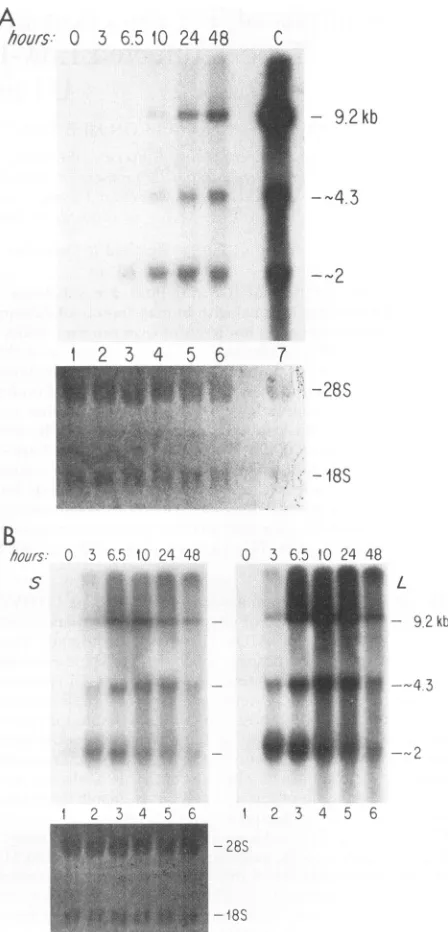
Tobecertain thatourACH-2and U 1 cellsdoindeed harbor latent HIV, we have examined the kinetics of HIV RNA
expression inthese cells at various time points after treatment
with50 ngof phorbolmyristateacetate (PMA [Sigma]) per ml.
Without stimulation by PMA, Ul and ACH-2 cells produce
few virus particles and a small amount of supernatant p24
(about100 to 200pg/ml),and RNA expression is at a very low level (Fig. IA and B, lane 1). (Note that the levels of
subgenomic RNAs are so lowthat the RNAbands are visible
onlyafter alongexposuretime,which is notshown here.) This
pattern of RNA expression is similar to the situation in the
earlytranscriptional phase ofaproductive infection (16). HIV RNAlevels began to risedramatically3 to 6h afterstimulation
with PMA, and the pattern of transcripts produced switched
from the multiply spliced to the singly spliced and unspliced species (Fig. 1), as has been reported by others (12, 21, 24). Thesechanges in the pattern ofRNA expression after PMA
stimulation faithfully recapitulate the transition from early to
late phases of transcription that occursduring asynchronous infection by cell-freevirus. Following PMAstimulation, there wasalsoacorresponding increasein viral p24production from the Ul and ACH-2 cells (data not shown). These results
confirm that the Ul and ACH-2 cells used in this study do indeedharbor latent HIV-1provirus, which ishighlyinducible byPMA.
An HIV LTR-chloramphenicol acetyltransferase (CAT) plasmid, p938 (17),canbeconsideredtobe a reporter plasmid for Tat function, because maximum transcription from a
TAR-containing LTRrequires thepresenceofTatprotein.By
comparing the levels of p938 CAT activity, both with and
withoutcotransfectionof a Tatexpressionplasmid, pCMVTat
(provided byD. Trono[2]),wecanobtainsomemeasureof the level of endogenous Tat activityin an infectedcell line. Ifthe
levels of Tat are high, then p938will not require supplemen-tation with pCMVTat to achieve maximum transcription.
However,ifthecellularlevels ofTat are so low as tolimit p938
expression, then cotransfection of pCMVTat will still boost
CAT activity. We can therefore use thissystem to quantitate endogenousTat activity.
Transfectionofplasmidswasby theDEAE-dextran method
(15).Tenmillioncells weretransfected by2
[Lg
ofp938and by 4pLg
ofpCMVTatwhenrequired. Cellswereplatedat 2 x105
cells per ml and harvested after 48 h, and CAT assays wereperformed by standard procedures (2, 13). As controls for these experiments, we also transfected CEM and U937 cells,
whicharethe uninfectedparentalcelllines for ACH-2and
Ul,
respectively. When p938alone was transfected into CEM andU937cells, therewasminimal CATactivity (Fig. 2, lanes1 and 3). However, when pCMVTat wasalso cotransfected, a
con-siderable increaseinCAT activitywasobserved because of the presenceof Tat. The same procedure was then applied to the
latently infected Ul and ACH-2 cell lines. CAT activity from the LTR construct alone was higher in these infected cell lines than in the uninfected parental cell lines because of the presence ofvirally produced Tat protein (lanes 5 and 7; note also the different assay conditions used for the different cell
lines). Even so, cotransfection of the Tat expression plasmid could boost CATactivityin Ul cells by afurther20-fold (lane
6), suggesting that, although Tatis present in U1 cells, it is at
suboptimal levels for maximum transcription from an endog-enous LTR. In contrast, LTR transcription from p938 in ACH-2 cells was not increased by cotransfection of the Tat
A
hours:
0
3 6.510 24
48
C
c*0
0~w~:
fl
-9.2
kb
*0,
--2
1
2 3 4 5 6
7
-28S
-
18S
B
hours:
S
0 3 6.5 10 24 48
i
0 3 6.5 10 24 48
L
- 9.2kb
---2
1 2 3 4 56 1 2 3 4 5 6
-28S
-18S
FIG. 1. Changes in the pattern of RNA expression inUI (A) and ACH-2 (B) cells on PMA induction.Ul and ACH-2 cellswere treated with 50 ng of PMA per ml, and RNA was harvested at several time points,asindicatedabove the lanes. Twenty micrograms of total RNA was electrophoresed and probed for HIV-1-specific transcripts as previouslydescribed (16). S and L are short and long exposures of the same Northern blot, respectively. Lane I is RNA fromunstimulated cells whose bands arevisible only after along exposure; lanes 2 to 6 contain RNAs from PMA-treated cells. (C) RNAs prepared from productivelyinfected H9 cells (lane 7 in panel A). Quantities of RNA weredetermined at an optical density at 260 nm, and 20
p.g
of total cellular RNA was loaded in each lane. Equivalent loading was confirmedby the intensity of theethidium bromide staining of the gel. 28S and18S indicate the positionsofrRNAs.on November 9, 2019 by guest
http://jvi.asm.org/
[image:2.612.326.550.92.558.2]NOTES 1995
*
_,
X*
S t__*.
G
002~~-SV0
9'0
* t1
2
3
4
5
6
7
8
0.2
3.2 0.3
90.6
1.4
30.1 12.2 8.5
L31
C1
L1 L1U937
GEM
UI
AGH2
1
6
53
0
-cd
AC
CZ.
1 C0
;_ 3
FIG. 2. Effect of cotransfection with Tat expression vector on
LTR-CAT activity. An HIV LTR-CAT construct, p938 (17), was
transfected intoUl and ACH-2cells and theiruninfected parentalcell lines U937andCEM, respectively,in thepresence(+)orabsence(-) ofaTatexpressionvector,pCMVTat.The resultsshowninthisfigure
representonly one ofmany independent transfections. CATactivity
wasmeasuredat37°C under the followingincubation conditions: U937 and CEM, 50 pLg ofprotein for 1 h; U1, 25 ,ug ofprotein for 1 h;
ACH-2, 10 ,ugofproteinfor 20min. Differentassayconditionswere
used with each cell line to keep the assays in the linear range for
[image:3.612.319.561.76.244.2]quantitation. The most important parameter in each case is the magnitude ofinduction upon cotransfection with the Tatexpression
vector.C, unconverted ['4C]chloramphenicol substrate; Ac, acetylated products.
expression plasmid (compare lanes 7 and 8). This implies that the level or activity of Tat protein is not the primary factor limiting LTR expression and viral replication in ACH2 cells
andsuggests thatadifferent mechanismmay underlie the low level of virusproduction in these twocell lines.
Wenextexamined theabilityof Tatprotein toactivateviral production in these twocell lines by monitoring the effecton
viralp24production of incubation with purified Tat protein. It haspreviously been demonstrated that incubation of cells with Tat protein in the presence of protamine sulfate will allow
uptake of the protein and subsequent transactivation of an
HIV LTR(5). We hypothesized that if virus production from U1 cellswaslimited in partby insufficientlevels of Tat protein,
then the addition ofexogenousTat shouldbe able to
upregu-late virusproduction. Similarly,ourtransienttransfection data ledustopredict that ACH-2 cells wouldnot be stimulatedby the additionofmoreTat.
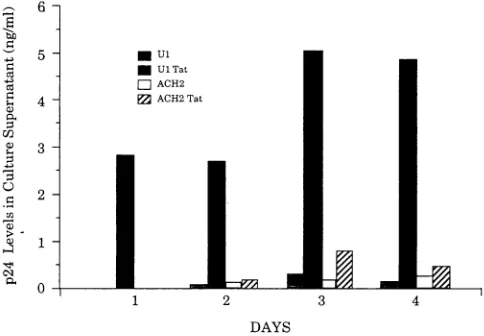
Ten millionUl andACH-2 cellswere incubated with 10 pLg
of purified Tat protein (provided by Alan Frankel at the Gladstone Institute)permlin I mlof medium containing 100
,ug ofprotamine sulfate per ml, as previously described (5).
Control cellsweretreated withprotamine sulfate alone. Cells wereplatedat2 x 105cellsperml, andsupernatantp24 levels
were measured atvarious times postaddition with a
commer-cially available kit (Abbott Laboratories, Chicago, Ill.). Viable cell numbers were monitored throughout to ensure that the
procedure didnotkillthe cells(datanotshown). The results of
onesuchexperimentareshown inFig.3.p24 productionfrom U1 cells incubated with Tatproteinwas increasedby
approx-imately 20-fold over that of the control population, whereas
*U1
*Ul Tat
z ACH2
E ACH2Tat
1 2 3 4
DAYS
FIG. 3. Effect of incubationwithpurified Tat proteinonviralp24
production. Ten million Ul and ACH-2 cellswereincubatedwith 10
,ugof Tatproteinin thepresenceofprotaminesulfate(UITat,ACH-2 Tat),asdescribedbyFeinbergetal.(5).Thecontrolpopulations (UI,
ACH-2) weretreated with protamine sulfate alone. Supernatant p24 levelsweremeasuredat I to4daysposttreatment.The resultsshown in thisfigurerepresentonlyone of fiveindependent experiments.
the ACH-2 cellswere not significantly induced. These obser-vationsaretherefore in goodagreementwith the data from the LTR-CAT transfections andsuggestthat the low level of virus production from U1 cells canbeboosted simply by increasing
the levelsof Tat. Incontrast,virusproduction in ACH-2 cells
appears tobe regulated bysome other mechanism.
We have also analyzed the viral RNA after addition of Tat protein. The levels of viral RNA induced by Tat were lower
than those induced by PMA and were not detectable by conventional Northern (RNA) blot hybridization analysis. Therefore, cDNAs were synthesized and amplified by PCR
with total RNAs prepared from Ul and ACH-2 cells treated with Tat. An 800-bp DNA fragment representing 1-actin mRNAwasalsoamplified from thesameRNAas acontrol for
the cDNA synthesis and subsequent amplification. We have previously reported that three major DNA bands are found with primer pair ART7-US (Fig. 4A) and that they are
approximately 400, 220, and 200 bp in length, representing cDNAsformRNAsspecifictoTat, Rev, and Nef,respectively (18). (In the gel shown inFig. 4B, the two DNAfragments of 200 and 220bp ran as one band. The specificity of each band was confirmed by blot hybridization analysis, as described
previously [18].) Untreated U1 and ACH-2 cellsproducedvery
little viral RNA, although a number of PCR analyses
repro-ducibly showed that ACH-2 cells expressed higher basal levels ofmultiply splicedmessagesthan U1cells(Fig. 4B, lanes1and
4; alsosee lane 1 ofFig. lA and B).The levels of viral RNAs
weresignificantly increased aftertreatmentwith PMA inboth cell lines (Fig. 4B, lanes 7 and 8). (Note that because the magnitude of induction by PMA is muchgreater thanthatby purified Tat protein, asmaller number ofcycleswasused for amplification ofcDNAprepared from PMA-treated cells. See the legend to Fig. 4 for details.) Treatment of Ul cells with purified Tat protein for 24 h strongly increased the level of viral RNAs, while there was no significant change in the controlculturecontainingnoTatprotein (Fig. 4B,lanes 2 and
3). On the contrary, addition of Tat protein did nothave any
significant effectonthelevel of viral RNA in ACH-2 cells(Fig.
4B, compare lanes 4and 5 with lane 6). These results again
TAT
%
Conversion
VOL.68, 1994
on November 9, 2019 by guest
http://jvi.asm.org/
[image:3.612.58.297.76.270.2]1996 NOTES
-M Tat,Rev,Nef mRNA
~NetmRNA
II
B r-U1= rACH2i Ul AC
Tat Tat PMA
_-+ _+
-buis 0 24 24 0 24 24 24 24
:% wi;a02
.:~~~~
*--_
I
to:H2
_400bp(Tat) -200/220bp (Nef,Rev)
1 2 3 4 5 6 7 8
~
g
. e Xi--800bp(Actin)Tat RevNef
X O u
IX 0 S, S
|0 -. -.
06
21 n1
FIG. 4. PCR analysis of HIV mRNAs in UI and ACH-2 cells. (A) Locations of oligonucleotideprimers ART7 and US. The thin linerepresents
viral DNA with the flanking LTRs, and thick linesrepresent exons. Solidarrowheads indicate splice donors; openarrowheads indicate splice
acceptors. Nucleotidesare numbered according tothe numbering systemofRatneret al. (25). Specificities ofDNA bandswere confirmed as
previously described in detail (18). (B) PCR analysis of HIV mRNAs. TotalRNAswerepreparedfromU1 andACH-2cells priortoor24 h after
induction with Tat protein orPMA. Following cDNA synthesis with ART7, specificHIV mRNAsequences were amplifiedwith the upstream
primer US. In addition, ,B-actinmRNAwasalso amplifiedtocontrol for the cDNA synthesis and PCR reactions with thesameRNA samples. (The
oligonucleotide primers for ,3-actinwerepurchased fromClontech [Palo Alto, Calif.].) ThesamePCR conditions (30 cycles)wereusedtoamplify thecDNAs from untreated andTat-induced cells (lanes1to6), while only 25 cycleswereusedwith the cDNAs from the PMA-treated cells(lanes
7 and8). The results shown in thisfigureweretaken fromoneof fourindependent experiments, which produced virtually identicaldata.
suggested that Tat might be limiting in Ul cells but not in ACH-2 cells.
In thesimplest interpretation ofourresults, the Ul cell line
appears to direct low levels of LTR transcription because of
suboptimal levels of Tat. Transcription from a transfected
LTR-CAT reporter plasmid can be enhanced 20-fold by
co-transfection withaTat-expression plasmid.Furthermore, viral
p24 production from these cells can be boosted to a similar
degree by incubation with purified Tat protein. Incontrast,the ACH-2 cell line appears tobe abletodirect reasonable levels oftranscription fromatransfected LTR-CAT, andthiscannot be further increased by cotransfection of a Tat expression
plasmid. In addition, virus production is not stimulated in ACH-2 cellsby addition of Tat protein.
Addition ofexogenousTatprotein toUl cells may cause a
temporary increase intranscription, leadingtoincreased
pro-duction ofvirally encoded Tat. This would initiate a positive-feedback loop, and as a result, levels of other viral proteins would increase. The net result would be a shift in viral
transcription toaproductive infection. The underlying mech-anismmaintaining latency is therefore thestatusquolow-level
transcription, which could be broken by increasing levels of Tat. Insupportofthishypothesis,wenotedalarge increasein overall transcription levels in Ul cells upon induction,which
wasnotjust ashift in the types of RNA produced (Fig. IA). While some investigators have found asimilarincrease inthe overall levels of RNA after stimulation (24), others have described a more qualitative than quantitative change in the RNAproduced (21).
This description of Ul latency therefore argues against a
modelof virallatencybasedsimplyonsuboptimal levels of the
Revprotein, asbothwe andothershavepreviously suggested (16, 21, 24).Revprobably dependsonthepresence ofTat for itshigh-level expression, whereasTatexpressionisnot depen-dent on Rev. The apparent Rev- phenotype of latent cells couldalso be describedasalow-level-transcription phenotype.
Enhancing transcription byincreasinglevels of Tatallows the virus toescape from this situation andtriggersacascade into productive infectionwhich involvesother viral geneproducts.
Theunderlyingreasonfor the low level ofactivityof Tat inU1 cells, and therefore the reason for the establishment of the
latent state inthe first place, is still opento speculation.
The blocking ofproductive infection in the ACH-2 cell line isprobably also caused by subcritical levels of LTR transcrip-tion, because RNA levels are low. However, there is clearlya
fundamentally different underlying cause, because the
inte-grated proviral LTR does not behave in the same way as an
exogenous transfected LTR. The reasonable level of CAT
activity obtained with the transfected LTR argues against a
lack ofeither viral or cellularfactorsnecessary for high-level
viral transcription, yet the endogenous proviral LTR is only capable ofproducing small amountsofviral RNA in unstimu-lated cells (Fig. IB). We therefore think that ACH-2 cells probably contain reasonable levels of the cellular and viral factors necessary forefficient LTR transcription but that the
ACH-2proviral LTR somehowcannotusethem effectively. It
has been proposed that transcription from the proviral LTR
can be affected by local conditions at the site of integration, such aschromatin condensationormethylation (1, 9). In such
circumstances, the blocking of proviral transcription mayonly
beovercomeby general cellular activation, and the addition of
moreTatprotein alone hasnoeffect. Insummary,wepropose
thatthe latentstateinbothU1 and ACH-2cell linesis caused by low activity of the endogenous LTRs. The bases for this low-level activity, however, appear to be different in the two cell lines.
We thank Alan Frankel for his generous supply of purified Tat
protein.
This work was supported by the Korea Science and Engineering Foundation(S.K.)and Public Health ServicegrantAl 30897 (S.K.).
REFERENCES
1. Bednarik, D. P., and T. M. Folks. 1992. Mechanisms of HIV-1
latency. AIDS 6:3-16.
2. Cannon, P. M., D. G. Tenen, M. B. Feinberg,H.S.Shin, and S. Kim. 1993. Human immunodeficiency virus-I infection of the humanpromyelocytic cell line HL-60: highfrequency oflow-level
infection and effect of subsequent cell differentiation. Blood 81:437-445.
3. Clouse,K.A.,D. Powell, I.Washington, G. Poli,K. Strebel,W. Farrar, P. Barstad, J. Kovacs, A. S. Fauci,and T.M.Folks. 1989.
Monokine regulation of humanimmunodeficiencyvirus-I
expres-sion in a chronically infected human T-cell clone. J. Immunol.
142:431-438.
4. Duh, E. J., W. J. Maury, T. M. Folks, A. S. Fauci, and A. B.
A
226-250 US
J. VlROI
7982-8002 AR17
m
t
AI
on November 9, 2019 by guest
http://jvi.asm.org/
[image:4.612.83.542.81.204.2]NOTES 1997 Rabson. 1989. Tumor necrosis factor alpha activates human
immunodeficiency virus type 1 through induction ofnuclear factor binding to the NF-KB sites in the long terminal repeat. Proc. Natl. Acad. Sci. USA 86:5974-5978.
5. Feinberg, M. B., D. Baltimore, and A. D. Frankel. 1991. The role of tat in the human immunodeficiency virus life cycle indicates a primary effect on transcriptional elongation. Proc. Natl. Acad. Sci. USA 88:4045-4049.
6. Feinberg, M. B., R. F. Jarrett, A. Aldovini, A. Gallo, and F. Wong-Staal. 1986. HTLV-III expression and production involve complex regulation at the levels of splicing and translation of viral RNA. Cell 46:807-817.
7. Folks, T. M., K. A. Clouse, J. Justement, A. Rabson, E.Duh, J. H. Kehrl, and A. S. Fauci. 1989. Tumor necrosis factor a induces expression of human immunodeficiency virus in a chronically-infected T cell clone. Proc. Natl. Acad. Sci. USA 86:2365-2368. 8. Folks, T. M., J. Justement, A. Kinter, C. A. Dinarello, and A. S.
Fauci. 1987. Cytokine induced expression of HIV-1 in a chroni-cally-infected promonocyte cell line. Science 238:800-802. 9. Folks, T. M., D. M. Powell, M. M. Lightfoote, S.Benn, M. A.
Martin, and A. S. Fauci. 1986.Induction ofHTLV-III/LAVfrom anonvirus-producingT-cell line:implications forlatency. Science 231:600-602.
10. Frankel, A. D. 1992.Activationof HIVtranscriptionby Tat. Curr. Opin.Genet. Dev. 2:293-298.
11. Garcia-Blanco, M. A., and B. R. Cullen. 1991.Molecular basis of latency in pathogenic human viruses. Science 254:815-820. 12. Golden, M. P., S. Kim, S. M. Hammer, E. A. Ladd, P. A. Schaffer,
N. DeLuca, and M. A. Albrecht. 1992. Activation of human immunodeficiency virus by herpes simplex virus. J. Infect. Dis. 166:494-499.
13. Gorman, C. M., L. F. Moffat, and B. H. Howard. 1982. Recombi-nantgenomeswhich expresschloramphenicol acetyltransferasein mammaliancells. Mol. Cell.Biol.2:1044-1051.
14. Griffin,G. E., K. Leung, T. M. Folks, S. Kunkel, and G. J. Nabel. 1989.Activation of HIV geneexpression duringmonocyte differ-entiationbyinduction of NF-KB. Nature (London)339:70-73. 15. Grosscheld, R., and D. Baltimore. 1985. Cell-type specificityof
immunoglobulin gene expression is regulated by at least three DNAsequenceelements. Cell 41:885-897.
16. Kim, S.,R.Byrn, J.Groopman, andD.Baltimore. 1989.Temporal aspectsofDNAand RNAsynthesis during human immunodefi-ciency virus infection: evidencefor differential geneexpression.J. Virol. 63:3708-3713.
17. Kim, S.,K.Ikeuchi, J.Groopman,and D.Baltimore. 1990. Factors affecting cellular tropism of human immunodeficiency virus. J. Virol. 64:5600-5604.
18. Klotman, M. E., S.Kim,A.Buchbinder,A.DeRossi,D.Baltimore,
and F. Wong-Staal. 1991. Kinetics of expression of multiply spliced RNA in early human immunodeficiency virus type 1 infection of lymphocytes and monocytes. Proc. Natl. Acad. Sci. USA 88:5011-5015.
19. Lagakos, S. W., and V. De Gruttola. 1989. The conditional latency distribution of AIDSfor persons infected by blood transfusion. J. Acquired ImmuneDefic. Syndr. 2:84-87.
20. Lifson, A. R., G. W. Rutherford, and H. W. Jaffe. 1988. The natural history of HIV infection. Rev. Infect.Dis.158:1360-1367. 21. Michael, N. L., P. Morrow, J. Mosca, M. A.Vahey, D. S. Burke,
andR. R.Redfield. 1991. Induction of humanimmunodeficiency virus type 1 expression inchronically infected cells is associated primarily with a shift in RNA splicing patterns. J. Virol. 65:1291-1303.
22. Michael, N. L., M. Vahey, D. S. Burke, and R R. Redfield. 1992. Viral DNA and mRNA expression correlate with the stage of humanimmunodeficiencyvirus(HIV)type 1infectionin humans: evidence for viralreplicationinall stages of HIV disease. J. Virol. 66:310-316.
23. Pomerantz, R. J., M. B. Feinberg, D. Trono, and D. Baltimore. 1990. Lipopolysaccharideis apotent monocyte/macrophage-spe-cificstimulator ofhumanimmunodeficiencyvirus type 1 expres-sion. J. Exp. Med. 172:253-261.
24. Pomerantz, R. J., D. Trono,M. B. Feinberg, and D. Baltimore. 1990. Cellsnonproductivelyinfected with HIV-1 exhibitan aber-rant pattern of viral RNA expression: a molecular model for latency. Cell 62:1271-1275.
25. Ratner, L., W. Hasetine, R Patarca, K. J.Livak, B. Starcich, S. F. Josephs, E. R Doran, J. A. Rafalski, E. A. Whitehorn, K. Baumeister, L.Ivanof, S. R.Petterway, Jr., M. R. Pearson,J.A. Lautenberger, T. S.Papas, J. Ghrayeb, N. T.Chang,R.C.Gallo, and F.Wong-Staal. 1985. Complete nucleotide sequence of the AIDSvirus,HTLV-III. Nature (London) 313:277-284.
26. Schnittman, S. M., J. J.Greenhouse, H. C.Lane,P. F.Pierce, and A. S. Fauci. 1991.Frequentdetection ofHIV-1specificmRNAsin infected individuals suggestsongoingactive viralexpressioninall stagesofdisease.AIDS Res. Hum. Retroviruses7:361-367. 27. Trono, D., and D. Baltimore. 1990. A human cell factor is
necessaryforHIV-1 Revaction.EMBO J. 9:4155-4160. 28. Zack, J. A., S. J. Arrigo, S. R. Weitsman, A. S.Go,A.Haislip,and
I.S. Y. Chen.1990. HIV-1 entry into quiescentprimary lympho-cytes:molecularanalysisrevealsalabile,latentviralstructure.Cell 61:213-222.
29. Zack, J. A., A. M. Haislip, P.Krogstad,andL.S. Y. Chen. 1992. Incompletelyreversetranscribed humanimmunodeficiencyvirus type 1 genomesinquiescent cellscanfunctionasintermediatesin theretrovirallifecycle.J.Virol. 66:1717-1725.
VOL.68, 1994