Copyrightq1997, American Society for Microbiology
Protein Interactions in the Herpes Simplex Virus Type 1
VP16-Induced Complex: VP16 Peptide Inhibition and Mutational
Analysis of Host Cell Factor Requirements
KENNETH A. SIMMEN,* ANNE NEWELL,† MICHAEL ROBINSON,‡ JOHN S. MILLS, GAIL CANNING,
RAJ HANDA, KEVIN PARKES, NEERA BORKAKOTI,ANDRAY JUPP†
Roche Research Centre, Welwyn Garden City AL7 3AY, England
Received 18 November 1996/Accepted 31 January 1997
The herpes simplex virus VP16 protein functions as a potent transcriptional activator and targets DNA sites with the consensus TAATGARAT present in all the viral immediate-early gene promoters. To do so, VP16 directs assembly of a multiprotein complex involving two cellular proteins, host cell factor (HCF) and the Oct-1 DNA-binding transcription factor. To investigate the importance of specific protein-protein interactions to formation of this VP16-induced complex (VIC), we used oligopeptides to prevent VIC assembly. Linear and cyclic peptides corresponding to a region of VP16 previously implicated in complex formation were potent inhibitors of VIC assembly. To further characterize the protein interactions involved, we cloned a human cDNA encoding the minimal VP16 interaction domain of HCF, containing amino acids 1 to 380 [HCF (1-380)]. The REHAYS-based peptides active in preventing VIC assembly were found to specifically block binding of VP16 to HCF (1-380), without affecting VP16–Oct-1 binding. The inhibitory activity of these VP16 peptides was strictly sequence specific for the EHAY residues. Site-directed mutagenesis of the HCF (1-380) domain revealed residues E102 and K105 to be critical determinants in support of VIC formation. Alteration of a single residue in HCF, K105, was shown to virtually abolish complex assembly. Interestingly however, none of the HCF mutants that were impaired in their ability to support complex formation exhibited defects in direct VP16 binding, supporting loss of function at a higher order in complex assembly.
Upon infection of permissive cells, herpes simplex virus (HSV) genes are expressed in a temporally regulated manner (18). Induction of the immediate-early (IE) genes requires the action of VP16 (also known as Vmw65 oraTIF), a 490-residue viral late protein which is packaged into the virion tegument (5, 30). VP16 mediates transcriptional activation through DNA motifs with the consensus TAATGARAT (where R is a pu-rine) present in the upstream promoters of all the HSV-1 IE genes (reviewed in references 9 and 32). To associate with the TAATGARAT motif, VP16 forms a heteromeric complex with the ubiquitously expressed cellular proteins, host cell factor (HCF) (46), also referred to as VCAF, C1, and CFF (21, 22, 50), and Oct-1. Oct-1 is a transcription factor of the POU family which recognizes octamer DNA sites (ATGCTAAT) in cellular gene promoters through a bipartite POU domain (17). Although unable to bind DNA independently, VP16 can bind to Oct-1 and the TAATGARAT element in vitro in the ab-sence of HCF. However, TAATGARAT association is greatly enhanced by the presence of HCF, which can bind directly to VP16, promoting formation of the VP16-induced complex (VIC) (24, 36, 44, 46). At the DNA level, recent work by Walker et al. (40) has helped explain the ability of VP16 to target specifically those octamer-related sites which bear a 39
flanking GARAT motif. Their study revealed that VP16 pref-erentially recognizes the Oct-1 POU domain in a conformation that is dependent on the 39 GARAT sequence. Upon VIC
formation, transactivation of the IE gene promoter requires the potent C-terminal acidic activation domain of VP16 re-ported to interact with components of the polymerase II tran-scription apparatus (10, 26, 37, 39). In addition to VP16, the activation domains of Oct-1 may also contribute to full VIC-dependent transactivation (49).
The full-length cDNA for HCF encodes a nuclear precursor protein of 2,035 amino acids which is proteolytically processed at a set of conserved 26-residue repeats to a family of shorter polypeptides which remain stably associated (23, 46, 48). This processing is not thought to be essential for the role of HCF in VIC formation, however, as an N-terminal domain of HCF is sufficient for VP16 interaction and VIC formation (45). In addition to acting as a VP16 accessory protein during viral infection, HCF presumably has an important cellular role, but so far this remains undefined. The many protein-protein and protein-DNA interactions in formation and function of VIC make it an ideal model for studying multiprotein-DNA tran-scriptional complexes. In addition, disruption of VIC forma-tion may offer an approach to blocking HSV IE gene activaforma-tion and, hence, viral replication. The importance of VP16 trans-activation in lytic HSV replication is highlighted by a virus mutant with an insertion in the VP16 transactivation domain (in1814), which is impaired in both IE gene activation and viral replication at low multiplicities of infection (2). Furthermore, propagation of HSV carrying a deletion of the entire VP16 coding region requires growth in VP16-complementing cells (41).
In this study, we have used a combination of approaches to investigate molecular interactions within the VP16-induced complex, looking in particular at VP16 and HCF requirements. Using VP16-based peptides we have demonstrated sequence-specific disruption of VIC formation and investigated which protein-protein interactions were being disrupted by using
re-* Corresponding author. Mailing address: Roche Research Centre, 40 Broadwater Rd., Welwyn Garden City AL7 3AY, England. Phone: 44-1707-366427. Fax: 44-1707-332053.
† Present address: Rhone-Poulenc Rorer, Dagenham, Essex RM10 7XS, England.
‡ Present address: Windeyer Institute for Medical Sciences, London W1P 6DB, England.
3886
on November 9, 2019 by guest
http://jvi.asm.org/
formed to amplify the DNA encoding the N-terminal 380 amino acids of HCF with primers based on the published sequence (47). The resulting 1.1-kb NheI/HindIII fragment was subcloned into plasmid pDS56/RBSII 63His (38), and then an EcoRI/HindIII fragment was further subcloned into M13mp18. Oligonucleotide-directed in vitro mutagenesis of HCF (1-380) was performed with the Sculptor II kit (Amersham) with single-stranded M13 template DNA and the following oligonucleotides (59–39): M1 (GCGGTGGCCGTGTTCGGG CCGGAACACCGGACCCGA), M2 (CGTAGAGGTCATTGCTGATATCCC CATACTCCACCATC), M3 (GTCTTTGCTTTGAGCGGCTTCCAGTGCCA CCGGCTCGC), M4 (CCGTTTTTGGGCGTCTCTGCCGGGAGTCTCTTCC ACTC), M5 (GTTGGTACACTTCCACCGCGGCTCGTGTGTGGCCAC), K105D (GAGGTCATTGCTGTAATCCCCATACTCCACC), Y106I (CGTAG AGGTCATTGCTGATTTTCCCATACTCCACC), E102A (GCTGTATTTCC CATACGCCACCATCCCACC), N108A (GAGTTCGTAGAGGTCCGCGCT GTATTTCCC), and D109A (CTGGAGTTCGTAGAGCGCATTGCTGTATT TCC).
The correct mutations in the resulting constructs were verified by DNA se-quencing. DNA fragments encoding the mutated HCF inserts were then sub-cloned as NdeI (Klenow-repaired)/HindIII fragments into pBSK1(Stratagene) digested with SalI (Klenow-repaired)/HindIII. The resulting plasmids facilitated T7 RNA polymerase transcription of sense-strand HCF (1-380) RNAs. The bacterial expression plasmid pVP16 encoded amino acids 1 to 490 of HSV-1 VP16 fused to an N-terminal six-histidine tag which facilitated metal chelate affinity purification of the recombinant protein. Similarly, the bacterial expres-sion plasmid pOct1-POU encoded residues 266 to 446 of human Oct-1 encom-passing both the POU homeodomain and POU-specific domain (17), fused to an N-terminal six-histidine tag after subcloning into pDS56/RBSII 63His (38).
Plasmids pRIT2T, pRIT-VP16, and pRIT-POU encoded the Staphylococcus aureus protein A alone (28), protein A fused in frame to HSV-1 VP16, and protein A fused in frame to the Oct-1 POU domain, respectively. Plasmids pet15bOct-1 and pT7-Jun were used as templates for in vitro transcription-translation of full-length human Oct-1 and c-Jun, respectively (27).
Protein purification.Histidine-tagged recombinant POU and VP16 proteins were induced and purified essentially as described previously under nondenatur-ing conditions (20). Purified fractions of POU and VP16 were judged to be
.90% pure by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and Coomassie blue staining. The protein A fusions were expressed and purified according to the manufacturer’s instructions (Pharmacia).
Nuclear extracts were prepared from HeLa cells according to the method of Dignam et al. (8).
Peptide synthesis.Cyclic peptide cyc6 (Ro 32-1297) was prepared by manual solution phase chemistry with a benzyloxycarbonyl (Cbz)/tBu protection strategy.
Successive couplings of 1.1 equivalents of Cbz amino acid to alanine methyl ester were performed with water-soluble carbodiimide (1.1 equivalents) in the pres-ence of 1-hydroxybenzotriazole (1.1 equivalents) and N-ethylmorpholine (1.15 equivalents), and the cyclization was achieved by slow addition of the N- and C-deprotected peptide to the same activating reagents. Side-chain protections for amino acids were Glu (tBu), Arg (Pmc), His (Bum), Tyr (tBu), and Ser (tBu).
Side-chain deprotections and purification were achieved under the same condi-tions as those used in the solid-phase peptide syntheses. Purity of the final product was assessed by analytical reverse-phase high-pressure liquid chroma-tography (HPLC), and the structure was validated by mass spectrometry and proton nuclear magnetic resonance.
cyc12 (Ro 32-1558) was also prepared by block coupling of N- and C-depro-tected portions of the Ro 32-1297 precursor, to give a 12-mer which was cyclized, deprotected, and purified in the same way.
Subsequently, cyc12 was also prepared by solid-state synthesis, as were the linear Ro 32 peptides, on a Milligen 9050 Pepsynthesizer under continuous flow conditions employing 9-fluorenylmethoxycarbonyl (Fmoc)/tBu chemistry. Pepsyn
KA resin was used for the synthesis of linear peptides. A fourfold excess of Fmoc amino acids was used, and the activation was carried out by TBTU (4 equiva-lents) in the presence of N-ethylmorpholine (8 equivaequiva-lents). Side-chain protec-tions for amino acids were Asp, Glu (OtBu), Arg (Pmc), His (Trt), and Ser (tBu).
For peptides cyc12 and cyc14 (Ro 32-1981), Pepsyn KH resin, which provided the linear precursors in the form of N- and C-deprotected fully side-chain-protected peptides which were cyclized in solution was used. Conditions for resin and side-chain deprotections were trifluoroacetic acid
(TFA):phenol:1,2-ethanedi-buffer (50 mM NaCl, 0.5% Nonidet P-40, 5 mM EDTA, 1 mM dithiothreitol, 20 mM HEPES [pH 7.9]) in a volume of 100ml. The volume was then increased to 1 ml with pull-down buffer, and 10ml of immunoglobulin G (IgG)-Sepharose beads (Pharmacia) (50% slurry) was added prior to mixing at 48C for 20 min. IgG-bound complexes were collected by centrifugation and washed three times in pull-down buffer to remove any residual radiolabelled proteins nonspecifically bound to the beads. Samples were resuspended in SDS-containing protein sam-ple buffer and separated by SDS-PAGE on a 12% polyacrylamide gel, followed by autoradiography of the dried gel.
In the peptide competition assay, peptides were preincubated with the trans-lations for 5 min prior to the addition of protein A-VP16, after which the binding reactions continued for a further 25 min. The peptides were used at final con-centrations of 1, 10, and 100mM.
Gel shift assay.Proteins (10mg of nuclear extract and 4 ng of VP16 [see Fig. 2] and 4 ng of VP16, 0.5 ng of POU, and 2 to 5ml of the HCF translations [see Fig. 6A and 7]) were incubated for 20 min at room temperature in 25ml of assay buffer [25 mM HEPES [pH 7.9], 5 mM dithiothreitol, 1 mM EDTA, 50 mM NaCl, 4% Ficoll 400, 0.05% Nonidet P-40, 100mg of bovine serum albumin per ml, 1mg of poly(dI-dC), 10 ng of pBSK1] with 0.5 pmol ofa-32P-labelled probe
DNA containing the TAATGARAT motif (59-CGTGCATGCTAATGATATT CTTT-39) of the HSV-1 ICPO gene. To assay for inhibition of VIC formation, VP16 peptides were preincubated for 10 min with HeLa cell extract prior to addition of VP16 and the probe. Protein-DNA complexes were resolved by electrophoresis for 90 min at room temperature on a 4% nondenaturing poly-acrylamide gel containing 0.53Tris-borate-EDTA. The gels were dried and subjected to autoradiography at2708C.
Protease cleavage assay.Proteolysis reactions were performed in 25ml of phosphate-buffered saline, using 3 to 5ml of radiolabelled HCF (1-380) in vitro translations, in the presence or in the absence of porcine trypsin or Enteroki-naseMax (Invitrogen). The concentrations of enzyme required to generate a cleavage pattern for HCFwt (1-380) were determined in preliminary assays, and these concentrations were used thereafter. Following incubation at 378C for 2 h (enterokinase) or 30 min (trypsin), SDS-containing protein sample buffer was added to halt the reaction, and samples were subjected to SDS-PAGE on 16% polyacrylamide gels followed by autoradiography of the dried gels.
RESULTS
Disruption of VIC formation with VP16 peptides. Earlier
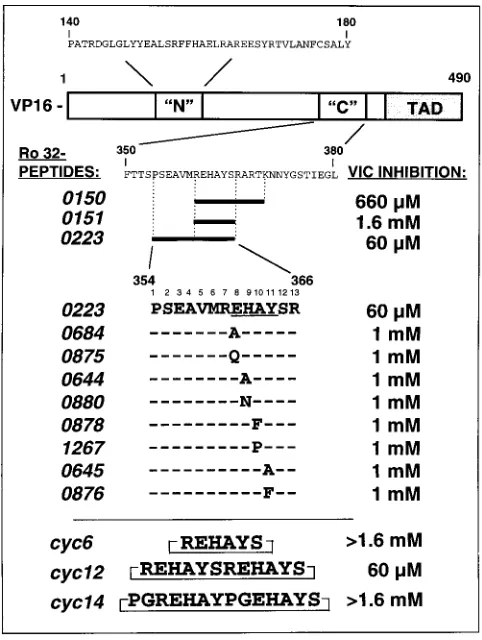
studies of the VP16 requirements for VIC formation and IE gene activation suggested two regions within the first 388 res-idues as likely to be important (Fig. 1). Deletion or mutation of residues within these regions led to concomitant loss of both VIC formation and TAATGARAT-dependent transactivation (1, 11, 12, 39, 42, 43). To further define their roles, we designed peptides based on VP16 and assayed them initially for their ability to disrupt VIC formation in gel shift assays. Peptides were assayed at concentrations of up to 1.6 mM by preincuba-tion with HeLa cell nuclear extract prior to addipreincuba-tion of VP16 and radiolabelled TAATGARAT-containing probe, and pro-tein-DNA complexes were resolved by gel electrophoresis. A series of peptides based on the N region (Fig. 1) were found to be inactive at blocking VIC formation (data not shown). In contrast, the 11-mer peptide Ro 32-0150 was reproducibly able to prevent VIC detection completely at 660mM and partially at 200mM (Fig. 1). This peptide corresponds to amino acids 360 to 370 of VP16 previously implicated in VIC formation (12– 14). Truncation of the Ro 32-0150 peptide resulted in a loss of VIC competition, with the 7-mer peptide Ro 32-0151 being active only at the highest concentration assayed, 1.6 mM. Strik-ingly, Hayes and O’Hare have shown that an 8-mer
on November 9, 2019 by guest
http://jvi.asm.org/
passing one more C-terminal residue (REHAYSRA) than our Ro 32-0151 peptide (REHAYSR) retains a potent inhibitory effect, pointing to a minimum-length or sequence-specific re-quirement for activity (14). N-terminal extension of the Ro 32-0151 sequence in peptide Ro 32-0223 generated a more potent competitor, exhibiting complete inhibition reproducibly at 60mM (Fig. 1; Fig. 2, lanes 3 to 7). Ro 32-0223 (PSEAVM REHAYSR) encompasses VP16 residues reported to be sur-face exposed and implicated in protein-protein interactions with either Oct-1 or HCF (14, 29). To address the importance of individual amino acids, single residue substitutions were introduced into a series of peptides based on Ro 32-0223. The sequences and activities of some of these peptides in VIC inhibition are shown in Fig. 1. Changing peptide residues S2, E3, R7, S12, and R13 to alanine had no deleterious effect on competitive activity, as those substituted peptides all blocked VIC formation at 60 mM (data not shown). By contrast, all substitutions at positions 8 to 11 in the peptide (Fig. 1) resulted in substantially (.15-fold) reduced competitive activity, impli-cating those residues as critical in VIC assembly. Our data confirm and extend other reports pointing to the importance of
this region of VP16 in assembly (13, 14, 49) and demonstrate that all four positions in the EHAY motif are critical.
To address the influence of peptide secondary structure, we synthesized a series of three cyclized REHAY-based peptides to determine whether conformational restriction would lead to enhanced efficacy in VIC competition. In addition, cyclization of short peptides is generally considered to enhance peptide stability. The cyc6 REHAYS peptide was inactive at concen-trations up to 1.6 mM in our gel shift competition assay (Fig. 1). However, presentation of the REHAY motif in the cyc12 peptide resulted in VIC inhibitory activity (100% at 60mM) identical to that of the linear Ro-32-0223 peptide (Fig. 1; and Fig. 2, lanes 3 to 12). Strikingly, the cyc14 peptide, in which additional glycines and prolines had been introduced, was in-active at up to 1.6 mM (Fig. 2, lanes 13 to 17) suggesting those alterations of the primary sequence to have resulted in presen-tation of the REHAY motif in an inactive conformation. In summary, although conformational restriction by cyclization did not result in enhanced competitiveness, the data do sup-port length- and sequence-specific requirements for peptide activity, which may be interpreted as evidence for a conforma-tional dependence.
VP16 REHAY peptides specifically disrupt HCF-VP16
inter-action. The ability of VP16 peptides to interfere with VIC
[image:3.612.58.299.68.388.2]assembly in nuclear extracts suggests that the corresponding region of VP16 contacts either Oct-1 or HCF. To address whether VP16-HCF or VP16–Oct-1 interactions were being disrupted, we first established in vitro protein-protein interac-tion assays with recombinant proteins. VP16 was expressed and purified from bacteria as a protein A fusion (pA-VP16), thus facilitating IgG-Sepharose pull-down assays to look at associated proteins. Following a recent report that only the first 380 amino acids of HCF were necessary and sufficient for VIC formation (45), we generated a HeLa cell cDNA that encoded HCF residues 1 to 380 by reverse transcription and PCR (see Fig. 5). To verify that our HCF (1-380) cDNA encoded a product competent for VP16 association, we incu-bated an [35S]methionine-labelled HCF (1-380) in vitro trans-lation (Fig. 3, lane 1) with a range of purified protein A (pA) fusions. Incubation with pA-VP16 (Fig. 3, lane 4) resulted in greater association with radiolabelled HCF (1-380) than with pA alone (lane 3), pA-POU (lane 5), or IgG-Sepharose beads alone (lane 2). Consistent with this binding to VP16, our in
[image:3.612.328.556.72.197.2]FIG. 1. The ability of VP16 peptides to block formation of the VP16-induced complex. The primary structure of VP16 is represented at the top, and the N and C regions to which peptides were designed are indicated. The numbering refers to residues of VP16. The transactivation domain (TAD), which is known to be dispensable for VIC formation, is also depicted. Peptides based on the C region were assayed for their ability to inhibit formation of VIC in gel shift assays with HeLa nuclear extract and recombinant VP16. A series of single amino acid substitutions were made in the Ro 32-0223 peptide sequence background. Shown at the bottom are the three cyclic peptides used. The stated concentration of peptide required to reproducibly inhibit VIC formation by 100% was derived from several assays performed with peptides used at concentrations of 1, 6, 20, 60, 200, and 660mM and 1 and 1.6 mM.
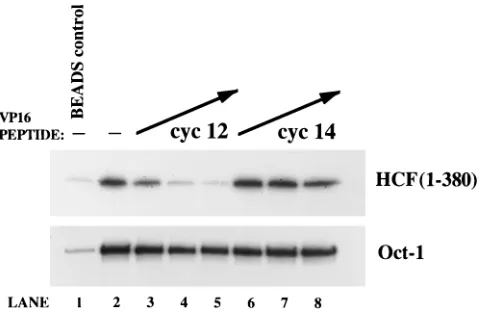
FIG. 2. Disruption of VIC formation by linear and cyclic VP16 peptides in vitro. Gel shift binding reactions were performed with HeLa cell nuclear extract (N.Xt) and recombinant VP16, except for lane 2 which contained only nuclear extract. The positions of the VIC and the Oct-1–DNA complexes are indicated. The VP16 peptides Ro 32-0223, cyc12, and cyc14 were assayed for their ability to disrupt VIC formation by preincubation with nuclear extract at final concentra-tions of 1, 6, 20, 60, and 200mM (lanes 3 to 7, 8 to 12, and 13 to 17, respectively).
on November 9, 2019 by guest
http://jvi.asm.org/
vitro-translated HCF (1-380) was also competent for VP16-induced complex formation in gel shifts (see below).
Having established that our HCF (1-380) protein was rec-ognized by pA-VP16, we utilized the same interaction assay to address whether the VP16 peptides disrupted VP16–HCF (1-380) or VP16–Oct-1 binding. In Fig. 4, the cyc12 and cyc14 peptides were assayed by preincubation with either radiola-belled in vitro-translated HCF (1-380) (upper panel) or Oct-1 (lower panel), prior to addition of pA-VP16 and subsequent IgG bead pull down. Relative to binding in the absence of competitor peptide (Fig. 4, lane 2), preincubation with increas-ing concentrations of cyc12 (Fig. 4, lanes 3 to 5) resulted in decreased binding of pA-VP16 specifically to HCF (upper pan-el), without affecting pA-VP16–Oct-1 binding (lower panel). Addition of 10 mM cyc12 peptide was sufficient to decrease VP16–HCF (1-380) binding to the level of background HCF pull down by the IgG beads alone in this assay (compare lanes 4 and 1, Fig. 4, upper panel). In contrast to cyc12, addition of the cyc14 peptide had no effect on either VP16-HCF or VP16– Oct-1 interactions (Fig. 4, lanes 6 to 8), in agreement with its inactivity in the gel shift competition assay.
That the observed peptide competition of binding of VP16 to HCF (1-380) is sequence specific to the particular region of
Identification of HCF residues critical for Mini-VIC
forma-tion. Having mapped the target of the VP16 peptides to the
N-terminal 380 amino acids of HCF, we then sought to further characterize this domain of HCF and identify amino acids critical to VP16-induced complex formation.
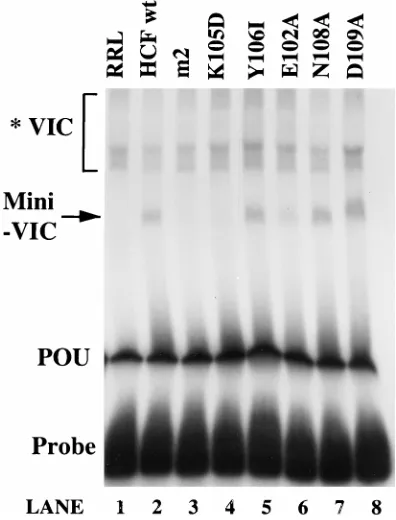
To allow a well-defined analysis of HCF requirements, we utilized a gel shift assay with recombinant VP16 and recombi-nant Oct-1 POU domain. Binding of the POU domain to radiolabelled TAATGARAT-containing probe generated a protein-DNA complex (see Fig. 6A). Like Walker et al. (40), under certain assay conditions we could detect binding of VP16 to the POU-DNA complex in the absence of HCF (data not shown). However, as we sought to probe HCF require-ments, we utilized buffer conditions and protein concentrations at which HCF was strictly required for complex formation. Under these conditions, POU binding was unaffected by the presence of VP16 (see Fig. 6A, lane 1). Addition of in vitro-translated HCF (1-380) in the preincubation reaction mixture resulted in a series of novel retarded complexes (see Fig. 6A; compare lane 1 with lanes 4 to 5). The complex indicated that Mini-VIC was dependent on the presence of POU, VP16, and RRL programmed with HCF (1-380) RNA, as it was absent when control unprogrammed RRL was used (see Fig. 6A; lanes 2 and 3) and could be supershifted by addition of anti-bodies directed against VP16 or the Oct-1 POU domain (data not shown). The term Mini-VIC highlights that this complex contains the minimal domains of both HCF and Oct-1 neces-sary for association with VP16 and is consequently smaller than complexes harboring full-length HCF. When using RRL in our gel shifts we consistently detected complexes of slower mobility than Mini-VIC, which could be similarly supershifted (data not shown); we conclude that these complexes, desig-nated *VIC, are dependent on HCF activity endogenous to the lysate (see Fig. 6A).
As an approach to mapping HCF amino acids crucial to VIC formation, surface exposure probabilities and secondary struc-ture analysis of the HCF (1-380) sequence were used to iden-tify potential protein interaction sites. To determine whether the regions thus identified are important for VIC assembly, we introduced a series of double residue substitutions by site-directed mutagenesis into the HCF (1-380) cDNA to generate the mutant HCF constructs m1 through m5 (Fig. 5). When assayed for their ability to support Mini-VIC formation in the gel shift assay, in vitro-translated m1, m2, and m3 HCF mu-tants exhibited severely reduced activity compared to the HCFwt (1-380) translation (Fig. 6A, lanes 4 to 11). In contrast, the m4 and m5 mutants supported wild-type or even enhanced Mini-VIC assembly (Fig. 6A, lanes 12 to 15). The minor vari-ations in detection of *VIC (see the legend to Fig. 6A) when different HCF translations were used suggest that HCF intrin-sic to the lysate and the translated HCF (1-380) proteins may be in competition for the supplied VP16 and POU.
To address whether the loss of function in the m1, m2, and m3 HCF proteins results from localized structural changes in
[image:4.612.59.300.496.652.2]radiolabelled HCF input; lane 2, control pull down with IgG beads in the absence of any protein A fusion; lanes 3 to 5, pull downs following incubation of HCF (1-380) with protein A (pA), pA-VP16, and pA-POU, respectively.
FIG. 4. Inhibition of VP16 binding to HCF (1-380) by VP16 peptides. The cyc12 and cyc14 peptides were assayed for their ability to disrupt the interaction of pA-VP16 with radiolabelled HCFwt (1-380) (4ml) (upper panel) and Oct-1 (2
ml) (lower panel) in pull-down assays. Lane 1, pull downs performed with no pA-VP16; lanes 2 to 8, pull downs following incubation with pA-VP16. Lanes 3 to 5 and 6 to 8, pA-VP16 pull downs following preincubation of the in vitro translations with VP16 peptides as indicated at final concentrations of 1, 10, and 100mM. Lane 2, pA-VP16 pull downs performed in the absence of any compet-itor peptides.
on November 9, 2019 by guest
http://jvi.asm.org/
HCF regions normally implicated in interactions necessary for Mini-VIC assembly or from a more global misfolding due to conformational changes induced by the substitutions, we used the technique of limited proteolysis to address the conforma-tion of the proteins (31). Figure 6B shows that incubaconforma-tion of radiolabelled HCFwt (1-380) with enterokinase at two differ-ent concdiffer-entrations generated a characteristic pattern with two prominent cleavage products. Comparison of the HCFwt pro-teolysis pattern with that obtained with equivalent amounts of the radiolabelled HCF mutants revealed that two of the three proteins defective in Mini-VIC assembly, namely m1 and m3, were refractory to proteolysis under the conditions active for HCFwt cleavage (Fig. 6B, lanes 4 to 6 and 10 to 12). However, the remaining HCF protein defective in VIC formation, m2, displayed a proteolysis profile with enterokinase identical to that seen with the wild-type protein (Fig. 6B, lanes 7 to 9). The two other HCF mutants found to be wild type in supporting Mini-VIC assembly, m4 and m5, generated a weak cleavage pattern similar, but not identical, to that of the HCFwt (1-380) pattern. These data are supported by similar findings obtained when proteolysis was performed with trypsin instead of en-terokinase (Fig. 6C). As with enen-terokinase, limited trypsin digestion of HCF m2 generated a cleavage pattern identical to that observed with the HCFwt (1-380) (Fig. 6C, compare lanes 1 to 3 with lanes 7 to 9). Although m1 and m3 were clearly susceptible to proteolysis at the higher concentration of trypsin used (Fig. 6C, lanes 4 to 6 and 10 to 12), they generated cleavage products of sizes differing from those seen with HCFwt and HCF m2.
We interpret these results to indicate that of the three pro-teins defective in Mini-VIC assembly, only m2 is probably folded in a wild-type conformation, while m1 and m3 have adopted non-wild-type conformations. Therefore, we consider
m2 to be the only HCF (1-380) protein in this series (Fig. 5) which displays a Mini-VIC assembly defect attributable to a local alteration in structure at the site of the substitutions.
Single residue substitutions in HCF (1-380) can
compro-mise Mini-VIC assembly.To further investigate the possibility
that the HCF region encompassing the residues mutated in HCF m2 is an important determinant in Mini-VIC assembly, we engineered a series of single amino acid substitutions be-tween HCF residues 100 and 110. To probe whether the m2 phenotype in Mini-VIC assembly could be attributed to either of its two changes, constructs HCF (1-380) K105D and Y106I were made. In addition, three alanine substitutions, in each case altering a charged residue (at E102, N108, and D109), were designed to address whether positions flanking the orig-inal m2 site were also sensitive to mutation.
[image:5.612.59.297.69.311.2]These in vitro-translated HCF mutants (called the m2 se-ries) were assayed for support of Mini-VIC formation by gel shift (Fig. 7). As seen in Fig. 6A, the HCF m2 translation was severely compromised for Mini-VIC formation compared to HCFwt (Fig. 7, lanes 2 and 3). This defect mapped to alter-ation of the lysine at HCF position 105, rather than the adja-cent tyrosine at 106, as Y106I displayed wild-type Mini-VIC
[image:5.612.319.552.69.352.2]FIG. 5. Targeted mutagenesis of HCF (1-380). HCF is depicted with the regions involved in VP16 interaction and proteolytic cleavage outlined. The 380-amino-acid N-terminal VP16 interaction domain is shown expanded. Using Genetics Computer Group secondary structure predictions from the primary sequence, regions expected to be surface exposed and involved in structural folds or turns were identified. A series of double amino acid mutations was introduced into the HCF (1-380) cDNA in constructs m1 to m5.
FIG. 6. Function and conformation of the HCF (1-380) mutants. (A) Unla-belled in vitro translations of HCFwt (1-380) or mutant constructs assayed with recombinant VP16 and Oct-1 POU for association with a radiolabelled TAAT GARAT-containing DNA probe by gel shift. Lane 1, probe, POU, and VP16. The complex designated POU is the same as that formed in the absence of VP16 (data not shown). Lanes 2 and 3, POU and VP16 supplemented with 2 or 5ml, respectively, of unprogrammed RRL. Lanes 4 to 15, 2 and 5ml, consecutively, of each HCF translation. The position of the complex formed with the HCF (1-380) proteins (Mini-VIC) is indicated by an arrow. A higher complex (*VIC), depen-dent on HCF activity endogenous to the RRL, is indicated by a bracket. (B) Proteolytic analysis of the radiolabelled HCF (1-380) proteins. Each of the HCF in vitro translations was incubated with buffer alone (2) or 0.1 (1) or 2 (11) U of enterokinase at 378C for 2 h and analyzed by SDS-PAGE. (C) Proteolytic analysis of the radiolabelled HCF (1-380) proteins. Each of the HCF in vitro translations was incubated with buffer alone (2) or 12.5 (1) or 50 (11) ng of trypsin at 378C for 30 min and analyzed as for panel B.
on November 9, 2019 by guest
http://jvi.asm.org/
formation (Fig. 7, lane 5), whereas K105D had considerably reduced activity (Fig. 7, lane 4). Introduction of alanines at positions 108 and 109 had no deleterious effect on Mini-VIC formation (Fig. 7, lanes 7 and 8). However, introduction of an alanine at residue 102 led to moderately decreased complex formation (Fig. 7, lane 6). Limited proteolysis analysis with enterokinase and trypsin confirmed that all of the HCF single amino acid substitutions retained cleavage patterns identical to those of HCF m2 and HCFwt (data not shown), consistent with similar protein folding. These data suggest that HCF residues 102 to 105 constitute at least part of a determinant critical for functional participation in VIC formation.
The defect in Mini-VIC formation of the HCF m2 mutants is
not at the level of VP16 association.Having generated mutants
of HCF (1-380) which exhibit defective complex formation, we then attempted to determine the nature of the interaction which was being prevented or disrupted. As HCFwt (1-380) binds pA-VP16 in vitro (Fig. 3), the simplest explanation would be that the HCF (1-380) m2 mutants are defective in VP16 association. Therefore, we tested the m2 series of HCF pro-teins for binding to pA-VP16 in our pull-down assay. Figure 8A shows aliquots of the radiolabelled inputs of HCFwt and the HCF m2 series, as well as human c-Jun used as a control protein. All of the HCF proteins were proficient in VP16 binding, as detected following incubation with pA-VP16 and IgG bead pull downs (Fig. 8B). Those differences between levels of different HCF mutants which were observed after pA-VP16 pull down paralleled minor differences in the corre-sponding inputs (compare Fig. 8A and B). That the pA-VP16 interaction with the HCF translations is specific was supported by the lack of binding to radiolabelled c-Jun. Furthermore, the levels of HCF pull down observed in Fig. 8B were dependent
on the presence of pA-VP16, as control pull downs in the absence of pA-VP16 resulted in considerably lower HCF levels (Fig. 8C). This ability of the HCF mutants to bind VP16 was further verified when the assay was repeated under different salt and detergent concentrations, with the same results (data not shown). From these assays, we conclude that the defect in productive Mini-VIC formation of HCF mutants m2, K105D, and E102A does not reflect any inability to interact with VP16. Possible functions of HCF which are compromised by alter-ation of the surface around residues 102 to 105 are discussed below.
DISCUSSION
[image:6.612.315.553.70.245.2]In this study, we have characterized several of the multiple interactions necessary for the HSV activator VP16 to mediate assembly of a transcription factor complex. We utilized two different approaches, (i) VP16 peptide competition of complex formation and VP16-HCF/Oct-1 binding and (ii) mutagenesis of HCF residues to study the requirements for HCF function in complex formation. Previous studies of VP16 had revealed the requirements for transactivation and VIC assembly to map to separable domains (7, 11, 12, 33, 39): the N-terminal 388 residues being sufficient for complex formation and the C-terminal transactivation domain being dispensable for complex formation. Using gel shift assays, we analyzed peptides based on VP16 for their ability to prevent VIC formation. Although residues 140 to 180 had been implicated in VIC formation and transactivation by mutational analysis (1, 42, 43), we found peptides based on this region to be ineffective competitors, in agreement with a previous report (13). In contrast, peptides corresponding to the surface-exposed region of VP16 around residues 360 to 370 were highly effective competitors, showing complete inhibition at 60 mM peptide, in accordance with other studies which support an important role for this VP16 region (13, 14, 49). Although cyclization of the REHAY se-quence did not enhance the inhibitory activity of the cyc12 peptide relative to the linear Ro 32-0223 peptide, we did gain support for a conformational requirement, as the cyc6 and cyc14 peptides, both of which contained the REHAY motif, were inactive in our assay. Substitution of single amino acids
[image:6.612.79.277.71.331.2]FIG. 7. Single residue changes in the HCF (1-380) m2 region are sufficient to disrupt Mini-VIC formation. The activity of the m2 series of HCF mutants in Mini-VIC formation. Unlabelled in vitro translations (3ml) of the HCF proteins carrying the substitutions indicated were assayed in the gel shift exactly as for Fig. 6A. Every lane contained POU and VP16. Lane 1, unprogrammed RRL. Mini-VIC and *Mini-VIC are as described in the legend to Fig. 6A.
FIG. 8. HCF mutants defective in VIC formation are competent for VP16 association. In vitro pull-down assays with pA-VP16 were performed with radio-labelled HCF (1-380) proteins or human c-Jun, as indicated above the lanes. (A) Aliquots of the in vitro translations (1/4 of the input into each assay). (B) IgG-Sepharose pull downs following incubation with pA-VP16. (C) Pull downs performed in the absence of pA-VP16.
on November 9, 2019 by guest
http://jvi.asm.org/
highlighted the importance of four positions to inhibitory ac-tivity: these corresponded to VP16 residues 361 to 364, EHAY. Although revealed as critical by their sensitivity to substitution, these four positions are not in themselves sufficient to bind to a cellular target(s), as our Ro 32-0151 REHAYSR and cyc6 REHAYS peptides were ineffective competitors compared to longer peptides. The EHAY residues presumably comprise part of a larger functional surface defined by positions 365 to 378, as single substitutions between 373 and 378 in VP16 (map-ping C-terminal to our peptides) virtually abolish both VIC formation and VP16 transactivation (12, 14).
Previous studies have suggested, but not proven, that the cellular target of REHAY-based peptides and, by extension, of the corresponding region of VP16 in VIC assembly, was HCF (13, 14, 49). However, an overlapping VP16 peptide (contain-ing residues 360 to 391) has been reported to bind the Oct-1 POU domain (36); but see also reference 14). To definitively address whether VP16-HCF or VP16–Oct-1 interactions were targeted by our peptides, we established in vitro pull-down assays with recombinant proteins. Following evidence that the N-terminal 380 amino acids of HCF were necessary and suffi-cient for VIC formation (45), we cloned a cDNA from HeLa cells encoding that domain and expressed the protein in RRL. Our data confirmed that HCF (1-380) was capable of VP16 interaction and participation in VIC formation. Strikingly, pre-incubation with VP16 peptides inhibitory to VIC formation was shown to specifically block HCF (1-380)–VP16 interaction, with no apparent effect on Oct-1–VP16 binding. Consistent with this observed inhibition being relevant to our VIC com-petition data, peptides inactive for VIC comcom-petition were found to have no effect on HCF (1-380)–VP16 binding. This, we believe, represents the first demonstration with recombi-nant proteins, as opposed to partially purified fractions, that REHAY peptides interfere specifically with HCF-VP16 asso-ciation, and we infer that this region of VP16 is bound to HCF in the course of VIC assembly. In support of HCF-VP16 in-teraction also being critical to VP16 transactivation, Wu et al. (49) recently demonstrated inhibition of VP16 transactivation by using VP16 peptides similar to ours. Their findings, together with our data, strongly suggest that disruption of the VP16-HCF interaction leads to loss of VIC function and contradict a recent report which argued that VP16-HCF binding was not essential for VP16 activation (34).
Although our cyc12 and Ro 32-0223 peptides, which did not compete for VP16–Oct-1 association, extended only to VP16 position R366, we note that the peptide reported by Stern and Herr (36) to bind the Oct-1 POU domain contained residues 360 to 391 of VP16. Thus, we suggest that if Oct-1 is a target for this adjacent region of VP16, the N-terminal boundary on VP16 would map C-terminal to position 366. If HCF and Oct-1 do interact with adjacent VP16 sequences, then they may be juxtaposed, raising the possibility of direct HCF–Oct-1 bind-ing. However, our finding that HCF (1-380) binds to pA-POU no better than to pA alone argues against HCF–Oct-1 inter-action. Although the pA-POU protein used had much of Oct-1 deleted, the POU domain would be a likely candidate for any potential HCF interaction surface on Oct-1, as the determi-nants of Oct protein specificity in support of VIC function map exclusively to that domain (25).
Having demonstrated that the association between the N-terminal domain of HCF and VP16 is a critical step in VIC assembly, we then addressed the HCF requirements in more detail. Many studies have used crude deletions or linker inser-tions or deleinser-tions to delineate regions important to protein function. However, a major disadvantage with this approach is the potential for changes in protein folding resulting from the
relatively large alterations made. Therefore, we used site-di-rected mutagenesis to introduce double substitutions into gen-erally charged regions of HCF (1-380) predicted to be both surface exposed and structured and assayed their effect on HCF (1-380) function. In addition, we addressed the effect of the mutations on protein folding. Of the five mutant HCFs tested, three were defective in our VIC gel shift assay. How-ever, limited proteolysis of radiolabelled proteins revealed that only one of these, m2, had retained a conformation indistin-guishable from that of HCFwt (1-380). The use of more than one protease helped to safeguard against any spurious differ-ences in cleavage profiles which may have resulted from direct mutation of cleavage sites within HCF. Such findings highlight the importance of assessing the folding status of proteins al-tered in primary sequence. We propose that the defect in VIC assembly with m2 reflects the local effect of the introduced mutations rather than an indirect effect on the global confor-mation of the protein. Analysis of single substitutions between HCF residues 102 and 109 encompassing the m2 positions confirmed the requirement for particular amino acids for effi-cient VIC formation. Positions E102 and K105 were both sen-sitive to alteration, perhaps implicating their side chains as critical determinants in VIC assembly.
Following our earlier demonstration of VP16 binding to HCFwt (1-380), the defect in the HCF mutants like m2, K105D, and E102A may have resulted from disruption of this interaction. Interestingly however, all of those mutant HCF proteins were competent for VP16 binding in vitro, implicating a loss of function at an event in VIC assembly subsequent to, or independent of, HCF-VP16 association. If the region be-tween residues 102 and 105 of HCF does not mediate VP16 binding, then how does its alteration by mutation prevent VIC formation? An obvious interpretation is that HCF contributes to VIC function in multiple ways, of which binding to VP16 is only one. Perhaps upon binding to VP16, HCFwt induces a conformational change in VP16 necessary for efficient Oct-1/ TAATGARAT association. If the HCF region around residues 102 to 105 is important for inducing this conformational change, then its alteration in the mutant HCF m2 proteins could result in defective complex assembly, without necessarily affecting VP16 interaction per se. Precedents for such
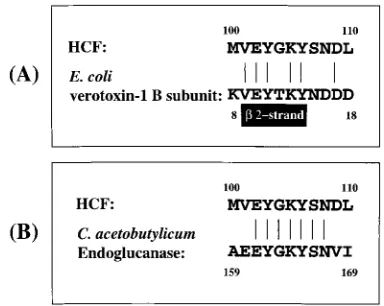
confor-FIG. 9. Sequence alignments to the HCF m2 region. (A) The m2 region of HCF (amino acids 100 to 110) is shown aligned to the cell-binding B oligomer of VT-1 of E. coli (35). This alignment was obtained from a search of the Brookhaven Protein Data Bank of proteins with known three-dimensional struc-tures (3). Positions of identity are indicated by a bar, and residues 9 to 14 forming ab-sheet in VT-1 are indicated (35). (B) The m2 region is shown aligned to the predicted sequence of endo-beta-1,4-glucanase from C. acetobutylicum (51). This alignment was obtained by a homology search of the SwissProt database.
on November 9, 2019 by guest
http://jvi.asm.org/
[image:7.612.339.531.503.656.2]form a b-sheet structure capable of protein-protein interac-tion. When this sequence search is extended to include primary sequences of all known proteins, a 100% identity across 7 amino acids located near the active site of the endo-beta-1,4-glucanase from Clostridium acetobutylicum (51) is obtained (Fig. 9B). This protein is a member of the extensive glycosyl hydrolase family, which also includes many bacterial and viral sialidases and oxidases (15, 16). The structures of several of these proteins have been studied by X-ray crystallography and reveal a superbarrel fold with extensive antiparallelb-sheets (4, 19). Although HCF (1-380) displays only 17% overall iden-tity with the 448-residue endo-beta-1,4-glucanase, it is tempt-ing to speculate that perhaps this region of HCF also adopts such a structure. Mutation of the m2 region could then be seen as disrupting a site of protein-protein contact, perhaps even between HCF (1-380) monomers. Such an interaction would be consistent with the observation that amino- and carboxy-terminal HCF polypeptides remain stably associated after cleavage of the HCF precursor (46, 48) and would invoke a functional role for this association.
ACKNOWLEDGMENTS
We thank Liz Keech, Liz Conway, and William Spurden for valuable help in peptide synthesis; Peter O’Hare and Angus Wilson for discus-sions; Harikrishna Nakshatri for the kind gift of pet15bOct1; and Margit Urban, Phil Wong Kai In, and Mary Graves for critical com-ments on the manuscript.
REFERENCES
1. Ace, C. I., M. A. Dalrymple, F. H. Ramsay, V. G. Preston, and C. M. Preston. 1988. Mutational analysis of the herpes simplex type 1 trans-inducing factor, Vmw65. J. Gen Virol. 69:2595-2605.
2. Ace, C. I., T. A. McKee, M. Ryan, J. C. Cameron, and C. M. Preston. 1989. Construction and characterization of a herpes simplex virus type 1 mutant unable to transinduce immediate-early gene expression. J. Virol. 63:2260– 2269.
3. Bernstein, F. C., T. F. Koetzle, G. J. Williams, E. F. Meyer, M. D. Brice, J. R.
Rodgers, O. Kennard, T. Shimanouchi, and M. Tasumi.1977. The protein data bank. A computer-based archival file for macromolecular structures. Eur. J. Biochem. 80:319–324.
4. Bork, P., and R. F. Doolittle. 1994. Drosophila kelch motif is derived from a common enzyme fold. J. Mol. Biol. 236:1277–1282.
5. Campbell, M. E. M., J. W. Palfreyman, and C. M. Preston. 1984. Identifi-cation of herpes simplex virus DNA sequences which encode a trans-acting polypeptide responsible for stimulation of immediate early transcription. J. Mol. Biol. 180:1–20.
6. Cleary, M. A., and W. Herr. 1995. Mechanisms for flexibility in DNA se-quence recognition and VP16-induced complex formation by the Oct-1 POU domain. Mol. Cell. Biol. 15:2090–2100.
7. Cousens, D. J., R. Greaves, C. R. Goding, and P. O’Hare. 1989. The C-terminal 79 amino acids of the herpes simplex virus regulatory protein, Vmw65, efficiently activate transcription in yeast and mammalian cells in chimeric DNA-binding proteins. EMBO J. 8:2337–2342.
8. Dignam, J. D., R. M. Lebowitz, and R. G. Roeder. 1983. Accurate transcrip-tion initiatranscrip-tion by RNA polymerase II in a soluble extract from mammalian nuclei. Nucleic Acids Res. 11:1475–1489.
9. Goding, C. R., and P. O’Hare. 1989. Herpes simplex virus Vmw65-Octamer protein interaction: a paradigm for combinatorial control of transcription. Virology 173:363–367.
10. Goodrich, J. A., T. Hoey, C. J. Thut, A. Admon, and R. Tjian. 1993.
Dro-15. Henrissat, B., and A. Bairoch. 1993. New families in the classification of glycosyl hydrolases based on amino acid sequence similarities. Biochem. J.
293:781–788.
16. Henrissat, B., and A. Bairoch. 1996. Updating the sequence-based classifi-cation of glycosyl hydrolases. Biochem. J. 316:695–696.
17. Herr, W., and M. Cleary. 1995. The POU domain: versatility in transcrip-tional regulation by a flexible two-in-one DNA-binding domain. Genes Dev.
9:1679–1693.
18. Honess, R. W., and B. Roizman. 1974. Regulation of herpesvirus macromo-lecular synthesis. Cascade regulation of the synthesis of three groups of viral proteins. J. Virol. 14:8–19.
19. Ito, N., S. E. V. Phillips, C. Stevens. Z. B. Ogel, M. J. McPherson, J. Keen,
K. D. S. Yadav, and P. F. Knowles.1991. Novel thioether bond revealed by a 1.7 Angstrom crystal structure of galactose oxidase. Nature 350:87–90. 20. Jupp, R., S. Hoffmann, A. Depto, R. M. Stenberg, P. Ghazal, and J. Nelson.
1993. Direct interaction of the human cytomegalovirus IE86 protein with the cis repression signal does not preclude TBP from binding the TATA box. J. Virol. 67:5595–5604.
21. Katan, M., A. Haigh, C. P. Verrijzer, P. C. van der Vliet, and P. O’Hare. 1990. Characterization of a cellular factor which interacts functionally with Oct-1 in the assembly of a multicomponent transcription complex. Nucleic Acids Res. 18:6871–6880.
22. Kristie, T. M., J. H. LeBowitz, and P. A. Sharp. 1989. The octamer-binding protein form multi-protein-DNA complexes with the HSV alpha TIF regu-latory protein. EMBO J. 8:4229–4238.
23. Kristie, T. M., J. L. Pomerantz, T. C. Twomey, S. A. Parent, and P. A. Sharp. 1995. The cellular C1 factor of the herpes simplex virus enhancer complex is a family of polypeptides. J. Biol. Chem. 270:4387–4394.
24. Kristie, T. M., and P. A. Sharp. 1990. Interactions of the Oct-1 POU sub-domains with specific DNA sequences and the HSVa-transactivator protein. Genes Dev. 4:2383–2396.
25. Lai, J.-S., M. A. Cleary, and W. Herr. 1992. A single amino acid exchange transfers VP16-induced positive control from the Oct-1 to the Oct-2 homeo domain. Genes Dev. 6:2058–2065.
26. Lin, Y. S., and M. R. Green. 1991. Mechanism of action of an acidic tran-scription activator in vitro. Cell 64:971–981.
27. Nakshatri, H., P. Nakshatri, and R. A. Currie. 1995. Interaction of Oct-1 with TFIIB. J. Biol. Chem. 270:19613–19623.
28. Nilsson, B., and A. Uhlen. 1985. Immobilization and purification of enzymes with staphylococcal protein A gene fusion vectors. EMBO J. 4:1075–1080. 29. O’Hare, P. 1991. Targets for antiviral chemotherapy: herpes simplex virus
regulatory protein, Vmw65. Antiviral Chem. Chemother. 2:1–7.
30. Post, L. E., S. Mackem, and B. Roizman. 1981. Regulation ofagenes of herpes simplex virus: expression of chimeric genes produced by fusion of thymidine kinase withagene promoters. Cell 24:555–565.
31. Price, N. C., and C. M. Johnson. 1989. Proteinases as probes of conformation of soluble proteins, p. 163–179. In R. J. Beynon and J. S. Bond (ed.), Proteolytic enzymes: a practical approach. IRL Press, Oxford, England. 32. Roizman, B., and A. Sears. 1993. Herpes simplex virus and their replication,
p.11–68. In B. Roizman, R. J. Whitley, and C. Lopez (ed.), The human herpesviruses. Raven Press. New York, N.Y.
33. Sadowski, I., J. Ma, S. Triezenberg, and M. Ptashne. 1988. GAL4-VP16 is an unusually potent transcriptional activator. Nature 335:563–564.
34. Shaw, P., J. Knez, and J. P. Capone. 1995. Amino acid substitutions in the herpes simplex virus transactivator VP16 uncouple direct protein-protein interaction and DNA binding from complex assembly and transactivation. J. Biol. Chem. 270:29030–29037.
35. Stein, P. E., A. Boodhoo, G. J. Tyrell, J. L. Brunton, and R. J. Read. 1992. Crystal structure of the cell-binding B oligomer of verotoxin-1 from E. coli. Nature 355:748–750.
36. Stern, S., and W. Herr. 1991. The herpes simplex virus trans-activator VP16 recognizes the Oct-1 homeo domain: evidence for a homeo domain recog-nition subdomain. Genes Dev. 5:2555–2566.
37. Stringer, K. F., C. J. Ingles, and J. Greenblatt. 1990. Direct and selective binding of an acidic transcriptional activation domain to the TATA-box factor TFIID. Nature 345:783–786.
38. Stueber, D., H. Matile, and G. Garotta. 1990. System for high-level
on November 9, 2019 by guest
http://jvi.asm.org/
tion in Escherichia coli and rapid purification of recombinant proteins: ap-plication to epitope mapping, preparation of antibodies, and structure-func-tion analysis, p. 121–152. In I. Lefkovits and B. Pernis (ed.), Immunological methods, vol. IV. Academic Press. N.Y.
39. Triezenberg, S. J., R. C. Kingsbury, and S. L. McKnight. 1988. Functional dissection of VP16, the trans-activator of herpes simplex virus immediate early gene expression. Genes Dev. 2:718–729.
40. Walker, S., S. Hayes, and P. O’Hare. 1994. Site-specific conformational alteration of the Oct-1 POU domain-DNA complex as the basis for differ-ential recognition by Vmw65 (VP16). Cell 79:841–852.
41. Weinheimer, S. P., B. A. Boyd, S. K. Durham, J. L. Resnick, and D. R.
O’Boyle.1992. Deletion of the VP16 open reading frame of herpes simplex virus type 1. J. Virol. 66:258–269.
42. Werstuck, G., and J. P. Capone. 1989. Identification of a domain of the herpes simplex virus trans-activator Vmw65 required for protein-DNA com-plex formation through the use of protein A fusion proteins. J. Virol. 63: 5509–5513.
43. Werstuck, G., and J. P. Capone. 1989. Mutational analysis of the herpes simplex virus trans-inducing factor Vmw65. Gene 75:213–224.
44. Werstuck, G., and J. P. Capone. 1993. An unusual cellular factor potentiates protein-DNA complex assembly between Oct-1 and Vmw65. J. Biol. Chem.
268:1272–1278.
45. Wilson, A. C. Personal communication.
46. Wilson, A. C., K. LaMarco, M. G. Peterson, and W. Herr. 1993. The VP16 accessory protein HCF is a family of polypeptides processed from a large precursor protein. Cell 74:115–125.
47. Wilson, A. C., J. E. Parrish, H. F. Massa, D. L. Nelson, B. J. Trask, and W.
Herr.1995. The gene encoding the VP16-accessory protein HCF (HCF1) resides in human Xq28 and is highly expressed in fetal tissues and the adult kidney. Genomics 25:462–468.
48. Wilson, A. C., M. G. Peterson, and W. Herr. 1995. The HCF repeat is an unusual proteolytic cleavage signal. Genes Dev. 9:2445–2458.
49. Wu, T.-J., G. Monokian, D. F. Mark, and C. R. Wobbe. 1994. Transcriptional activation by herpes simplex virus type 1 VP16 in vitro and its inhibition by oligopeptides. Mol. Cell. Biol. 14:3484–3493.
50. Xiao, P., and J. P. Capone. 1990. A cellular factor binds to the herpes simplex virus type 1 transactivator Vmw65 and is required for Vmw65-dependent protein-DNA complex assembly with Oct-1. Mol. Cell. Biol. 10:4974–4977. 51. Zappe, H., W. A. Jones, D. T. Jones, and D. R. Woods. 1988. Structure of an endo-beta-1,4-glucanase gene from Clostridium acetobutylicum P262 showing homology with endoglucanase genes from Bacillus spp. Appl. Environ. Mi-crobiol. 54:1289–1292.