Analysis of Sequence Diversity
in
Hypervariable Regions of
the
External Glycoprotein of
Human
Immunodeficiency
Virus
Type
1
PETERSIMMONDS,lt* PETERBALFE,' CHRISTOPHER A. LUDLAM,2 JOHN0. BISHOP,13
AND ANDREW J. LEIGH BROWN'
Department ofGenetics, University of Edinburgh, EdinburghEH9
3JN,l
andDepartment of Haematology, Royal Infirmary, Edinburgh EH3 gyW,2 Scotland, and Department of Biological Sciences, University ofMaryland-Baltimore County, Baltimore, Maryland212283
Received 13July 1990/Accepted 5 September 1990
Nucleotidesequencesin threehypervariable regionsof thehumanimmunodeficiency virustype1(HIV-1)env
gene were obtained by sequencing provirus present in peripheral blood mononuclear cells of HIV-infected
individuals. Singlemoleculesoftargetsequences wereisolatedbylimiting dilution andamplifiedintwostages
by thepolymerasechain reaction, usingnestedprimers. Theproductwasdirectly sequencedtoavoiderrors introduced by Taq polymerase during the amplification process. There was extensive variation between
sequences from the same individual as well as between sequences from different individuals. Interpatient
variabilitywasmarkedly less in individualsinfectedfromacommon source.Ahighproportionofamino acid substitutions in thehypervariable regionsaltered thenumber andpositionsofpotentialN-linkedglycosylation sites.Sequences intwohypervariable regions frequently containedshort(3-to15-bp) duplicationsordeletions, and by amplifying peripheral blood mononuclear cell DNA containing 102 or 103 proviral molecules and
analyzingtheproductby high-resolution electrophoresis, the total number and abundance of distinctlength
variants within anindividual could beestimated, providing a more comprehensive analysisof the variants presentthanwouldbeobtainedby sequencingalone.Sequencesfrommanyindividualsshowedfrequentamino
acid substitutions at certain key positions for neutralizing-antibody and cytotoxic T-cell recognition in the
immunodominant loop. Theratesofsynonymousandnonsynonymous nucleotide substitution in theregionof this and flanking regions indicate that strong positive selection for amino acid change is operating in the
generation of antigenic diversity.
Understanding the nature of sequence change in the
human immunodeficiency virus type 1 (HIV-1) genome is central to current theories of viral pathogenesis and the immune response to infection. In common with other
eu-caryotic viruses with RNAgenomes, HIV-1 shows
consid-erable sequence diversity between different isolations,
par-ticularly those from geographically distinct regions, where divergence has taken place over a number of years.
Se-quence diversity is seen within isolations from the same
individualaswellasbetween HIV strains infecting different individuals (11, 29). Therate of HIV sequence variation is not uniform throughout the genome. Comparison of pub-lished sequences has shown that thegagandpolgenes are more conserved than env. Furthermore, within env the
pattern of variation is unusual (3, 34, 38). Five regions in
gpl20,VltoV5,have beendesignatedashypervariable (23). Theseweredefinedasregionswith25%orlessconservation ofaminoacids betweenanumberof published sequences.In addition to high rates of amino acid substitution, the
pub-lished sequences ofsome hypervariable regions have been
previously reportedtocontain short deletions and insertions (3, 11, 23, 34). Several actualorpotential gpl20 epitopesare located in these regions (23, 34). Antigenic variation
conse-quent to sequence differences in V3 (the immunodominant
loop)hasbeen reported(19, 36).Thissequencecorresponds
tothepredominant neutralization epitope of gpl20(28), and ithas beenargued that the observed variabilityrepresentsan
* Corresponding author.
tPresentaddress:DepartmentofMedicalMicrobiology,Medical
School, University of Edinburgh, Teviot Place, Edinburgh EH3
9AG, Scotland.
adaptive responseby HIVto evade the immune system, as
proposedforequineinfectious anemia virus and visna virus
(4, 5, 24). Whether othervariable areasofgpl20are
impor-tant in neutralizing-antibody or T-cell recognition is not known.
In thiswork,variation in thegpl20sequencesofprovirus
present in circulating peripheral blood mononuclear cells
(PBMCs) was studied. Anumber ofindividuals included in thisstudywereinfected fromacommon source(20),
allow-ing variation between patients to be assessed. The hyper-variableregionsofenvnotonlyarepolymorphicinsequence but inmanycases also differ inlength (3, 11, 23, 34). Thus, thespacingof theconservedregionsofgpl20shows consid-erable variation. Using the polymerase chain reaction
(PCR), we have investigated length variation of amplified DNA in different regions of gpl20 and have visualized distinctive patterns ofcoexisting variants in each infected individual. Thedataonlengthvariationtogetherwith exten-sive data obtained by sequencing hypervariable regions of
single isolated molecules (30) indicate that the number of variants present in eachpatientis extremely large. Despite
this heterogeneity, HIV sequences from individuals in the
hemophiliac cohort, who were infected from a common
source, wereclearlymorecloselyrelated toeach other than
to published sequences of HIV-1 and to those ofa
hemo-philiac infected in the United States. In particular, the V4
sequence of the cohort memberswasdistinctive and served
to distinguishthem from other hemophiliacs.
MATERIALSAND METHODS
Clinical samples. Blood samples were obtained from 11
hemophiliacs infected with HIV-contaminated factor VIII
5840 0022-538X/90/125840-11$02.00/0
Copyright ©1990,American Society forMicrobiology
on November 10, 2019 by guest
http://jvi.asm.org/
ANALYSIS OF SEQUENCE VARIATION IN gpl20 5841
approximately
5 years ago(p12
to p95). For sequencingstudies, samples
were used from eight haemophiliacs whowere exposedabatch of factor VIII thatwasimplicated in infection
by
HIV-1 of 18 outof 32 recipients(20). Alleight individuals seroconverted for antibody between 3 and 10 months afterreceiving
the factor VIII (31). p82 is currentlyasymptomatic,
and p83 has thrombocytopenia but is other-wisewell;
all others have been classified as IVc and suffer from a range ofopportunistic
infections and constitutional symptoms of HIV infection. Samples from individuals in-fected from othersources includep12,
whowasinfectedin theUnited StatesfromcommercialfactorVIII,and 10drug abuse-related andpediatric seropositive
individuals (nl tonlO)
from J. Mok(Infectious
Diseases Unit, City Hospital,Edinburgh, Scotland).
HIV primers. Oligonucleotides were synthesized by the OswelDNA
Service,
DepartmentofChemistry,
University ofEdinburgh,
andwerepurified by high-performance
liquidchromatography.
Theprimers
werebasedontheconsensusof the
following published
HIV sequences: HIVHTLVIIIB(clones HXB2,
BH102, and BH8), HIVBRU, HIVCDC42, HIVELI, HIVMAL, HIVMN, HIVPV22, HIVRF, HIVsc,HIVSF2,
HIVWMJ22,
andHIVZ6.
Theprimer-binding
sitesingpl20
were chosen to be ashighly
conserved aspossible
between
published
sequences ofgeographical
variants of HIV-1. Nomore than one mismatchwith any of the NorthAmerican, Haitian,
orAfrican sequenceswaspresentin anyoneof the
primers.
The sequencesof theprimers
aregiven,
with their
positions
in HIV clone HXB2indicated in paren-theses(+,
sense; -,antisense):
V3(a)TACAATGTACACA TGGAATT(+, 6957),
(b)
TGGCAGTCTAGCAGAAGAAG(+, 7009), (c)
CTGGGTCCCCTCCTGAGG (-, 7331), and(d)
ATTACAGTAGAAAAATTCCCC (-, 7381); V4-V5 (e) TCAGGAGGGGACCCAGAAATT(+, 7316), (f)
GGGGAA TTTTTCTACTGTAAT(+,
7360), (g)
CTTCTCCAATTGT CCCTCATA(-, 7665),
and(h)
CCATAGTGCTTCCTGCT GCT(-,
7814).
Double-PCR method. DNA fromat least 5 x
106
PBMCswas
prepared
as describedby
Simmonds etal.(30).
Ampli-fication of DNA
by
double PCR andquantification by
limiting
dilutionwerecarriedout asdescribedby
Simmondsetal.
(30).
All DNAextractions andamplification
reactions carriedappropriate parallel negative
controls(blood
fromseronegative,
low-risk group blooddonors)
to detectcon-tamination at any stage in the
procedure.
Inprinciple,
atleast five outer
primer pairs
may be usedsimultaneously
in the firstamplification
reaction.However,
DNA sequencesamplified by
the outer V3 and V4-V5primers overlap.
Topermit
the samesample
to beamplified
in the tworegions,
the
following
combinations ofprimers
wereused. In the firstreaction, positive-sense,
outer V3primer (primer
a; seeabove)
was used with theantisense,
outer V4-V5primer
(primer
h),
toamplify
a858-bp
fragment;
in the secondreaction,
asample
of theproduct
wasamplified by using
the inner V3(primer b)
andinner V4-V5(primer g) primers (657
bp);
in the thirdreaction,
the finalamplification
used the innerV3(b plus c)
andV4-V5(f
and g)primers
in separate reactions. Thismodifiedprocedure
isequivalent
insensitiv-ity
andyield
to separateamplification
in two stagesby
double PCR
(data
notshown).
Length analysis of PCR products. To
investigate length
variation in
product
DNAamplified
in the threeenvregions,
the final PCR reactionwascarriedoutin the presence of0.05 to 0.1
,uCi
of[cI-355]thio-dATP
(1,000
Ci/mmol; Amersham)
and 8.3 mM each of unlabeled
deoxynucleoside
triphos-phates (dNTPs).
A1-,ul sample
of the PCRproduct
washeated to 95°C in 50% formamide for 3 min and electro-phoresed on a denaturing polyacrylamide gel (6% acryl-amide, 0.3% N',Nbisacrylamide, 8 M urea, 0.089 M Tris, 0.089Mboric acid,0.002MEDTA, pH 8.3). Standard DNA sizemarkers (1,635, 1,018, 516, 506, 394, 344, 298, 220, 200, 154, and 142 bp; BCL)were end labeled with [_y-32P]dATP. Gels were dried and exposed overnight on X-ray film. Analysis of migrationdistances and intensity of bands was assisted by theuseofadensitometer.
DNA sequencing ofPCR products. The double-PCR pro-cedure produces sufficient DNA from single isolated mole-cules oftarget sequencetopermit direct sequencing of the PCR product (30). Unreacted dNTPs and primers were
removedfrom product DNA by using Gene-Clean (Bio 101, Inc.). Direct sequencingwasthencarried out by usingoneof theprimersusedfor the finalamplification bytheSequenase protocol (U.S. Biochemical Corp.), with the following
mod-ifications: the standard annealing mix was adjusted to 10%
dimethyl sulfoxide (40), and the sample was denatured by boilingfor 5 min andimmediatelychilled onice. The labeling reactionwas performedatroomtemperaturefor5min with 0.25 ,uM two unlabeled dNTPs and 5 ,uCi ofal-35S-labeled dATPordCTP(1,000Ci/mmol; 37).Thechoice of unlabeled and labeled dNTPs wasbased onthe nucleotides present 3'
to the priming site used. Termination reaction mixtures contained a final concentration of 10% dimethyl sulfoxide. Sequences were analyzed on a 6% polyacrylamide wedge sequencing gel.
Nucleotidesequenceaccession number.All sequences used in this study have been submitted to GenBank (accession numberM36997).
RESULTS
Amplification of HIV provirus by PCR. Two different
approaches to PCR amplification were used. In the first, a
sample of DNA large enough to contain a representative sampleofproviruswas amplified. In this case, theproduct should be representative ofthe provirus population in the
sample (see below). In the second method (30), single moleculesofproviruswereisolatedbydilution beforebeing amplified. In this case, the product of each amplification derives fromasingleprovirus.In many cases,adouble-PCR reactionusing nested primers was used. In this
method,
asmall sample of the first PCR reaction is used to prime a
second(30). Consequently, theproductsofafirst
amplifica-tion can be usedto prime more than one
amplification,
for example, one with labeled precursor for visualization by autoradiographyandasecondwithout labeled precursor for directsequencing (see below).In published HIV-1 sequences, the length of the region amplified bythe V4-V5 primers
(spanning
V4,C3,
andV5;
23) varies by as much as 30 of around 300 nucleotides. Substantial size variationin this
region
is also found within theprovirus
population
presentinasingle
infected individ-ual. Forexample, a sample ofDNAfromhemophiliac p79,
containing about 60 molecules of
provirus
(30), wasampli-fied with the V4-V5
primers
and labeled with[a-35S]thio-dATPduringthe second
amplification
step. Whenanalyzed
by gel
electrophoresis,
theproduct
showedacomplex
setof size variants (Fig. 1, lane U). To confirm that these werenatural variants present in the DNA
sample
rather than artifacts of the PCRreaction, single provirus
moleculeswereisolated by dilution before
amplification
(30). A total of 31 replicate tubes, each containing 625cell-equivalents
of DNA, wereamplified
in the doublePCR,
with[a-35S]thio-VOL.64, 1990
on November 10, 2019 by guest
http://jvi.asm.org/
M U 1 2 3 4 5 6 7 8 9 10 11 12 13 N M
Wm.
394 _
344-_
220-.
FIG. 1. Size analysis of amplifiedproviral DNA in the V4 and V5 regions fromPBMCs ofanHIV-infected individual (p79). Positive
reactionsatlimiting dilution of PBMC DNA fromanHIV-infected
hemophiliac (p79)were amplified with V4-V5 primers and
electro-phoresedon apolyacrylamide gel (lanes 1 to13). Lane U, 1 ,ug of PBMC DNA from p79; lane M, product size estimated with DNA markers of sizes (in nucleotides) indicated (see Materials and Methods); lane N, 1 ,ug of PBMC DNA fromanuninfected
individ-ual.
dATPadded to the second reaction. Thirteen of the
reac-tions were positive. From the 18 negative reactions, the Poisson formula predicts that approximately 75% of the
positive reactions are due to the amplification ofa single
provirus molecule. Electrophoretic analysis of the positive reactions showed that the lengthvariants found by amplify-ing 60 provirus molecules togetherwerealso foundwhenthe
moleculeswereisolated before amplification (Fig. 1, lanes1 to 13). A single lane (lane 5) clearly contained two length variants, consistent with the prediction of the Poisson for-mula.
Todetermine the basis of the length variation, the prod-uctsof 12 of the 13 first PCRreactions (omitting reaction 5) were againamplified in the second PCR but without labeled
substrate and directly sequenced (30). The 12 nucleotide
sequencesare showninFig. 2. Thelengths of the amplified
sequences calculated from electrophoretic analysis agree
well with thelengths determined by sequencing (Fig. 2,last
two columns). Thus, the differences in electrophoretic
mo-bility reliably reflect differences inlength.
Theisolatedmolecules appearedtobearandom sampleof
the sequences visualized in the bulk amplification of 60
provirus molecules (Fig. 1, lane U). Thus, the four more
intense bands in lane U withlengths of 296, 299, 302 and 305 nucleotides were represented by one, one, six, and two
isolatedsequences,respectively. Faint bands with lengths of
284 and 293 nucleotides were represented by one isolated
sequence,and others withlengths of 287 and 281 nucleotides
were not represented. Thus, the analysis of bulk samples providesaconvenient and rapidassessmentof thespectrum
of size variation within the provirus populationpresentin a
DNAsample.
Amino acidsequencevariation in HIV-infectedindividuals.
PBMC DNA from eight other infected individuals was diluted andsequencedin theV4 andV5 regionsasdescribed
for p79, in all cases using low frequencies of positive
reactions at limiting dilutiontoavoid multiple positives. To simplify the presentation, the nucleotide sequences were
translated. Figure 3 shows alignments of such amino acid
sequences obtained from each individual. Considerable
di-versity in the V4 and V5 regions was seen within and between individuals. In many individuals, itwas necessary to insert notional gaps to preserve alignment of the
se-quences. These gaps were concentrated exclusively in the
V4andV5 regions,where therewasalsoahighrateofamino acid substitution. Eachgap was amultiple of 3 nucleotides,
maintainingthereadingframe of thesequence.Thedegree of
sequencevariationwithinanindividualvariedconsiderably,
the sequences from p77and p79 showing the most
hetero-geneity in this groupandthose from p84 showing theleast. A number of sequences showed short repeats of 3 to 6 amino acids in V4 and V5 (Fig. 3). The same repeated
sequences were found in different individuals. In V4,
se-quencescontaining (N)STWwererepeatedin p74,p77, and
p82, whilep77, p79, p83, p84, p87, andp91 showedrepeats
ofsequences TTGSN and TTESN. In V5, repeats ofNET
were found intwoindividuals, p77 and p12. The number of repeated sequences varied both between and within
sam-ples.This duplication ofsequence motifsaccountsforsome of the observedlength variation. SitesforpotentialN-linked
glycosylation sites were concentrated in the hypervariable
regions. In each individual, there was a concentration of
potential N-linked glycosylation sites in the V4 and V5 regions. The number and positions ofsuch sites were vari-able between individuals and within each patient sample (Fig. 3). Variation in the number of repeats changed the overall potentialforcarbohydrate addition in this region of
gpl20 (Fig. 3).
Lengthpolymorphism of DNAamplifiedin the V4 and V5 regions. With theexception of p82, few identicalsequences
Seq. < V4- > < C3 > <---V5 --->
<-Sequence Measured
C4 Length (bps) Length (bps)
1 51 bps+
2 51 bps+
3 51 bps +
4 51 bps+
6 51 bps +
7 51 bps+
8 51 bps +
9 51 bps+
10 51 bps +
11 51 bps +
12 51 bps +
13 51 bps +
Cons51 bps+
+ 132 bps+ + 132 bps+
+ 132 bps+
+ 132 bps+ + 132 bps+
+ 132 bps+
+ 132 bps+
132 bps+
+ 132 bps+ + 132 bps+ + 132 bps+ + 132 bps+ + 132 bps+
+41 bps =
+41 bps=
+41 bps=
+41 bps=
+41 bps=
+41 bps=
+41 bps=
+41 bps=
+41 bps =
+41 bps=
+41 bps=
+41 bps=
302 302 293 305 302 284 302 305 302 302 296 299 303.4 302.7 294.3 306.3 303.0 285.1 302.0 305.8 302.0 302.0 295.8 300.1
[image:3.612.70.289.75.232.2]+41 bps= 305
FIG. 2. Alignment ofsequences obtained by limitingdilution of PBMC DNA from p79. Sequence labels V4, C3, V5, and C4follow
previous usage (23). Symbols: ., unsequenced; -, gap introducedto preserve alignment; ?, no majority consensus. Sequences 1 to 13 correspondtolanes 1 to13inFig. 1. Differences from theconsensusareindicatedinlowercase.
a g t 9 9 a
c t- a
ttg a 9 a
t
t 9 a
t t a
.GATACTACAGGGTCAAACACTACAGGGTCAAATAAC ACTGAACCTATCACA
t g
t g .
tat a --- .
c ac tgacc
9 9 a
---gg a a g ----a
t 9
c ac g ac
t g a
---t c
---t ac c--- ..
ggg a a--- a
---AAAAACGTGAGCAAC G?GTCCACCGAGACC
on November 10, 2019 by guest
http://jvi.asm.org/
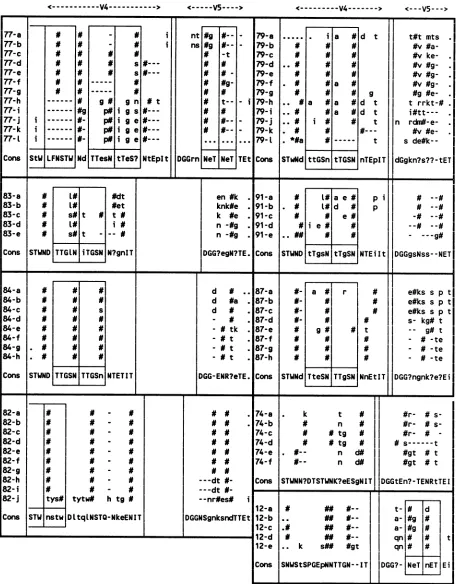
[image:3.612.64.552.571.686.2]< V4- > N LFNSTWI Ng N-P# P# g# p# pN p# s gn i 9s i ge i ge i ge
#---# t#---Nd TTesN tTeS? NtEplt
11
<---- V5--->
Int ns
DGGrn
ANALYSIS OF SEQUENCE VARIATION IN gp120
#g
NE
N#--t #9- t--N- -N- -INeT - i TEt I 79-a 79-b 79-c 79-d 79-e 79-f 79-g 79-h 79-i 79-j 79-k 79-L Cons<---V4--->
[image:4.612.77.537.92.676.2]STwNd * i a iN# Nf a a a a gd t 9 d t d t t
#---tttGSnI|
tTGSN|nTEpIT<---V5 ->
t#t mts . #v #a-#v ke-#v
#g-#v fg-#v #g
-Ng
#e-t rrkt-# .
i#tt---n
rdm#-e-N#v
e-s deNk--dGgkn?s??-tET
83-a N [T# ] #dt en#k .91-a N la e# pi # --#
83-b N LNj# #et knk#e .91-b. L#d P
83-c s# t t # k #e . 91-c N N eN
83-d N L# i # n -#g . 91-d Ni e N --# #
83-e N [ t --- # n -#gg91-e N#
Cons STWND TL TGSN N?gnIT DGG?egN?TE. Cons STWND tTgsN tTgSN NTEiIt DGGgsNss--NET
84-a N N N d N.87-a a# r e#ksspt
84-b N N N d #a . 87-b
eN-s
s p t84-c N N s d . 87-c - e#ks s p t
84-d N N N - . 87-d N- N N s- kg# t
84-e N N N -Ntk . 87-e N g N t -- g#t
84-f N N N -Nt . 87-f N N N -N-te
84-g . N N N t . 87-g N N N - N-te
84-h - N N N -Nt . 87-h N N N - #-te
Cons STWND TTGSN TTGSn NTETIT DGG-ENR?eTE. Cons STWNd TteSN TTgSN NnEtIT DGG?ngnk?e?Ei
-tytwN htgN#
DltqlNSTQ-NkeEN IT
# # # --n-dt #- ---dt#n---nr#es#N DGGNSgnksndTTEt 74-a 74-b 74-c 74-d 74-e 74-f Cons 12-a 12-b 12-c 12-d 12-e Cons k t n #tg #tg n d# n d# STWNN?DTSTWNK?eESgNIT
..- k sNN #gt
SNWStSPGEpNNTTGN--IT
#r- # s-#r- # s-#r- # # s---t
Ngt # t Ngt # t DGGtEn?-TENRtTEI t- a-qn qn DGG? -# #g Ng NeT d n nET t Ei
FIG. 3. Alignment ofsequencesfromninehemophiliacsinthe V4andV5 regions. Individualsequencesfrom eachindividualareindicated
as atoz.Symbols: notsequenced;-,gapintroducedtopreservealignment; #,asparagineresiduesiteofpotentialN-linkedglycosylation;
?,nomajorityconsensusatthis position;*,stopcodon.Conserved amino acidsareshowninuppercase;nonconservedaminoacidsareshown
inlowercase. Tandem repeatedsequencesare indicated by boxes. Potentialglycosylation sites shown whetherconserved ornot.
VOL. 64,1990 5843
77-a 77-b 77-c 77-d 77-e 77-f 77-g 77-h 77-i
77-j
77-k 77-L Cons stw 82-a 82-b 82-c 82-d 82-e 82-f 82-g 82-h 82-i 82-i ConsN#
tysN |nstw STU- I II
I
I
i
on November 10, 2019 by guest
http://jvi.asm.org/
n p
M11 2 3 4 5 6 7 8 9 10174 M 777982 87 91 89 95
394
344
298 _
[image:5.612.60.302.76.232.2]220
FIG. 4. Size analysis ofamplified proviral DNA in the V4 and VS regions from PBMCs ofHIV-infected individuals. Samples (1 jig) of PBMC DNA from HIV-infected nonhemophiliacs (nl to nlO) and hemophiliacs (p74 top95) wereamplified with V4-V5 primers and electrophoresed on a polyacrylamide gel. Lane M, Product size
estimated withDNAmarkers of sizes (in nucleotides) indicated (see Materials andMethods).
were isolated from the same individual (Fig. 3). No two
identicalsequences wereisolated from p12, p79, p83, p87,or
p91; p74 and p77 each hadtwoidenticalsequences,p84 had
three, and only p82 and p87 had multiple incidences oftwo
sequences. Clearly, the sequencesdetermined donot
repre-sent the full range of variation present in most of the
individuals studied. It would be possible in principle to
sequenceeach DNAsample exhaustively. However,a
sim-pler approach to the question of variation within and
be-tween individuals is to use the PCR technique described
abovetoassesstheprofile of length variation in each sample.
The length analysis can be carried out with a standard amount ofPBMC DNA, sothatmany samplescanbe dealt withrapidly.
Todemonstrate therangeoflength variation within PBMC
samples, 1-,ug samples of DNA from 18 HIV-infected indi-viduals were amplified by using the V4-V5 primers as
described above(Fig. 4). The samples comparedwerefrom
p74, p77, p79, and p82, and the number of provirus
mole-culespresentin the1-,ug samples ranged from 60(p79)to200
(p82; 30). A close concordance was observed between the
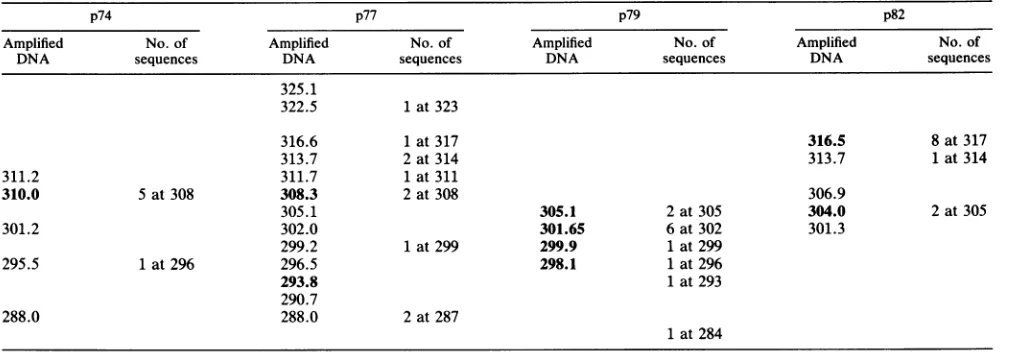
sizesof the bands measured by gelelectrophoresis and sizes determined by sequencing isolated molecules from thesame
sample of DNA(Table 1). For example, theprominentbands in the p74and p82 samples with estimated sizes of 308.0 and 316.5 bp were paralleled by a predominance ofsequences
with overall lengths of 308 and 317 bp. Similarly, the wide
rangeof band lengths observeduponamplicationof thep77
samplewasparalleled byawiderangeoflengthsamongthe
individually determinedsequences. These data show thatan
analysis of length variation serves as an approximate mea-sureof the overall sequencevariationinaprovirus
popula-tion. The sequence data(Fig. 3) show thata lengthvariant may comprise several different sequence variants. For
ex-ample,sequences74-a, -b, and-chad thesameoveralllength as 74-e and -f, although the two groups differed in the
positions of thegaps and anumber of aminoacid
substitu-tions. An impression ofthelarge range ofvariation which
canoccurwithinasamplemaybegained by consideringthe
range of size variation and the number of sequences of a
given lengthtogether. In severalcases,thismustrepresenta
tremendousamount ofvariation inthe V4-V5 region alone. Band lengths overved in the V4 and V5 regions with
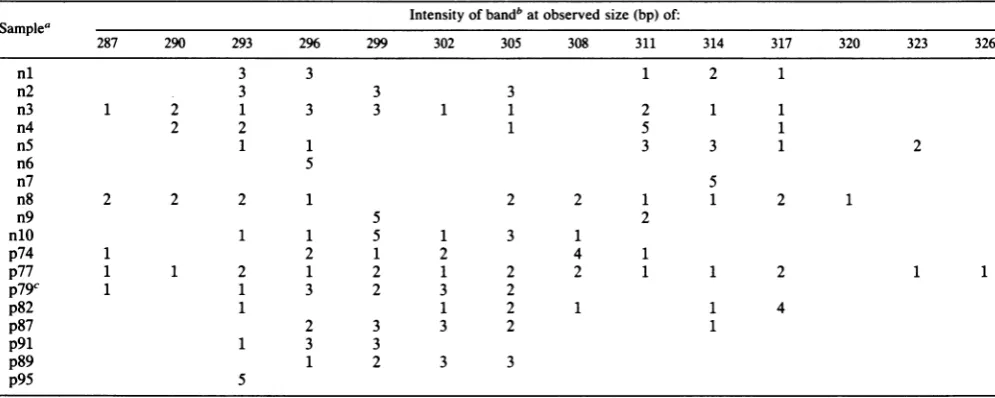
samples from eight hemophiliacpatientsare summarizedin
Table 2. Data from 10drug abuse andpediatric patientsare
alsolisted forcomparison. The overallrangeoflengths and patterns were similar in the two groups. The majority of
samples contained bandsin therange of293 to 317bp, but
there was considerable variation in the diversity of bands presentinasample. Atoneextreme,n3 andp77 had 10 and 13bands,respectively, with rangesof 287to317 and 287to
326 bp. At the other extreme, n6, n7, and p95 had single bands of296, 314, and 293 bp. Therangeoflengths of theV4
and V5 regions of geographically diverse isolated HIV-1
sequence variants, from 293 (HIVSF2) to 311 (HIVCDC42),
lies within the range observed in this study and suggests
possible selectiveconstraintson thesize oftheseregions.
Sequence variation in the V3 hypervariable region. To
investigate sequence variability of the immunodominant
loop (28),sequences of individualprovirus molecules in the
[image:5.612.62.567.539.715.2]PBMC DNAsampleswere determined with the V3 primers
TABLE 1. Comparison of size ofamplifiedDNAwithoveralllengths ofsequencesobtainedbylimiting dilutiona
p74 p77 p79 p82
Amplified No. of Amplified No.of Amplified No.of Amplified No.of
DNA sequences DNA sequences DNA sequences DNA sequences
325.1
322.5 1 at323
316.6 1 at317 316.5 8at317
313.7 2at314 313.7 1at314
311.2 311.7 1 at311
310.0 5at308 308.3 2 at308 306.9
305.1 305.1 2 at305 304.0 2 at305
301.2 302.0 301.65 6 at302 301.3
299.2 1 at299 299.9 1at299
295.5 1 at296 296.5 298.1 1at296
293.8 1 at 293
290.7
288.0 288.0 2at287
1 at284
a Prominent bandsareinbold type; all sizes aregivenin basepairs.
on November 10, 2019 by guest
http://jvi.asm.org/
ANALYSIS OF SEQUENCE VARIATION IN gpl20 5845 TABLE 2. Distribution of length variants in theV4and V5 regions
Intensity of bandb at observed size (bp) of: Samplea
287 290 293 296 299 302 305 308 311 314 317 320 323 326
nl 3 3 1 2 1
n2 3 3 3
n3 1 2 1 3 3 1 1 2 1 1
n4 2 2 1 5 1
n5 1 1 3 3 1 2
n6 5
n7 5
n8 2 2 2 1 2 2 1 1 2 1
n9 5 2
nlO 1 1 5 1 3 1
p74 1 2 1 2 4 1
p77 1 1 2 1 2 1 2 2 1 1 2 1 1
p79C 1 1 3 2 3 2
p82 1 1 2 1 1 4
p87 2 3 3 2 1
p91 1 3 3
p89 1 2 3 3
p95 5
anltonlO,HIV-infected nonhemophiliacindividuals; p74 to p95, hemophiliacs.
bScored on a scale of 1 (weak) to 5 (strong). c Bands (intensity= 1)also at 284 and 281 bp.
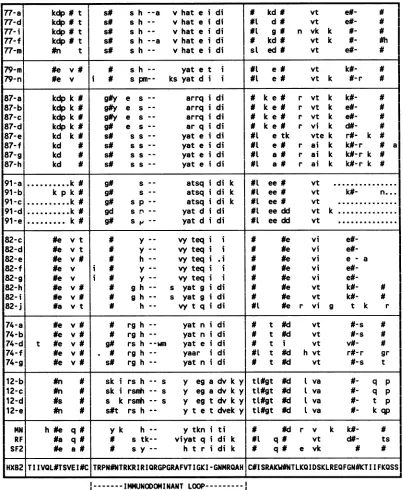
(Fig. 5). Sequences bearing the same identification letter in Fig. 3 (V4and V5 sequences) and Fig. 5 (V3 sequence) were derived from thesameprovirus molecule(seeMaterialsand Methods). Sequence variation accompanied by little length variation was the dominant feature of the sequences of V3 andflankingregions(Fig. 5). The sequencediversity within a sample was no less than in the V4 and V5 regions. However, in theV3 region, each sample showed particular sequence features, oftencommon toall sequences withina sample, whichdistinguished it fromothersamples. Overthe V3 region as a whole, only 23 of 83 amino acids were conserved in all of the sequences listed in Fig. 5. The immunodominant loop sequences all differed considerably from theprototypeHIVMNand
HIVHTLVIIIB
(clone HXB2) sequences and from those of other geographical HIV-1 variants (Fig. 5). The core sequence GPGRwas well con-served between individuals, although a numberof variants existed. However, in one individual (p12), the majority of sequences wereGSGR.The flanking regions of V3 contain a large number of potential N-linked glycosylation sites. Therearesix of these in theprototype HIVHTLVIIIB sequencein Fig. 5, and there are six potential sites for N-linked addition lying to either side of the V3 loop structure. No glycosylation sites are common to all of the sequences. By contrast, the two cysteine residues spanning the immunodominant loop are absolutely conserved (Fig. 5).
DISCUSSION
Diversityof envsequences withinHIV-infectedindividuals. Astriking feature of the proviralsequencesreportedhereis the diversity of env sequences within a number of the individuals studied. Inmany cases,there isacomplete lack of
homology
between variantsover anumber of amino acid residues (for example, the V5 region ofp79). It is clear thatavery considerable amountofsequencing would be neces-sary to describe fully the range of variation in proviral sequences within PBMCs. Thevisualization of length
vari-ants in the V4andV5 regions provides a rapid method for the partial characterization of sequence variants within a
sample. Analysis of the amplified DNA from samples con-taining large numbers of proviralsequencesshows therange and relative abundance of sequences that differ in length. The niethodprovides evidence for the existence of relatively scarce sequences that were not detected by conventional sequence analysis of arelatively small number of variants. The method does not provide comparative sequence infor-mation on the different length variants, and each length variant comprises an unknown number of distinct se-quences. However, itprovides a simple description of the provirus population whichallowslarge numbers of individ-ual samplestobecompared. Applications include the anal-ysis of sequence change over time in an individual (P. Simmonds, unpublished data) and comparisons between proviralvariants inPBMCsand viral RNAinplasma(L.-Q. Zhang, personal communication). It also allows genuine positive PCR results from patient screening to be distin-guished from those due to contamination by cloned se-quences(39).
Phylogenetic significance of amino acid changes in env. Many
hemophiliacs
treated withcommercial factor VIII in the early 1980s became infected with HIV-1. The rate of infectionwasconsiderably
lower in those whoweretreated with factor VIII prepared from plasma ofvolunteer blood donors inalow-prevalenceareafor HIV-1infection,
suchasScotland (21). However, somehemophiliacs treated in Ed-inburgh solely with locally produced factor VIII were in-fectedwith HIV-1in 1984(20).Asingle batch offactor VIII has been implicated in the infection of 18 hemophiliacs, including hemophiliacs p74, p77, p79, p82, p83,
p84,
p87, andp91 studiedhere. p12wasinfected intheUnited States from commercial bloodproducts.The V4 regions of six of the eight designated cohort members(p77, p79, p83,p84, p87, and p91)arevery similar toeach
other,
and allcontaintworepeatsofarelatively
well conserved 5-amino-acid sequence, TTGSN. p77 and p79 contain variantproviral
sequences thatlack one ofthetwocopies
of thissequence, whilesomevariants fromp77havea second copyofthe
preceding
LFNSTW sequence. How-ever, p82andp74, who werealsoconsideredtobeinfectedVOL.64,1990
on November 10, 2019 by guest
http://jvi.asm.org/
77-al
kdp
# ts#
s h --a v hat e i di # kd # vt e#- N77-di
kdp
# t s# s h-- v hat e i di #L d # vt e#- N77-i kdp # t s# s h-- v hat e i di #L g # n vk k N- #
77-f kdp # t s# s h --a v hat e i di N kd # vt k N- #h
77-m #n t s# s h-- v hat e i di sL ed # vt e#- #
79-m #e v # s h-- yat e t i #l e # vt k#- #
79-n #e v i N s pm-- ks yat d ii l e # vt k #-r #
87-a kdp k # g#y e s-- arrq i di k e # r vt k k#- NI
87-b kdp k # g#y e s-- arrq i di k e # r vt k e#- NI
87-c kdpk# g#y e s-- arrq i di k e # r vt k e#- #
87-d kdp k g# e s-- ar q i di k e # r vi k d#- #
87-e kd k# s# ss-- yat e i di #l e tk vte k r#- k #
87-f kd s# s s-- yat e i di #L e # r ai k k#-r # a
87-g kd s# s s-- yat e i di UL a # r ai k kf-r k #
87-hi kd s# s s-- yat e i di #L a # r ai k k#-r k #
91-a . k g# s-- atsqi di k #L ee# vt
91-b kp k # g# s-- atsqidi k #L ee# vt k#- n..
91-c . k # g# sp-- atsqi di k # ee# vt
91-d . k # gd srn-- yatd i di #L eedd vt k
91-e . k # g# s P-- yat didi #L ee dd vt ...
82-c #e v t y-- vy teq ii #e vi
e#-82-d #e v t y vy teq i i U #e vi
e#-82-e #e v# h-- vy teq i U #e vi e - a
82-f #e v i y v teq ii N #e vi
eN-82-g #e v i # y vy teq ii #e vi
e#-82-h #e v # g h-- s yatg i di #e vt k#- #
82-i #e v# # g h-- s yat g i di N #e vt k#- U
82-j #a v t h-- vy t q i di #L #e r vi g t k r
74-a #e v # rg h-- yat n i di t #d vt #-S #f
74-b #e v # rg h-- yat n i di t #d vt #-s
74-d t #e v # g# rs h --wm yat e i di Ut i vt V#- #f
74-f #e v # # rgh-- yaar i di #L t #d h vt r#-r gr
74-g #e v # s# rg h-- yat n i di t #d vt #-s t
12-b #n U sk i rs h --s y eg a dv k y tL#gt #d lvav- q p
12-c #n # sk i rsmh --s y eg a dv k y tL#gt #d Lvav- q p
12-d #s N s k rsh --s y eg t dv k y tL#gt #d L va - t p
12-e #n s#t rs h-- y t e t dveky tL#gt #d L va - k qp
MN h #e q # y k h-- y tkn i ti # #d r v k
k-RF #a q # N s tk-- viyat q i di k UL q # vt d#- ts
SF2 #e aN # s y -- h t r i di k q # e vk # UJ
HXB2 TIIVQL#TSVEI#C TRPN#NTRKRIRIQRGPGRAFVTIGKI -GNMRQAH C#ISRAKW#NTLKQIDSKLREQFGN#KTIIFKQSS|
I--- IMMUNODOMINANT
LOOP---FIG. 5. Alignment of sequences obtained fromsevenhemophiliacs in theV3andflanking regions. Individual sequencesareindicatedas
ato z. Symbolsare asforFig.3. Differences from thepHXB2 sequenceareshown in lowercase. Allpotential N-linkedglycosylationsites are shown.
from the same source, contained
proviral
sequences in thisarea that were distinct from each other, from those of the other cohort members and
p12,
and from publishedse-quences(Fig. 6A). However, atthe time ofseroconversion, the V4 sequences of both individuals were similar to those
reported
here for the twoindividuals(Zhangand Simmonds, unpublished observations). This finding rulesout thepossi-bility
thatahigher rate of sequence change ofHIV in thesetwo individuals was responsible for the current dissimilari-tiesin V4 sequencesfromtherestof the cohort. At present, itisnot clear whether several strains ofHIV-1 werepresent
in the same contaminated batch of factor VIII orwhether p82andp74wereinfectedfromadifferentsourcealtogether. There isnoevidence for other risk factors forHIVinfection in either hemophiliac. The most likely sources are other infectious batches oflocally prepared factor VIII concen-trate. V4 sequencessimilartothoseofp82have beenfound in anotherhemophiliac (datanotshown)whowasnottreated with theimplicatedbatch of factor VIII but who didreceive several otherlocally producedbatches thatwere alsogiven
top82.
Incontrast tothesimilarities observed in the V4
region,
noon November 10, 2019 by guest
http://jvi.asm.org/
[image:7.612.106.509.69.559.2]ANALYSIS OF SEQUENCE VARIATION IN gpl20 5847
A
p77 |StWLFNSTW|Nd TTesN tTeS? NtEpIt p79 STw Nd ttGSn tTGSN nTEpIT p83 STW ND TTGlN iTGSN N?gnIT p84 STW ND TTGSN TTGSn NTETIT p87 STW Nd TteSN TTgSN NnEtIT p91 STW ND tTgsN tTgSN NTEiIt p82 STW nst| DltqtNSTQ-NkeEN IT p74 STW NN?DTSTWNK?eESgN IT
p12 SNW StSPGEpNNTTGN-- IT HXB2 STUWFNSTW|S S NN ji DT IT
RF STW NSTEGSNNTGGNDT IT
SF-2 NTW RLNHTEGTKGNDT II
B
p77 DGG
rn[N
eieI
TEtI
p79 dGg kn?s??-t ET p83 DGG ?egN?T E. p84 DGG -ENR?eT E. p87 DGG ?ngnk?e? Ei p91 DGG gsNss--N ETp82
p74
p12
HXB2
RF
SF-2
DGG NSgnksndTT Et DGG tEn?-TENRtT El DGG
?-Ne iI
Ei DGG NSNNES ETDGG E DTT NTT El
DGG
T NVT NT EVC
p77 TIIVQLkdpVnITC TRPSNNTRKSIHI--gPGRVFHATGEIIGDIRQAH
CnLSR?dWNNTLkQIVtKLrEQFeN-KTIIFNqSSI
p79 TIIVQLNESWINC ?RPNNNTRKSI??--GPG??FYATG?I?GNIRQAH
CNLSRAEWNNTLKQIVTKL?EQF?N-?TIIFNQSSI
p87 TIIVQLKDpVkINC TRPgNnTRerISI--GPGRAF?A?g?IIGDIRQAH CN?S?AeUnnTLrQIv?kLKEQF?N-kTiIFNQSs p91 TIIVQLKNPVKINC TRPGnNTRKsIpI--GPGRAFvATsqIIGDIRkAH CNLSREEWnnTLKQIVTKLrEQFKN-KTIIFN...Cons TIIVQLkdpV?InC tRP?nnTRksl?i--gPGraF?Atg?IiGdlRqAH CnLSraeWnnTLkQIvtkLkEQF?N-kTiiFNqSs
Cons TIiVqLNeSVvInC tRPnrntRk?ihi--gpGrafyttg?IiGdirqAh CnisrakWn?TLkqIv?KLrEQF?n-ktliF?qSS
p82 TIIVQLNeSWInC tRPNNNTRKrI?I--GPGrAvYtTeqIIGnIRQAH CNiSRAKWNETLkQIViKLrEQFen-KtIVFkqSS;
p74 TIiVQLNESWINC TRPnNNTRRgIHI--gpGRAFYAtgnIIGDIRQAH CNiSRTKWndTLKqIVTKLREQFgN-sTIVFnqSSI p12 TIIVQLNnSVEINC TRPS?n?RRS?HI--GsGRAFYTiegI?GDVrKAY CTLNGTKWNDTLKLIVAKLREQFGN-KTI?FkPSSI
MN TIIVHLNESVQINC TRPNYNKRKRIHI--GPGRAFYTTKNIIGTIRQAH CNISRAKWNDTLRQIVSKLKEQFKN-KTIIFNQSS RF TIIVQLNASVQINC TRPNNNTRKSITK--GPGRVIYATGQIIGDIRKAH CNLSRAQWNNTLKQIVTKLREQFDN-KTIIFTSSS SF2 TIIVQLNESVAINC TRPNNNTRKSIYI--GPGRAFHTTGRIIGDIRKAH CNISRAQUNNTLEQIVKKLREQFGNNKTIIFNQSS
HX82 TIIVQLNTSVEINC TRPNNNTRKRIRIQRGPGRAFVTIGKI-GNMRQAH CNISRAKWNNTLKQIDSKLREQFGNNKTIIFKQSSJ
FIG. 6. Comparison of hemophiliac and published sequences in the env hypervariableregions V4 (A), V5 (B), and V3 (C). Consensus sequencesfrom individuals believedto havebeeninfected from a common source (p77, p79, p83, p83, p87, andp91)are compared with those ofapparently unrelatedsequences(p82andp74), thoseof a hemophiliac infectedfrom commercial factorVIII, and published sequences. Differences fromtheconsensus within infected individual are indicated in lowercase. ?, No majority consensus at this position in an individual orgroup consensussequence.
relationship between thecohort members is apparent in V5 (Fig. 6B), where sequence variation is much greater. The
rateofsequencechangein the short V5region appears more rapidand does notindicateanyrelatednessbetween the six individuals who have similar V4 sequences. The V3 regions of the members of the hemophiliac cohort differ from the reference HXB2 sequence by a number of amino acids. However, no clear relationship distinguishes the cohort sequences. This is shown bythesimilarity in the consensus of the four confirmed cohort members and the combined
consensus of noncohort members p12, p74, and p82 and published sequences (Fig. 6C). There are only five
differ-encesbetween theconsensus sequences of thetwogroups, and at all but one of these sites, variants within the
con-firmed cohort members exist that match the noncohort sequences (Fig. 6C).
Nature of variation in the hypervariable regions of env.
Both amino acid substitutions and gaps contribute to
varia-tion in the V4 and V5 regions. Many of the gaps in V4 involve repeated sequences suchasTTGSN (p77 and p79). In such cases, some variants have one copy while others have two. Similar variation in the numbers ofcopies of the sequence (F)NSTWmay also befound inp77. Indeed, the existenceof repeatsofthesetwo sequencesinmost individ-uals indicates thatsome sort ofduplicationeventhas taken place. In many sequences, there are minor differences
be-tweenthe twocopies, suggestingthat somesequencechange has taken place after duplication. The exact sequence in-volved in a duplication event may differ. Forexample, the
block NSTW isrepeatedinp82, while inp77thereis oftena
repeat of the longer sequence LFNSTW. In published
se-quences of the viral isolated HIVHTLVIIIB, the BH8 clone has one copy of the sequence FNSTW, while others
(for
example BH10) have two. The widespread occurrence of these repeated sequences (3, 11, 23, 34)and the likelihood that they occur independently in different HIV-infected
VOL.64, 1990
I
on November 10, 2019 by guest
http://jvi.asm.org/
[image:8.612.104.510.78.473.2]individuals suggest that this sequence is predisposed to duplicationduring eitherreverse transcriptionorRNA syn-thesis. Furthermore, ifduplicationisoccurring repeatedlyin thesamplesthat we haveexamined,it is alsolikelythatonce
formed, theduplicationsare predisposedtodeletion. Alloftheinsertionsordeletions in the V4 andV5regions aremultiples of3 nucleotides,thus
maintaining
thereading frame downstream. Similarly, only one chain termination mutation (p79, sequence 1; Fig. 3) was found in 37 V3 sequences and 71 V4 and V5 sequences. The low rate of inactivating mutations is consistent an absence of pheno-typic mixing. Thisfinding mayreflectthe low copy number (closeto one) ofprovirus in infected PBMCs(30).Positive selection for sequence change in hypervariable regions of env. There has been nosatisfactoryexplanationof the high rates of mutation in localized regions of the env
gene. It could be argued thatthe cause is
simply
a lack of functional constraints which might limit the amount of variation in regions such as the CD4 binding site (6, 16). However, this view isnotsupportedbyacomparison ofthe rates ofsynonymousandnonsynonymoussubstitutionrates in the different regions of env. Published data and data collected in thislaboratory (P. Balfeetal., unpublished data) give a ratio of synonymous to replacement substitutions(KsIKa
ratio; 18) for the CD4 binding site of 1.24. For comparison, the Ks/Ka ratio for gag sequences (17) and those ofsamples studiedhereis about6.7, andtheratio for 42 eucaryoticgenes is5.28(18). Thus, theCD4bindingsite does not appear to be understringent constrainttomaintain its amino acid sequence, and the much higher substitution frequency of the hypervariableregionscannotsimply be due to lack of constraint. The KsIKa ratio for the sequences reported here ofthe V3 loop and flanking regions is 0.67, lowerthananypreviouslyreported. Onaverage, the survival ofareplacement mutation is almost twiceasprobable
asthe survival of a synonymous mutation.Overall,
thissignifies
that selection favors change in this region, although the absence of stopcodonsand the conservationofanumberof aminoacids, includingthecysteineresidues
spanning
the V3 loop, indicate that the extent ofchange possible is limited. Although positive selection for changeisunusual,it has been observed in majorhistocompatibility complex proteins (13) and in mammalian and avianserineprotease inhibitors(12, 15). For the specific requirements ofincreaseddiversity of antigenic recognition by the majorhistocompatibility
com-plex molecule, and for defense againsta range of bacterial proteases in the latter example, positive selection appears to confer a selective advantage for the mutated sequences. Given theknown involvement ofV3 in virus neutralization (28), selection forchange in V3 suggests that the selective forceis the immune defense system.
Consequences of sequence change in the hypervariable regions of env. Several areas of the env gene product have beenshown to beantigenicupon natural infection and upon vaccination (8, 9, 19,26-28,36). The V3 loop and regions in gp4l have been positivelyidentified as targets of antibody-mediated neutralization or cytotoxic T-lymphocyte (CTL) killing. Both immune effector functions are sensitive to sequence variation around the crown of the V3 loop, i.e., either side of the relatively well conserved GPGR central sequence. The specificity of the CTL response can be determined in large part by the amino acids immediately downstream of GPGR (36), while residues immediately upstream of GPGR have been shown to be important in antibody-mediated neutralization (10, 19, 22). Considerable sequence variation is found in both of these sites in the
sequences obtained in this
study.
Bothtyrosine
and valinearefoundat a
position
criticalfor CTLrecognition (36),
notonly in different
samples
but also within the samesample
(e.g.,p87 and p91;
Fig. 6C).
Ahistidine substitution atthis site (p77) would also beexpected
tomodify
theepitope.
Other amino acid
changes
are concentrated at the site of B-cellrecognition,withmostsamplescontaining
atleasttwodifferentsequences. Therearealsomutations in the central GPGRmotif
(p12,
p74,andp77)thatcoulddisrupt
the,Bturn at this site (22) and probably abolish immunerecognition.
Thishighlevelofvariationin V3
predicts
the existence ofawide range ofneutralization serotypes in many of the pa-tients. Sera and CTLs from such individuals may be
ex-pected
to be reactiveagainst
a range of standard viral isolates. Thebroadening specificity ofneutralizing
antibod-ieswithtimeseeninHIV-seropositve individuals(1)
maybe due to the de novo appearance of V3 serotypes uponlong-term
infection.Neutralizing
antibodies that bind to V4 and V5regions
have notbeendescribed, and thecontribution ofthesetwo
regions to the overall antigenicity of
gpl20
is uncertain. However,bothregions
have beenidentifiedasbeing
poten-tially antigenic on the basis of surfaceprobability
and hydrophilicity (23). The absence of linearepitopes
ineitherregion
is shown by the poorserological reactivity
withsynthetic oligopeptides containing
V4andV5sequences and the lowimmunogenicity
of suchpeptides
uponvaccination (25). This does notrule outthepossibility
that V4 and V5 form conformationalepitopes
that are not mimickedby
synthetic peptides.
Furthermore, theantigenicity
of bothareasin vivomay bealtered
by
posttranslational
additionsof N-linkedoligosaccharide
groups.gpl20
isheavily
glycosy-lated at N-linked but not at 0-linked sites
(14),
and all 24potential
N-linked sites areglycosylated
whenrecombinant HIVHTLVIIIBgpl2O
isexpressed
in mammalian cells(T.
J.Gregory,
C. K.Leonard,
L.Riddle,
J. R.Thomas,
R.J.Harris,
and M. W.Spellman,
J. Cell. Biochem.14D:151,
1990). N-linked glycosylation can mask
potential
peptide
epitopes
(2, 32)orthemselves formconformationalepitopes
(2, 7, 33, 35).
Glycosylation
ofV4 and V5 may thereforeserve to mask the
relatively
invariantintervening
CD4binding region.
The absence ofmonoclonal antibodiestoV4,
C4,
andV5 may thus be areflection oftheeffectiveness ofglycosylation
inmasking potential epitopes
rather than asupposed
lowantigenicity
of theunderlying
peptide
se-quences. The
preponderance
ofglycosylation
sites in the hypervariable regions, and the major alterationin theposi-tionand numberofsuchsites
by
aminoacidsubstitutionand sequencereduplication,
could therefore beinterpreted
asanevolutionary
responseby
HIVtoevade the immunesystem.ACKNOWLEDGMENTS
We thank F. McOmish andA. Cleland for technicalassistance.
Samples were collected by the staff ofthe Haemophilia Centre, Edinburgh Royal Infirmary.
Theworkwassupported bytheMedicalResearchCouncilAIDS DirectedProgramme.
LITERATURECITED
1. Albert, J.,B.Abrahamson,K.Nagy,E.Aurelius,H.Gaines,G. Nystrom,and E. M.Fenyo. 1990.Rapid developmentof
isolate-specific neutralising antibodies afterprimary HIV-1 infection andconsequent emergenceof virus variants which resist neu-tralisationby autologoussera. AIDS4:107-112.
2. Alexander, S.,andJ.H. Elder.1984.Carbohydrate dramatically
influences immune reactivity of antiserato viral glycoprotein antigens. Science 226:1328-1330.
on November 10, 2019 by guest
http://jvi.asm.org/
ANALYSIS OF SEQUENCE VARIATION IN gp120 5849 3. Alizon, M., S. Wain-Hobson, L. Montagnier, and P. Sonigo.
1986. Genetic variability of the AIDS virus: nucleotide sequence analysis of two isolates from African patients. Cell46:63-74. 4. Carpenter, S., L. H. Evans, M. Sevoian, and B. Chesebro. 1987.
Role of the host immune response in selection of equine infectious anemia virus variants. J. Virol. 61:3783-3789. 5. Clements, J. E., F. S. Pedersen,0. Narayan, and W. A.
Hasel-tine. 1980. Genomic changesassociated withantigenicvariation of visna virusduring persistent infection. Proc. Natl. Acad.Sci. USA 77:4454 -458.
6. Cordonnier, A., L. Montagnier, and M. Emmerman. 1989. Single amino-acidchanges inHIVenvelope affectviraltropism and receptor binding. Nature (London) 340:571-574.
7. Feizi, T., and R. A. Childs. 1987. Carbohydrates as antigenic determinants. Biochem. J. 245:1-12.
8. Gnann, J. W., P. L. Schwimmbeck, J. A. Nelson, A. B. Truax, and M. B. A. Oldstone. 1987. Diagnosis of AIDS by using a 12-amino acid peptiderepresenting animmunodominantepitope of the human immunodeficiency virus. J. Infect. Dis. 156:261-267.
9. Goudsmit, J., C. A. B. Boucher, R. H. Meloen, L. G. Epstein, L. Smit, L. Van Der Hoek, and M. Bakker. 1988. Human antibody response to a strain-specific HIV-1 gpl20 epitope associated with cell fusioninhibition. AIDS 2:157-164. 10. Goudsmit, J., M. C. Debouck, R. H. Meloen, L. Smit, M.
Bakker, D. M. Asher, A.V. Wolff, C.J. Gibbs, and D. C.
Gaidusek.1987. Human immunodeficiency virustype 1 neutral-isation epitope with conserved architecture elicits early type-specific antibodies in experimentally infected chimpanzees. Proc. Natl.Acad. Sci. USA 85:4478-4482.
11. Hahn, B., G. M. Shaw, M. E. Taylor, R.R. Redfield, P. D. Markham, S. Z. Salahuddin, F. Wong-Staal, R. C.Gallo, E. S. Parks, and W. P.Parks. 1986. Genetic variation in HTLV-III/ LAV over time in patients with AIDS or at risk for AIDS. Science 232:1548-1553.
12. Hill, R. E., and N.D.Hastie. 1987.Accelerated evolution in the reactive centre regions of serine protease inhibitors. Nature (London) 326:96-99.
13. Hughes, A. L., and M. Nei. 1989. Nucleotide substitution at major histocompatibility complex class II loci: evidence for overdominant selection. Proc. Natl. Acad. Sci. USA 86:958-962.
14. Kozarsky, K., M. Penman, L. Basiripour, W. Haseltine, J. Sodroski, andM.Krieger. 1989.Glycosylationandprocessingof thehumanimmunodeficiency virus type 1 envelope protein. J. AIDS 2:163-169.
15. Laskowski, M.,I. Kato, W. Ardelt, J. Cook,A.Denton, M. W. Empie, W. J.Kohr, S.J.Park, K.Parks,B. L.Schatzley,0. L. Schoenberger, M.Tashiro, G.Vichot, H. E.Whatley, A. Wiec-zorek, and M.Wieczorek. 1987. Ovomucoid thirddomains from 100 avianspecies: isolation,sequences, and hypervariabilityof enzyme-inhibitor contact residues. Biochemistry 26:202-221. 16. Lasky, L. A., G. Nakamura, D. H. Smith, C. Fennie, C.
Shi-masaki, E. Patzer, P. Berman, T. Gregory, and D. J. Capon. 1987. Delineation of aregion of the humanimmunodeficiency virus type1gpl20glycoprotein criticalforinteraction with the CD4receptor. Cell50:975-985.
17. Leigh Brown, A., and P. Monaghan. 1988. Evolution of the structuralproteinsofhumanimmunodeficiency virus: selective constraintsonnucleotide substitution. AIDS Res. Human Ret-roviruses 4:399-407.
18. Li, W.-H., C.-I.Wu, and C.-C. Luo. 1985. A new method for estimatingsynonymous andnonsynonymous ratesofnucleotide substitution consideringtherelative likelihoodofnucleotideand codonchanges. Mol. Biol. Evol. 2:150-174.
19. Looney, D. J., A. G. Fisher, S. D.Putney, J. R. Rusche, R. R. Redfield, D.S. Burke, R. C. Gallo, and F. Wong-Staal. 1988. Type-restricted neutralization of molecular clones of human immunodeficiency virus. Science241:357-359.
20. Ludlam, C. A., J. Tucker, C. M. Steel, R. S. Tedder, R. Cheingsong-Popov, R. A. Weiss, D. B.McClelland, I. Philip,and R. J. Prescott. 1985. Human T-lymphotropic virus type III (HTLV-III) infectioninseronegative haemophiliacs after
trans-fusion of factor VIII. Lancetii:233-236.
21. Melbye, M., K. S. Froebel, R. Madhok, R.J. Biggar, P.S. Sarin,S. Sternbjerg, G. D. 0. Lowe, C. D.Forbes, J. J. Goed-ert,R.C.Gallo,and P.Ebbeson.1984.HTLV-IIIseropositivity
inEuropeonhaemophiliacsexposedtoFactor VIIIconcentrate imported from the USA. Lancet ii:1444 1446.
22. Meloen, R. H.,R. M. Liskamp,andJ. Goudsmit. 1989. Speci-ficityandfunctionof the individual amino acids ofanimportant
determinant of HIV-1 that induces neutralising antibody. J. Gen. Virol. 70:1505-1512.
23. Modrow, S., B. H.Hahn, G. M. Shaw,R.C. Gallo,F. Wong-Staal, and H. Wolf. 1986. Computer-assisted analysisof enve-lopeproteinsequencesofsevenhumanimmunodeficiency virus isolates: prediction of antigenic epitopes in conserved and variableregions. J. Virol. 61:570-578.
24. Narayan,O.,D. E.Griffin,and J.Chase. 1977.Antigenic shift of visna virus inpersistentlyinfected sheep. Science 197:376-378. 25. Neurath, A.R., N.Strick,and E.S. Y.Lee. 1990. Bcellepitope
mapping of human immunodeficiency virusenvelope
glycopro-teins with long (19- to 36-residue) synthetic peptides. J. Gen. Virol. 71:85-95.
26. Palker, T. J., T. J. Matthews, M. E. Clark, G. J. Cianciole, R. R.Randall, A.J. Langlois, G. C.White, B. Safai,R. Snyder-man, D. P. Bolognesi, and B. F. Haynes. 1987. A conserved region at theCOOHterminusof human immunodeficiencyvirus gpl20envelope protein contains an immunodominant epitope. Proc. Natl. Acad. Sci. USA 84:2479-2483.
27. Reitz, M. S., C. Wilson, C. Naugle, R. C. Gallo, and M. Robert-Guroff. 1988. Generation of a neutralization-resistant variant ofHIV-1 is duetoselectionforapointmutation in the envelope gene. Cell54:57-63.
28. Rusche,J. R.,K.Javaherian, C. McDanal, J. Petro, D. L.Lynn, R. Grimaila, A. Langlois, R. C. Gallo, L. 0. Arthur, P.J. Fischinger, D. P. Bolognesi, S. D. Putney, and T. J. Matthews. 1988.Antibodies that inhibit fusion of human immunodeficiency virus-infected cells bind a 24-amino acid sequence oftheviral envelope, gpl20. Proc. Natl. Acad. Sci. USA 85:3198-3202. 29. Saag, M. S., B. H. Hahn, J. Gibbons, Y. Li, E. S. Parks, W. P.
Parks, and G. M. Shaw. 1988. Extensive variation of human immunodeficiency virus type-1 in vivo. Nature (London) 334: 440-444.
30. Simmonds, P., P. Balfe, J. F. Peutherer, C. A. Ludlam, J. 0. Bishop, and A. Leigh Brown. 1990. Human immunodeficiency virus-infected individuals contain provirus in small numbers of peripheralmononuclearcells andatlow copy numbers. J.Virol. 64:864-872.
31. Simmonds, P., F. A. L. Lainson, R. Cuthbert, C. M. Steel, J. F. Peutherer, and C. A. Ludlam. 1988. HIV antigen andantibody detection: variable responses to infection in the Edinburgh haemophiliac cohort. Br. Med. J. 296:593-598.
32. Skehel, J., D. J. Stevens, R. S. Daniels, A. R. Douglas, M. Knossow,I. A. Wilson, and D. C. Wiley. 1984. Acarbohydrate side chainonhaemagglutinins ofHongKonginfluenza viruses inhibits recognition by a monoclonal antibody. Proc. Natl. Acad. Sci. USA 81:1779-1783.
33. Sodora, D. L., G. H. Cohen, and R. J. Eisenberg. 1989. Influ-ence of asparagine-linked oligosaccharides on antigenicity, processing, and cell surface expression ofherpes simplex virus type 1glycoprotein D. J. Virol. 63:5184-5193.
34. Starcich, B. R., B. H. Hahn, G. M. Shaw, P. D. McNeely, S. Modrow, H. Wolf, E. S. Parks, W. P. Parks, S. F. Josephs, R. C. Gallo, and F. Wong-Staal. 1986. Identification and char-acterization of conserved andvariable regions in the envelope gene ofHTLVIII/LAV, the retrovirus of AIDS. Cell 45:637-648.
35. Sugwara, K., F. Kitame, H. Nishimura, and K. Kakamura. 1988. Operationaland topological analysesofantigenic sites on influ-enza C virus glycoprotein and their dependence of glycosyl-ation. J. Gen. Virol. 69:537-547.
36. Takahashi, H., S. Merli, S. D. Putney, R. Houghten, B. Moss, R. N. Germain, and J. A. Berzofsky. 1989. Asingleamino acid interchangeyieldsreciprocalCTL specificities for HIV-1gpl60. Science246:118-121.
VOL.64, 1990
on November 10, 2019 by guest
http://jvi.asm.org/
37. Tsang, T. C., and D. R. Bentley. 1989. An improved method using Sequenase (tm) that is independent of template
concen-tration. Nucleic Acids Res. 16:6238.
38. Willey, R. L., R. A. Rutledge, S. Dias, T. Folks, T. Theodore,
C. E. Buckler, and M. A. Martin. 1986. Identification of
con-served anddivergent domains within the envelopegeneof the
acquired immunodeficiency virus syndrome retrovirus. Proc. Natl. Acad. Sci. USA 83:5038-5042.
39. Williams,P.,P.Simmonds, P. L. Yap,P. Balfe, J.0.Bishop,R.
Brettle,R.Hague, D.Hargreaves, J. Inglis, A. Leigh Brown,J. Peutherer, S. Rebus,andJ. Mok. 1990. Thepolymerase chain reaction in the diagnosis of vertically transmitted HIV infection. AIDS 4:393-398.
40. Winship, P. R.1989. An improvedmethod for directly
sequenc-ing PCR amplified material ussequenc-ing dimethyl sulphoxide. Nucleic Acids Res. 17:1266.