Vol.62,No.6 JOURNALOFVIROLOGY,June1988, p. 1898-1906
0022-538X/88/061898-09$02.00/0
Copyright C)1988,AmericanSociety forMicrobiology
A
Single Point Mutation Has
Pleiotropic
Effects
on
pp60vsrc
Function
MELANIE J. WELHAM* ANDJOHN A. WYKEt
Imperial CancerResearch FundLaboratories, St. Bartholomew's Hospital, Dominion House,Bartholomew Close, LondonECIA 7BE, UnitedKingdom
Received 25 November1987/Accepted 22February1988
TheRoussarcoma virus mutant tsLA29 encodesapp6ov-src moleculethat istemperaturesensitiveforboth tyrosinekinase activityand itsabilitytolocateatthe cellperiphery.Thedefect inlocalizationappearsto bedue toaperturbationin eventsfollowing complexdissociation, since themutantenzymeshowsarapidlyreversible
association with the cytoskeleton when shifted between permissive and restrictive temperatures. Although tsLA29
pp60v-s'c
differsfrom the wildtype
atthree amino acidresidues,
studies withchimericproteins
show that only one of the mutations, an alanine-for-proline substitution at residue 507, accounts for all the temperature-sensitive characteristics. Moreover,asinglesecond sitemutation, atresidue427,canrestorethewildphenotype. Cells infectedwithachimeric virusencoding onlythealaninesubstitutionatposition507have
aconspicuouslyfusiformmorphology, suggestingthat this mutation also hassubtle effectsonpp6O-srcfunction thatare apparently compensatedforby theother mutations in native tsLA29.
The product of the v-src gene of Rous sarcoma virus (RSV) isa60-kilodalton phosphoprotein,
pp6Ov-sr,
whichis associated with the cytoplasmic face of the plasma mem-brane and with the cytoskeleton (5, 20, 21, 39). pp6Ov-src alone iscapableofinducing andmaintaining transformation of cells in culture, and, in common with many other viral oncogene products and growth factor receptors, its only well-documented function isthat ofatyrosine-specific pro-tein kinase.Anumberof potential substrates for thisactivityare
phosphorylated
in RSV-transformed cells (14, 22, 38,43), but in no case hastheirphosphorylationbeen shownto beasufficient precondition for cell
transformation,
suggest-ingthat crucial features ofpp6Ovsr" activity have yet to be identified.Amajor contribution to the understanding ofv-src func-tion has been obtained from the physiological and genetic analysis ofa wide range ofRSV mutants (52). It has been possible as aresult tobuild up a consensus model defining domainsof thepp6Ov-srcprotein that affectdifferent aspects ofits biogenesis andfunction. Newly synthesized
pp6Ov-src
immediately enters acomplex with two cytosolic proteins, pp9OandppSO(2, 3).pp9Oisanalogoustothe90-kilodalton heat shock protein and the steroid hormone receptor-binding protein (41), whereas ppSO is a rare protein of no known function(1). This complex has been implicatedin the trans-portofpp6Ov-srctotheplasma membrane (1, 11), but it is not clearwhich regions ofpp6Ov-srcareimportantfor its forma-tion (52). Mutant proteins whose lesions lie within both C-terminal andN-terminal portions display abnormal kinet-ics ofcomplex formation or dissociation, whereas studies with antipeptide antibodies suggest that the carboxy-ter-minal region may be important in association with pp9O and ppSO (46, 52). Subsequent myristylation of the pp6Ov-sr N-terminal glycine residue is an absolute requirement for localizationattheinner face of the plasma membrane (7, 13, 18). Replacement of the N-terminal glycine with either
* Correspondingauthor.
t Present address: BeatsonInstitute for Cancer Research, Bears-den, Glasgow G611BD, Scotland.
alanine or glutamic acid results in proteins that are not myristylated, are unable to associate with the membrane (although retaining a fully functional kinase), and as a consequence are unable to transform cells (6, 25). Other mutants, which retain the N-terminal glycine residue but have extensive deletions in the amino-terminal region, also displayaninability to localize at the cellperiphery, suggest-ing that the amino-terminal 8- to 15-kilodalton region is important for attachment (18, 29, 31).
The catalytic or kinase domain, located in the carboxy-terminal half of the protein, wasinitially defined by proteo-lytic digestion (31) and more recently by the demonstration that many mutations mapping within this region affect the tyrosine kinase activity in vitro and in vivo (4, 52). The carboxy-terminal region may also play a role in amitogenic activity ofpp6vsrc, but this does not depend on a fully activetyrosine kinase(8, 23).
Finally, a region in the amino-terminal half ofpp60vsrc, with minimum boundaries fromamino acids 81 and 169, is thought to play an ill-defined modulatory role in pp6Ov-src functioning. Many mutants with lesions in this region pos-sess kinase activity, but induce an aberrant morphology in transformed cells, possibly reflecting an impaired or altered substratespecificity (12, 24).
We havepreviously studied severaltemperature-sensitive (ts) mutants of RSV Prague A to elucidatethe relationships between structure and function in pp6Ovsrc. One such mu-tant, tsLA29, falls into the commonclass of mutants with a defective tyrosine kinase (44). Membrane association is ts for this mutant, whereas myristylation occurs normally at both permissive and nonpermissive temperatures (45). De-letion analysis has mapped a crucial mutation within the carboxy-terminalregion of the pp6Ov-src protein (16). In this study we have extended the physiological characterization of tsLA29 andprecisely defineditsmutations. Construction of chimeric v-src genes and analysis of the biochemical properties of the resultant mutant proteins have identified a critical tspoint mutation which alters the extreme carboxy terminus of pp60vsrc. This single amino acid substitution appearsto havepleiotropic effectsonv-src function. 1898
on November 10, 2019 by guest
http://jvi.asm.org/
MATERIALS AND METHODS
Cells, viruses, and antisera. Chicken embryo fibroblasts (CEF)wereprepared from fertileeggscultured and infected as previously described (47). Infected cells were grown at 35°C (permissive temperature) or41°C (nonpermissive tem-perature). Isolation of tsLA29 RSV and itsrevertants tothe wildtype were described previously (49-51).
tsLA29-transformed Rat-1 cellswere isolated by
coinfec-tion of Rat-1fibroblastsathigh multiplicity with tsLA29 and Carr Zilber-associatedvirus, followed by single-cell cloning (48a). tsLA29 Rat-1 cells were grownin Dulbecco modified Eagle medium supplemented with antibiotics and 5%
new-born calfserum at 35°C (permissive temperature) or39.5°C
(nonpermissive temperature). Tumor-bearing rabbit sera wereused as described previously (15, 44). The monoclonal
antibody to pp60vsrc, JB327, was kindly provided by J.
Brugge (32). The rabbit polyclonal anti-pp60vsrc antiserum
wasraised against the bacterial fusion protein by the method
of Gilmer and Erikson (19) with an expression plasmid
kindly provided by R. Erikson.
Biosynthetic labeling and cell fractionation.Plates (35 mm) of cellswereincubated for 30minto1 h withmethionine-free Dulbecco modified Eagle medium. For studies to localize pp60v-src, cells were labeled with [35S]methionine (100 ,uCi/ml) for 3 h. For pulse-chase analyses, cellswerepulsed for 15min with 200 ,uCi of[35S]methionine perml, washed twice with cold medium, and chased with cold complete medium forvarious time intervals atthe requiredincubation temperature.
Cells for fractionation were washed three times with ice-cold phosphate-buffered saline and incubated on ice.
Lysates were made in situ by adding 0.5 ml of modified cytoskeletal extractionbuffer(MCSK; 20 mM N-2-hydroxy-ethylpiperazine-N'-2-ethanesulfonic acid [HEPES, pH 6.8], 3 mM MgCl2, 50 mM NaCl, 0.3 M sucrose, 0.5% Nonidet P-40 [NP-40], 1 mM CaCl2) to each dish. After 5 min, with occasional gentle rocking, the lysates were collected and a
further 0.5 ml of MCSK was addedto each dish, incubated for2 min, andremoved, andafinal 0.5-ml portion of buffer
was added and again incubated for2 min. All three lysates were pooled to give the soluble fraction. The residual insolublestructureswere solubilized with modified radioim-munoprecipitation assay buffer (RIPA; 1% NP-40, 1%
so-dium deoxycholate, 0.1% sodium dodecyl sulfate, 0.15 M NaCl, 0.01 M sodium phosphate [pH 7.0], 2 mM EDTA, 2 mM phenylmethylsulfonyl fluoride) or NP-40 buffer (1% NP-40, 150 mM NaCl, 20mM Tris [pH 6.8], 20 mM sodium PPi) and used asthe cytoskeletalfraction.
Immunoprecipitation and gel electrophoresis. Immunopre-cipitation with the monoclonal antibody JB327 and the polyclonal rabbit anti-pp60vsrc serum and sodium dodecyl-sulfate polyacrylamide gel electrophoresis were performed
aspreviously described(27, 30);in allcases 10% polyacryl-amide gelswere used. Gelswere impregnatedwithAmplify (Amersham Corp.), dried, and exposed on Kodak XAR-5 autoradiographic filmat -70°C.
Sedimentation analysis. Cells were labeled for 3 h with
[35S]methionine (100
,uCi/ml),
andlysateswere prepared byusingMCSK and NP-40 buffers butin smaller volumes (0.5 ml total). Lysates were layered onto 10 to 30% glycerol gradientsin NP-40 buffer. Centrifugation wascarried out in
a Beckman SW50 rotor at 230,000 x g at 4°C for 17 h.
Fractions(0.2 ml)werecollectedfrom thegradients, diluted
to 1 ml with NP-40 buffer, immunoprecipitated withJB327,
and
analyzed by
sodiumdodecyl sulfate-polyacrylamide gel
electrophoresis
on a 10%polyacrylamide
gel.
Kinase assays.Cells seededon60-mm disheswerewashed three times with
phosphate-buffered
saline andlysed
in NP-40 buffer. Kinase assays were carried out withequal
amountsof
protein
foreachsample
aspreviously
described(10, 44).
Tumor-bearing
rabbit serum no. 5whichdisplays
cross-reactivity
with c-src, was used forimmunoprecipita-tion.
Molecular
cloning
ofproviral
DNA.High-molecular-weight
DNAwasisolated from tsLA29-transformed Rat-1cells and from CEF transformedby
the revertant virus tsLA29 R2.The
genomic
DNA was restricted with EcoRI andelectro-phoresed through
0.8%
agarosegels,
and DNAbetween the sizes of2.2and 4.4kilobases(kb)
waselectrophoresed
ontoDEAE81-cellulose paper
(previously
soakedovernight
in 2.5 M NaCl and washed with distilledwater).
The DNA waselutedfromthe paperat
37°C
with1.5 MNaCl
in TE(10
mM Trishydrochloride [pH 8.0],
0.01 mMEDTA),
with exten-sivevortexing,
for 1 to 2 h. The paper was removedby
centrifugation,
and the DNAwas recovered from the super-natantby
phenol-chloroform
extraction and ethanolprecip-itation. Size-selected DNA was
ligated
to agarose gel-pu-rifiedAgtwes.XB
EcoRI arms(tsLA29)
or XNM1149 arms(tsLA29
R2)
packaged
in vitro and recombinantbacterio-phages
wereplated
onLE392by using
standardtechniques
(33, 34).
Plaques
werescreened,
insitu,
witha32P-labeled
nick-translatedv-src-specific probe prepared
from the612-base-pair
PstIfragment
(nucleotides
8054 to 8666[see
Fig.
5];
nucleotidenumbering
follows that of Schwartz et al.[42]).
Twopositive
clones were identified for the tsLA29library,
and fourpositive
clones were identified for thetsLA29 R2
library.
Sequencing
of tsLA29 and tsLA29 R2 DNA. The 3.1-kbEcoRI
fragment
waspurified
fromonepositive
lambda clone foreachvirus,
subclonedintotheEcoRIsiteofpUC13,
andsubsequently
digested
with a combination ofPstI, EcoRI,
andBglII.
src-specific
sequences were subcloned into M13vectors and used for
sequencing by
the chain terminationprocedure (36,
40).
Construction of chimeras. Chimeric src genes were con-structed
by
using
standardtechniques.
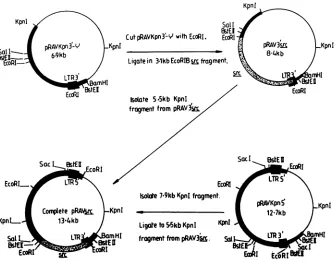
Thevectorusedwas based on achimera between Rous-associated virus type 1(RAV-1)
and avianerythroblastosis
virus(43a, 48),
whose salient features arerepresented
inFig.
1. Retroviralcon-structs were made
by
ligation
of the 3.1-kbEcoRI B v-srcfragment
to EcoRI-cut and calf intestinalphosphatase-treated
pRAVKpn3'-q.
Plasmids denotedpRAV3'src
weremade which contained the v-src genes of the
Prague
A,
tsLA29,
and tsLA29 R2 strains. Each of the constructscontained a
single
3'long
terminalrepeat.
Isolation ofthe 5.5-kbKpnI
fragment
from thesevectorsandligation
tothe 7.9-kbKpnI
fragment
ofpRAVKpn5'
(Fig.
1),
containing
the 5'long
terminal repeat, leads to formation of acomplete
retroviral construct. When such constructs are transfected
intoCEF
(26),
thecellsproduce
replication-competent
virus which canbe collected in theculture medium and used for furtherrounds of infection.ThepRAV3'src
constructscon-taining
the 3.1-kbEcoRI Bfragments
of tsLA29 andPrague
A were
digested
withSphI
and treated with calfintestinalphosphatase,
which removed 584 basepairs
of v-srcse-quence from nucleotides 8571 to 9155. The
SphI
584-base-pair
fragments
purified
from each vector were used togenerate chimeric v-src genes
containing
substitutions ofwild-type
and tsLA29v-srcsequences(see
Fig.
5).
Complete
constructswere madefor the chimerasasdescribed
above,
on November 10, 2019 by guest
http://jvi.asm.org/
1900 WELHAM AND WYKE
KpnI
Kpnl So(I
WtEI
Cut
pRAVKpn3-V
withEcoRI. EcoRApRAVKpn3-Y KpnI _
PRAVA3bM
KpnISatl= 6.9kb 8-4kb
lco_RI-R
/ Ligate in 31kbEcoRlB fragment.EccWil
StEI EcoRIKoate 5.5kb KpnI
fragment
frompRAV3s.
SacI
EcoRIs
Sac!
WENEcoRI... LTR
LTRE
EccRI EcoRIIsoWte
719kb KpnI
fragment. pRAVKpn nComplete pRA\tM Kpnl Kpnl
127kb
KpnL. 134kb Ligateto
5-5kb
KpnI KpniSal R fragment from
pRAV3i
sLTR53
mHI
BSt-
OstEl
sEI
EccoR
tornFIG. 1. Construction of recombinant viruses.ThepRAVvectorsystembasedonRAV-1and avianerythroblastosis virus(43a, 48)was
used to reconstructreplication-competent virus. Theimportant featuresareshown in thefigure;detailsaregivenin thetext.
the mutations being checked in each construct by DNA sequencing.
RESULTS
pp6Ov-src is synthesized on cytosolic polyribosomes and entersacomplex withtwocellularproteins, pp9O andppSO. The half-life of the complex is 9 to 15 min, which is also approximately the time taken for newly synthesized pp6Ov-srctobecome localizedatthe innerface of theplasma membrane (2, 3, 11). This has led tothesuggestion that the complex plays a role in the transport of pp6Ov-src to the membrane (1). We previously found that pp6Ov-src of tsLA29 is tsin itsability tolocalize at the cell periphery (45). This apparent defect in membrane association is not due to a
failure of myristylation, which occurs normally at both temperatures. On initial analysis of thepp6Ov-src:pp9O:pp5O complex, someinstabilitytoRIPA bufferwasobserved, but
thiswasapparently independent oftemperature (45). These observations raise thepossibility thatadefect in theputative
transportmechanism is responsible for the reduced levels of pp60v-src associated with the plasma membrane in tsLA29-transformed cells.
Glycerol gradient sedimentation analysis of complex. To investigate the possibility of a transport defect, we first
examined the nature of the complex. Although we have
foundthecomplex formed between tsLA29 pp6Ov-src, pp9O,
andppSOtobe unstable in buffers containing ionic detergents (e.g., RIPA buffer), it is possible to demonstrate complex formation in buffers containing nonionic detergents (e.g., NP-40 and Triton X-100) by coimmunoprecipitation of pp60v-src and pp9O with JB327 (45; data not shown). There-fore, the behavior of the complexwasexamined by glycerol
gradient sedimentation analysis oftsLA29Rat-1cell lysates in bufferscontaining nonionic detergents.
Comparison of the sedimentation profilesatboth permis-sive(35°C) and nonpermissive (39.5°C)temperaturesfor the
soluble fractions shows both monomeric
(slower-sedi-menting) and complexed
(faster-sedimenting)
forms ofpp6O-vsrc
(Fig. 2).Thecytoskeletal
fractionatthepermissive
temperature showsonly monomeric pp60-src. Thereare no bands
corresponding
topp9O
andpp5O,
because these pro-teinshave along half-life and henceincorporation
over3 his low. They can be identified, however, afterlabeling
with [35S]methionine for 18 h (data not shown). The results indicatethat attherestrictive temperature tsLA29pp60-src
isstill capable offorming acomplex withpp9O
andpp5O.
It isdifficult, however, tobe certain abouttherelative propor-tions ofmonomeric versuscomplexed forms oftheprotein
because of variability ingradients and the absence of two distinctpeakscorresponding solelyto thesedifferent forms. There does, however, appear to be a shift toward the monomeric form at the restrictive temperature. Since an increased amount ofpp6Ov-src
is present in the soluble fraction at this temperature (see below), the increase in monomeric form isprobablyaneffectrather than a causeofpp6Ov-src
temperature sensitivity. It seems unlikely,there-fore,
that theputative
defect in tsLA29pp6O-src
transportcanbe totally explained bythebehavior of the complex. Pulse-chase analysis. Several reports have indicated that pp60 -src is associated with the plasma membrane and the cytoskeleton (5,20,29).Inthis setofexperiments, fractions correspondingtosolubleandcytoskeletalcomponentsofthe cell were prepared in situ on the tissue culture dish. This technique allows more rapid processing of pulse-chased samples. The preparationof crude membranes is somewhat slower and thus not so amenable in this study. We have found that both membranelocalization(45) and cytoskeletal association (seebelow) are ts for tsLA29
pp6Ov
src,although it is notpossibletoemphaticallyequate thetwo. To examine thekinetics oftranslocation, wetherefore decided to moni-tor the transport ofpp6Ov-src
to the cytoskeleton, since earlier events could be studied.J. VIROL.
on November 10, 2019 by guest
http://jvi.asm.org/
[image:3.612.140.476.70.330.2]350A 350B 3950A
1 3 5 7 9 11 13 15 17 19 21 1 3 5 7 9 11 13 15 17 19 21 23 1 3 5 7 9 11 1 115 17 1921 23
FIG. 2. Glycerolgradient sedimentation analysis oftsLA29cellfractions and immunoprecipitationofpp604-src. tsLA29Rat-1cellswere
labeled with [35S]methionine for 3 h at the permissive (35°C) and nonpermissive (39.5°C) temperatures. Cell fractions were made and
sedimentedthrough 10to30%glycerol gradients.pp6v-srcwasimmunoprecipitated fromalternatefractions withmonoclonal antibody JB327.
The direction ofsedimentation is from lefttoright in eachcase. (A) Soluble cell fraction; (B)cytoskeletal cell fraction. Molecularmass
markersareindicated and expressed in kilodaltons.
The results (Fig. 3) demonstrate that at the permissive temperature there is a gradual increase in the amount of pp60v-srcappearing in the cytoskeletal fraction, correspond-ing to transport ofpp60v-src from the cytosol to the cyto-skeleton. We could identify pp6Ov-src in the cytoskeletal fraction aftera15-min chase. When pulse-chase analysesare
carriedoutwithPrA-transformed Rat-1 cells,somepp6Ov-src isapparentinthecytoskeletal fraction afternochase period at both permissive and nonpermissive temperatures (data notshown). It isnotclear whetherthis is duetomore rapid transportortothefact thatagreaterproportion of pp6Ov-src islocalized atthe cytoskeleton in these cells. At the
nonper-missive temperature, little or no tsLA29 pp6Ov-src became associated with the cytoskeletal fraction. However, upon shiftfrom thenonpermissivetothepermissive temperature, the soluble material accumulated under restrictive condi-tionsefficiently shiftedtothecytoskeletal fraction (Fig. 4A). Itthereforeappearsthat pp6Ovsrcis transported normally at 35°C, but thatat39.5°C (nonpermissive temperature) there is
areversible defect,independent of complex formation, that
either prevents the protein from associating at the cell periphery orrenders this association extremely labile. This raises the question of whether conformational changes within pp6Ov-src itself are responsible for its behavior or
whether it is unableto interact with other, as yet unidenti-fied, proteins which may be important for anchorage of
35C
pp6fi-vsr to the cytoskeleton and plasma membrane. To address these questions further, we analyzed prelabeled
pp60-src in tsLA29 Rat-1 cells by shifting them from the permissive tothenonpermissive temperature. The distribu-tion ofpp6O-src altered dramatically after this shift, alarge
proportion being found in the soluble fraction after 1 hatthe nonpermissive temperature(Fig. 4B). This shows that
asso-ciation with the cytoskeleton is a reversible phenomenon
and suggests that conformational changes occurring within the pp6Ov-src protein itselfare responsible for the tempera-turelability. Theapparentbiastoward monomericpp6Ov-src in the soluble fraction at the restrictive temperature in the glycerol gradients (Fig. 2) also suggests that many of these
defective moleculesarenotcomplexed with ppSO and pp9O. Weare currently investigating whether pp6Ov-src prelabeled at 35°C and shifted to 39.5°C becomes associated with the complex after the shift.
Genetic lesion in tsLA29. With the physiological data accumulated fortsLA29, itwasofgreatinteresttosequence the mutant src gene and to correlate the functional abnor-malities observed with structural alterations inv-src itself.
The nucleotide sequence of the entire v-src gene of tsLA29wasderived fromcodingsequencescontained within the3.1-kb EcoRI B fragment cloned into Agtwes.AB.
The nucleotide sequence revealed three pointmutations which lead to alterations in the predicted amino acid
se-395C
LENGTH OFCHASE
(minutes) 0 15 , 30 , ,60
A B A B A B A B A B
S
t
4 3---- 45A B A B A B A B A B
pp6O-FIG. 3. Pulse-chase analysisofpp6O-vrctransport. tsLA29Rat-1cells weregrownat thepermissive(35°C)andnonpermissive(39.5°C)
temperatures,pulse-labeledwith[35S]methioninefor15min,and chased withcompletemedium for the timeintervalsindicated; cellfractions
werethenmade,andpp6ov-srcwasimmunoprecipitatedwith the monoclonalantibodyJB327. Lanes:A, soluble cell fraction; B, cytoskeletal
cellfraction.
92-5-
69-pp6O
45-
on November 10, 2019 by guest
http://jvi.asm.org/
[image:4.612.74.567.74.214.2] [image:4.612.153.480.569.688.2]1902 WELHAM AND WYKE
A tB
S z S C S C S C' S S
C._ _ _ _s ' . ..
FIG. 4. Temperature shift analyses of pp6O-src localization in labeled tsLA29 Rat-1 cells. In each case cells were pulse-labeled for 15 min with [35S]methionine and chased for1 hat the original incubation temperature, after which the samples were temperature shifted and incubated forafurther 1or2 h. Identical samples weremaintained at the original temperature. At the appro-priate time points fractions were made corresponding to soluble (lanesS) and cytoskeletal (lanes C) cell fractions, andpp6Ov-srcwas immunoprecipitated with polyclonal rabbit anti-pp6O antisera. (A) Temperature shift from the nonpermissive to the permissive tem-perature; (B) shift from the permissive to the nonpermissive tem-perature. Lanes: 1, cells maintained at theoriginal temperature for a1-h chaseperiod; 2, cellsmaintainedattheoriginal temperature for a3-h chase period; 3, cells maintained attheoriginal temperaturefor a1-hchase and then shifted and incubated forafurther1-hchase; 4, cells maintained at the original temperature fora 1-hchase and then shifted and incubated for a further 2-h chase. Molecular mass markersareexpressed inkilodaltons.
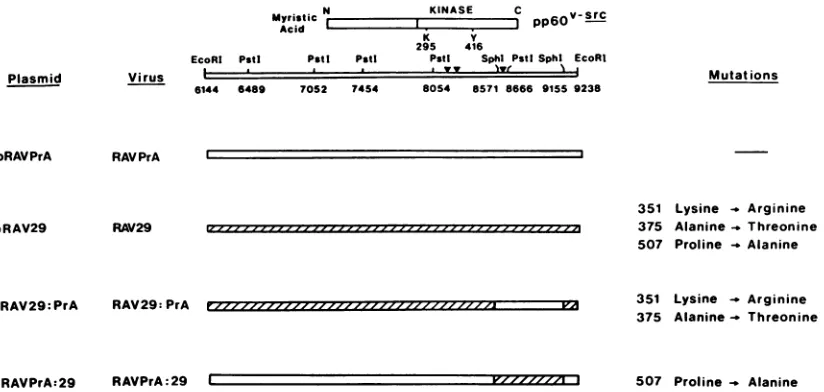
quence; all three were within the kinase domain. These changesaresummarized in Fig. 5. Thealterationatposition 8180, which changes the coding of amino acid 351 from lysine to arginine, is probably conservative. The tyrosine kinases v-erbB,v-abl,and v-ros all have an arginine instead oflysineat thecomparableposition,andit islikely that this mutation does not play a crucial role in determining the functional abnormalities of tsLA29. The alteration at nucle-otide 8251 (amino acid 375), which changes alanine to threonine,lies in the centerof the kinasedomain.Thethird
Plasmid
pRAVPrA
pRAV29
Virus
6I
alteration, at nucleotide 8647, which leadstosubstitution of alaninefor proline at amino acid 507, lies in the regionwhere genetic complementation studies predicted a crucial muta-tion (16). To test the relative contribumuta-tion of each ofthese mutations to the phenotype of tsLA29, we constructed chimeric v-src genes by usinganRAV-1-avian erythroblast-osis virus (AEV10) vector system (Fig. 1).
Construction of chimeras. The SphI 584-base-pair frag-ment, containing the coding region for amino acids 482 to 526, was exchanged between tsLA29 sequences and PrA wild-type sequences (Fig. 5). The resultant chimeras were checked by DNA sequence analysis. The parental and chimeric pRAV derivatives were transfected into CEF, transformed foci were picked and expanded, and recombi-nant viruses were recovered from the culture fluid of the infected CEF. These recombinant viruses were used in further studiesonmorphology,localizationofpp6Ov-src,and invitro kinase activity.
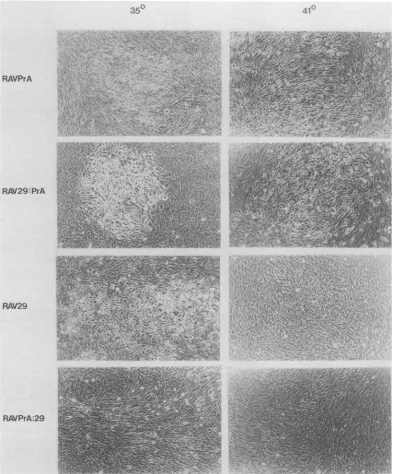
Morphology. The titers of the recombinant viruses ob-tained were all comparable. The wild-type RAVPrA pro-ducedfociatboth temperatures, whereas RAV29 was ts for focusformation(Fig. 6).ThepRAV29:PrAconstruct, which contains the coding sequences for the mutant amino acids 351 and 375, was able to induce focus formation at both temperatures, thefocitendingtobeveryroundin morphol-ogy. The pRAVPrA:29 construct, containing the coding sequencefor the mutant amino acid at position 507 alone, inducedfocus formationat thepermissivetemperatureonly. Thesefociwereratherfusiformin theirmorphology, as were thecells whentransformed in mass culture. Thus, notonly did it appear that the mutation at amino acid 507 was responsible for the ts phenotype, but it also had subtle effects on morphology. A region in the amino-terminal portion ofpp6O-src has previously beenconsidered impor-tant in determining morphology, so the observation that a carboxy-terminal mutation also has effects on morphology raises the possibility that interactions between domains of pp6Ov-src arerequiredfor normal functioning.
Myristic N KINASE C v-SrC
Acid I I C
pp60-K Y
295 416
EcoRI Pstl PstI Pstt Pstt Sphlt PstlSphl EcoRI
I a yy lyr~~v N I
144 6489
Mutat ions
RAV PrA
RAV29
351 Lysine _ Arginine 375 Alanine_ Threonine 507 Proline -_ Alanine
pRAV29:PrA RAV29: PrA 351 Lysine - Arginine
375 Alanine- Threonine
[image:5.612.57.291.76.181.2]pRAVPrA:29 RAVPrA:29 C V'/ZZZ 3 507 Proline Alanine
FIG. 5. RSV variants used in this study. The nucleotide numbering follows that of Schwartz et al. (42). Chimeric v-src genes were
constructedbetween PrAwild-typeand tsLA29sequences. The SphIsitesusedaredenoted by the lines bisectingthe chimericgenes.The
positions ofthe nucleotidemutationsaredenoted (V). The 584-base-pairSphI fragmentwas used inamix-and-match approachto generate
pRAV29:PrAand pRAVPrA:29. The parental and chimeric genes wereconstructed into thesamereplication-competentplasmid and then transfectedinto CEF. Viruseswere recoveredas described in thetext.
70S2 7454 8054 8571 8666 9155 9238
I
-j
Izz--- , 1- -III I ZZ-J..
rzzzzz-z ZZZ,/'i P71
J. VIROL.
on November 10, 2019 by guest
http://jvi.asm.org/
[image:5.612.100.512.485.681.2]350
RAVPrA 0A
iz\
RAV29:PrA
RAV29
..
4.4-'X''.
42 4... q. ..; ~ [image:6.612.118.513.77.551.2]RAVPrA:29
FIG. 6. Morphologies ofvirus-infected CEFatthe permissive(35°C)andnonpermissive(41°C)temperatures.Fociwerephotographed6
to8dayspostinfection.
Subcallular localization. Having shown tsLA29tobetsfor membrane and cytoskeletal association (45; see above), it
was importanttodetermine the localization of the chimeric pp6jv-src proteins. Infected CEF were plated onto 35-mm dishes and metabolically labeled with [35S]methionine, and MCSK and NP-40 buffers were used to make lysates as
described above. TheJB327monoclonalantibodyandrabbit anti-pp6Ovsrc antiserum were used to immune precipitate pp60v-srcfrom thelysates. Both RAV29 and RAVPrA:29are
tsforassociation with the cytoskeleton, but RAVPrA and RAV29:PrA behave in atemperature-independent manner
(Table 1). Hence, it appearsthat the change atamino acid 507 is sufficient toconfer temperature sensitivity on cyto-skeletalassociation.
Kinaseactivityofchimericproteins.All three mutations lie withinthepredicted region for the kinase domain, andhence
we needtoevaluatetheireffectonpp6Ov-src kinaseactivity.
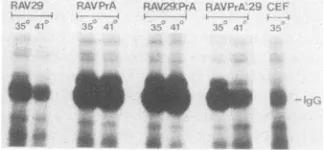
In vitro kinaseassayswereperformedonNP-40lysates from CEF infected with the chimeric recombinant viruses.Equal amounts of protein were assayed in each case, and the resultsarerepresented in Fig. 7. Thelevel of
immunoglob-ulinheavy chainphosphorylationobserved in the uninfected CEF track is due to cross-reactivity of the tumor-bearing rabbit serumused withc-srcand assuchispresent in each sample assayed. The pp6Ov-src proteins of RAVPrA (wild type) and RAV29:PrA both display a high level of kinase activitythatisindependentoftemperature.The RAV29and
RAVPrA:29 recombinant pp6Ov-src proteins, on the other
on November 10, 2019 by guest
http://jvi.asm.org/
1904 WELHAM AND WYKE
TABLE 1. Subcellularlocalization ofpp6o-src by cellfractionation
% ofpp6vOSrcatfollowing
Infectingvirus Cellfractiona growth tempb
35°C 410C
RAVPrA S 5 24
C 95 76
RAV29 S 10 60
C 90 40
RAV29:PrA S 5 15
C 95 85
RAVPrA:29 S 30 70
C 70 30
aS,Soluble cellfraction; C, cytoskeletalfraction.
b Each value represents theproportion ofpp60v-sr,determinedby densi-tometricscanning,in each cell fractionexpressedas apercentageofthetotal solubleplus cytoskeletal fractions.Theresultspresentedarethe average of twoindependent experiments.
hand, have ahighactivityatthepermissivetemperature,but thekinaseactivity is reducedatthe nonpermissive temper-ature.Thisreductionis dueto adecrease in
specific
activity at restrictive temperature rather than to adifference in the half-life of the pp6Ov-src protein, which is the same at the permissive and nonpermissive temperatures (data not shown).Itthereforeappears thatthe mutationatresidue 507 is also the crucial lesion in determining the temperature sensitivity ofthe tyrosine-specificprotein kinase.Nucleotide sequence of revertant tsLA29 virus-src gene. Previous studies have shown that the reversion of otherts mutants to the wild phenotype resulted from second-site lesionsthat wereeither close to, or remotefrom,theforward mutation (17). The nucleotide sequence of the complete v-src gene encoded by tsLA29 R2 was derived from se-quencescontainedwithinthe3.1-kbEcoRIfragment cloned into XNM1149. The revertant contained all the forward mutations of tsLA29, but a single compensating mutation was located at nucleotide 8409. This distant second-site mutation changes lysine to arginine in the center of the kinasedomainataminoacid427andeffectivelyrestoresthe behavior ofpp60v-srctothatofwild-type PrA RSV (data not shown).
DISCUSSION
The mutant tsLA29 is ts for tyrosine kinase activity and also in itsability to locate at the cellperiphery (44, 45).We
§* * : *,: *
*FIG. 7. Invitro kinase assaysperformed on lysates from CEF infected withthe RSV variants used in this study. The products of thereactionswere separated on a sodium dodecyl sulfate-polyacryl-amidegelelectrophoresisgel(10%
pojyacrylamide)
and autoradio-graphed. Abbreviation: IgG, 42P-labeled immunoglobulin heavy chains.showherethat itsaberrantlocalizationisnotattributableto
defective
complex
formation withpp90
andppSO
or to aberrantdissociation from thesetwoproteins, although
thecomplex
does show evidence ofslight abnormality
(45).
These
findings
areincontrast tothose foranumber of other RSV mutants, in which defects in subcellular localization canbe attributedalmostentirely
toabnormal behavior of thecomplex (23,
35).
Furtherexaminationby using
pulse-chase
experiments
demonstratesareversibletsdefect in theasso-ciation of tsLA29
pp60v-src
with thecytoskeletal
cell frac-tion. Thispresumably
reflects aperturbation
in eventsfollowing complex
dissociation. A defect in amino-terminalmyristylation
might
produce
thisphenotype,
butmyristyla-tion oftsLA29
pp60v-src
is not ts(45).
Wetherefore suspect that the mutantprotein
displays
atemperature-dependent
conformational
change
that reduces its kinaseactivity
andprecludes
either an inherentability
topersist
at thecyto-skeleton or its
capacity
to interactwith unidentified mole-culesatthatlocation. Inany event, it appears thatmyristy-lation of
tsLA29
pp60v-src
alone is notsufficietit
to ensurestable associationwith the
plasma
membrane andcytoskel-eton.
The defects observed in
physiology
canbe attributedtoasingle-point
mutation resulting
in thesubstitution of alaninefor
proline
at amino acid 507. The coordinate effect of thissingle
mutation on both kinaseactivity
andlocalization
isdemonstrated
by
studies with chimericpp60v-src
moleculesand
emphasized by
the factthat asingle
second-sitemuta-tionat
position
427, close totheconservedalanine,
proline,
and
glutamic
acid motif(residues
430to432),
fully
restoresthefeatures
of
wild-type
pp60v-sr.
The crucialtslesionis atthe
carboxy-terminal
end ofthe conserved kinase domainadjacent to a region which has been suggested to have a
regulatory
role inpp60v-src
function(37).
The observationthatthe mutated
residue,
proline 507,
ishighly
conservedin a wide range oftyrosine
kinases also suggests that it may itselfplay
a role in theregulation
of the kinase or other activities ofpp60v-src.
The Chou and Fasman(9)
rulespredict
a coil/turn structure in theregion
ofthe mutation.Substitution of alanine for
proline
may decrease theflexi-bility
ofthe molecule andsodisrupt
therelationships
oftheregulatory C terminus to the
kinase
or other domains.Alternatively,
such a decrease inflexibility
could affectinteractions withother
proteins
which may beimportant
inpp6fiv-src
function andbiogenesis.
Itcan beenvisaged
that this mightcoordinately
affect both kinaseactivity
andpp60V-srC localization,
but thefact thatadistant
second-sitemutation,
also at ahighly
conserved residue(lysine
427)within thekinasedomain,compensatesforthis defect shows that such an
explanation
isinevitably
facile.Itis
interesting
that the chimericprotein containing onlythe mutationat amino acid 507 has subtle effects uponcell morphology. Infected cells are markedly
fusiformn,
in con-trast to the rounded cell morphology seen when CEF are infected with either the parental ts virus, the chimera RAV29:PrAcontaining justthe 351and 375mutations,or the wild-type revertantRAV29R2. It appears that the two mu-tations at amino acids 351 and375,
although having
no detectable effectonpp6Ovsrc
physiology, are in some waycompensating
in theparentaltsLA29virus for the effect thelesionatamino acid 507 has on
cell
morphology. Alterationsin
the amino-terminal region are more usually associated withdefects in cellmorphology.CEFtransformedby mutantviruses
which have amino-terminal deletions, as well asthose infected
by
many recovered avian sarcoma viruses, exhibit fusiformmorphologies
(13, 27, 28). However, other J.VIROL.on November 10, 2019 by guest
http://jvi.asm.org/
[image:7.612.93.255.597.672.2]mutations mapping to the carboxyterminus also appear to influence cellmorphology. Our
wild-type
Prague A clone has a different residue at amino acid 502 compared with other cloned v-src genes (17), and this clone tends to transform cells with a slightly fusiform morphology (Fig. 6). Other reports have also mentioned such phenomena, although the effect issubtle (35). This suggests that the C-terminal amino acids spanning residues 500 to 510 may play a modulatory role in determiningcell morphology, perhaps byinfluencing the access ofthe
pp6Ov-src tocertain targets. Moredetailed studies on the location and molecular interactions of the mutant pp60-src should be informative.ACKNOWLEDGMENTS
We are grateful to all members ofthe laboratory for technical advice, to S. Kellie, M.Owen, and G. Petersforcriticalcomments; andtoNikkiPinchen for her endless patience duringthe preparation of the manuscript.
LITERATURECITED
1. Brugge, J. S. 1986. Interaction ofRous sarcomavirus protein
pp60v-src
with cellular proteinspp5O
andpp9O.
Curr. Top. Microbiol. Immunol. 123:1-21.2. Brugge, J. S.,E.Erikson, and R. L. Erikson. 1981.Thespecific interaction of the Rous sarcoma virus transforming protein pp60src withtwocellular proteins. Cell 25:363-372.
3. Brugge, J., W. Yonemoto, and D. Darrow. 1983. Interaction between the Rous sarcomavirustransformingprotein andtwo
cellularphosphoproteins: analysis of theturnoverand distribu-tion of this complex. Mol. Cell. Biol. 3:9-19.
4. Bryant, D., and J. T. Parsons. 1984. Amino acid alterations within a highly conserved region ofthe tyrosineprotein kinase activity. Mol. Cell. Biol. 4:862-866.
5. Burr,J. G.,G.Dreyfuss,S.Penman,andJ.M. Buchanan.1980. Association of thesrcgeneproductofRoussarcomaviruswith cytoskeletalstructures ofchickembryo fibroblasts. Proc. Natl. Acad. Sci. USA 77:3484-3488.
6. Buss, J. E.,M. P.Kamps,K.Gould,and B. M.Sefton.1986.The absence ofmyristic acid decreases membrane binding ofthe
p6Osrc but does not affect tyrosine protein kinase activity. J. Virol. 58:468-474.
7. Buss, J. E.,M. P.Kamps,and B. M. Sefton.1984. Myristicacid is attached tothe transforming proteinofRous sarcoma virus during or immediately after synthesis and is present in both solubleand membrane-bound forms of the protein. Mol. Cell. Biol. 4:2697-2704.
8. Calothy, G.,D.Laugier,F. R.Cross,R.Jove,T. Hanafusa,and H. Hanafusa.1987.Themembrane-bindingdomain and myristy-lation of pp6Ov-src are not essential for stimulation of cell proliferation. J. Virol.61:1678-1681.
9. Chou,P.K.,andG. D. Fasman. 1978. Empiricalpredictionsof protein conformation. Annu. Rev. Biochem. 47:251-276. 10. Collett, M.S., andR. L. Erikson. 1978.Protein kinase activity
associated with the aviansarcomavirussrcproduct.Proc.Natl. Acad.Sci. USA75:2021-2024.
11. Courtneidge,S. A.,andJ.M.Bishop. 1982. Transit ofpp60-src to the plasma membrane. Proc. Natl. Acad. Sci. USA 79: 7117-7121.
12. Cross, F.R.,E.A. Garber,and H.Hanafusa. 1985.N-terminal deletions in Rous sarcoma virus pp60v-sr; effectson
tyrosine
kinaseandbiologicalactivities andonrecombination in tissue culture with the cellularsrcgene.Mol. Cell. Biol.5:2789-2795. 13. Cross,F.R.,E. A. Garber,D.Pelman,andH. Hanafusa. 1984. A short sequence in the pp6osrc N terminus is required for pp60src myristylation and membrane association and for cell transformation. Mol. Cell. Biol. 4:1834-1842.14. Declue, J. E.,andG. S. Martin. 1987.Phosphorylationoftalinat
tyrosine in Rous sarcoma virustransformed cells. Mol. Cell. Biol. 7:371-378.
15. Enrietto,P.J.,L.N.Payne, andJ.A.Wyke. 1983.Analysisof thepathogenicityoftransformation defective
partial
mutantsofavian sarcoma virus: characterisation of recovered viruses which encode novelsrcspecific proteins. Virology127:397-411. 16. Fincham,V.J.,D.J. Chiswell,andJ.A.Wyke. 1982.Mapping of nonconditional and conditional mutants in the src gene of PraguestrainRous sarcomavirus. Virology 116:72-83. 17. Fincham,V.J.,andJ. A.Wyke. 1986. Localization of
temper-ature-sensitivetransformation mutations and back mutations iti theRous sarcomavirussrc gene. J.Virol. 58:694-699. 18. Garber,E.A., F. R.Cross,andH. Hanafusa.1985.Processing
ofp60v-srctoitsmyristylatedmembrane-bound form. Mol. Cell. Biol. 5:2781-2788.
19. Gilmer, T. M., and R. L. Erikson. 1983. Development of
anti-pp60src
serumwith antigenproducedin Escherichiacoli.J.Virol.45:462-465.
20. Hamaguchi,
M.,
and H. Hanafusa. 1987. Association ofp6Osrc with triton X-100 resistant cellular structure correlates with morphological transformation. Proc. Natl. Acad. Sci. USA 84:2312-2316.21. Hanafusa,H.1977.Cell transformationbyRNAtumourviruses, p. 401-483. In H. Fraenkel-Conrat and R. P. Wagner (ed.), Comprehensive virology, vol. 10. Plenum
Publishing Corp.,
NewYork.22. Hirst, R., A. Horwitz, C. Buck, and L. Rohrschneider. 1986. Phosphorylation ofthe fibronectin receptor complex in cells transformedby oncogenesthat encode tyrosinekinases. Proc. Natl. Acad. Sci. USA 83:6470-6474.
23. Jove, R., E. A. Garber, H. Iba, and H. Hanafusa. 1986. Biochemicalproperties ofp60v-srcmutantsthatinduce different cell transformationparameters. J.Virol. 60:849-857.
24. Jove, R.,B.J.Mayer,H.Iba,D.Laugier,F.Poirier,G.Calothy,
T.Hanafusa,and H. Hanafusa.1986.Geneticanalysisof p60vsrc domains involved in the induction ofdifferent cell transforma-tion parameters.J. Virol.60:840-848.
25. Kamps,M.P.,J.E.Buss,and B. M. Sefton.1986.Roussarcoma
virustransformingprotein lacking myristicacidphosphorylates knownpolypeptidesubstrates withoutinducingtransformation. Cell 45:105-112.
26. Kawai, S., and M. Nishizawa. 1984. New
procedure
ofDNA transfection withpolycationanddimethyl
sulfoxide. Mol. Cell. Biol.4:1172-1174.27. Kellie, S.,B. Patel,N. M. Wigglesworth, D. R. Critchley, and J.A.Wyke.1986. TheuseofRoussarcomavirustransformation mutants with differing tyrosine kinase activities to
study
therelationshipsbetween vinculinphosphorylation,
pp6Ov-rc
local-isation and adhesion plaqueintegrity.
Exp. Cell Res. 165: 216-228.28. Krueger,J.G.,E.A.Garber,S. S. M.Chin,H. Hanafusa,and
A. R.Goldberg.1984.Size-varient
pp6Osrc proteins
ofrecovered avian sarcoma virus interact with adhesionplaques
asperiph-eral membrane
proteins:
effects on cell transformation. Mol. Cell. Biol. 4:454-467.29. Krueger,J. G.,E.Wang, and A. R. Goldberg.
1980;
Evidence thatthesrcgeneproductofRoussarcomavirus ismembrane-associated.Virology 101:25-40.
30.
Laemmll,
U.K. 1970.Cleavage
ofstructuralproteins during
theassembly ofthe head of
bacteriophage
T4. Nature (London) 227:680-685.31. Levinson, A. D., S. A. Courtneidge, andJ. M.
Bishop.
1981. Structural and functionaldomainsofRoussarcomavirustrans-forming protein
(pp6osrc).
Proc. Natl. Acad. Sci. USA 78: 1624-1628.32. Lipsich,L.A.,A.J.Lewis,andJ.S.
Brugge.
1983. Isolation of monoclonal antibodiesthatrecognize
thetransforming
proteins
of avian sarcomavirus. J. Virol. 48:352-360.33. Loenen, W. A., and W. J. Brammar. 1980. A
bacteriophage
lambda vector forcloning large
DNAfragments
made with several restrictionenzymes.Gene20:249-259.34. Maniatis, T., E.F. Fritsch,andJ. Sambrook. 1982. Molecular
cloning:
alaboratorymanual. ColdSpring
HarborLaboratory,
ColdSpringHarbor,N.Y.35. Mayer,B.J.,R.Jove,J.F. Krane,F.Poirier,G. Calothy, and
H. Hanafusa. 1986. Genetic lesions involved in temperature
sensitivity
ofthesrcgeneproducts
of fourRous sarcomaviruson November 10, 2019 by guest
http://jvi.asm.org/
1906 WELHAM AND WYKE
mutants. J. Virol.60:858-867.
36. Messing, J., and J. Vieira. 1982. A new pairofM13 vectorsfor selecting either DNA strand of double-digest restriction frag-ments. Gene19:269-276.
37. Parsons, J. T., D. Bryant, V. Wilkerson, G. Gilmartin, and S. J. Parsons.1984. Site-directedmutagenesis ofRous sarcomavirus pp60src: identification of functional domains required for trans-formation,p. 37-42.InG.F.VandeWoude,A.J.Levine,W.C. Topp,andJ. D. Watson(ed.),Cancercells: oncogenesand viral genes. Cold Spring Harbor Laboratory, Cold Spring Harbor, N.Y.
38. Pasquale, E. B., P. A. Maher, and S. J. Singer. 1986.Talin is phosphorylatedontyrosinein chicken embryofibroblasts trans-formed by Rous sarcoma virus. Proc. Natl. Acad. Sci. USA 83:5507-5511.
39. Purchio, A. F., E. Erikson, J. S. Brugge, and R. L. Erikson. 1978. Identification of a polypeptide encoded by the avian sarcoma virus src gene. Proc. Natl. Acad. Sci. USA 75: 1567-1571.
40. Sanger,F., S. Nicklen, and A. R. Coulson. 1977. DNA sequenc-ing with chain-terminatsequenc-ing inhibitors. Proc. Natl. Acad. Sci. USA 74:5463-5467.
41. Schuh,S., W. Yonemoto, J. Brugge, V. J. Bauer, R. M.Riehl, W. P.Sullivan, and D.0. Toft. 1985. A 90,000-dalton binding proteincommon tobothsteroidreceptors and the Rous sarcoma virus transforming protein pp60v-src. J. Biol. Chem. 260: 14292-14296.
42. Schwartz,D. E., R.Tizard, and W. Gilbert. 1983. Nucleotide sequence ofRous sarcomavirus. Cell 32:853-869.
43. Sefton, B. M. 1986. The viral tyrosine protein kinases. Curr. Top.Microbiol.Immunol. 123:39-72.
43a.Stoker, A. W., and M. J. Bissell. 1988. Development of avian sarcomaand leukosis virus-basedvector-packaging cell lines. J. Virol. 62:1008-1015.
44. Stoker, A. W., P. J. Enrietto, andJ.A.Wyke.1984. Functional domains of the pp6O-src protein as revealed by analysis of temperature-sensitive Roussarcoma virusmutants. Mol. Cell. Biol. 4:1508-1514.
45. Stoker, A. W., S. Kelie, and J. A. Wyke. 1986. Intracellular localization and processing of pp6ov-src proteins expressed by two distinct temperature-sensitive mutants of Rous sarcoma virus. J. Virol. 58:876-883.
46. Tamura, T., H. Bauer,C. Birr, and R.Pipkorn. 1983. Antibod-iesagainst syntheticpeptidesas atQol for functional analysis of thetransforming protein pp60src. Cell 34:587-596.
47. Tato, F., J. A. Beamand, and J. A. Wyke. 1978. Amutantof Rous sarcoma virus with a thermolabile defect in the virus envelope.Virology 88:71-81.
48. Vennstrom, B., L. Fanshier, C.Moscovici, andJ. M. Bishop. 1980. Molecular cloning of the avian erythroblastosis virus genome and recovery of oncogenic virus by transfection of chicken cells. J. Virol. 36:575-585.
48a.White,M.K., and M.J.Weber.1988.Transformationby thesrc oncogenealtersglucosetransportintoRatandchicken cellsby different mechanisms. Mol. Cell. Biol. 8:138-144.
49. Wyke, J. A. 1973. The selective isolation of temperature-sensitivemutantsofRous sarcomavirus.Virology 52:587-590. 50. Wyke, J. A. 1973.Complementation of transforming functions bytemperature-sensitivemutantsof aviansarcomavirus. Virol-ogy54:28-36.
51. Wyke, J. A., and R. Kurth. 1978. Reversion of temperature-sensitivetransformationmutantsofRous sarcomavirus andits effectontheexpression oftumourspecific surface antigen. J. Gen. Virol. 40:701-704.
52. Wyke, J. A., and A. W. Stoker. 1987. Genetic analysis of the form and function of the viralsrconcogeneproduct. Biochim. Biophys.Acta907:47-69.
J. VIROL.