Copyright © 2000, American Society for Microbiology. All Rights Reserved.
Virus-Specific Cofactor Requirement and Chimeric Hepatitis C
Virus/GB Virus B Nonstructural Protein 3
NANCY BUTKIEWICZ,1NANHUA YAO,2WEIDONG ZHONG,1JACQUELYN WRIGHT-MINOGUE,1
PAUL INGRAVALLO,1RUMIN ZHANG,2JAMES DURKIN,2DAVID N. STANDRING,1
BAHIGE M. BAROUDY,1DAVID V. SANGAR,3STANLEY M. LEMON,3
JOHNSON Y. N. LAU,1ANDZHI HONG1*
Departments of Antiviral Therapy1and Structural Chemistry,2Schering-Plough Research Institute,
Kenilworth, New Jersey 07033-0539, and Department of Microbiology and Immunology,
The University of Texas Medical Branch at Galveston, Galveston, Texas 77555-10193
Received 22 November 1999/Accepted 22 January 2000
GB virus B (GBV-B) is closely related to hepatitis C virus (HCV) and causes acute hepatitis in tamarins
(Saguinusspecies), making it an attractive surrogate virus for in vivo testing of anti-HCV inhibitors in a small
monkey model. It has been reported that the nonstructural protein 3 (NS3) serine protease of GBV-B shares similar substrate specificity with its counterpart in HCV. Authentic proteolytic processing of the HCV polypro-tein junctions (NS4A/4B, NS4B/5A, and NS5A/5B) can be accomplished by the GBV-B NS3 protease in an HCV NS4A cofactor-independent fashion. We further characterized the protease activity of a full-length GBV-B NS3 protein and its cofactor requirement using in vitro-translated GBV-B substrates. Cleavages at the NS4A/4B and NS5A/5B junctions were readily detectable only in the presence of a cofactor peptide derived from the cen-tral region of GBV-B NS4A. Interestingly, the GBV-B substrates could also be cleaved by the HCV NS3 protease in an HCV NS4A cofactor-dependent manner, supporting the notion that HCV and GBV-B share similar NS3 protease specificity while retaining a virus-specific cofactor requirement. This finding of a strict virus-specific cofactor requirement is consistent with the lack of sequence homology in the NS4A cofactor regions of HCV and GBV-B. The minimum cofactor region that supported GBV-B protease activity was mapped to a central region of GBV-B NS4A (between amino acids Phe22 and Val36) which overlapped with the cofactor region of HCV. Alanine substitution analysis demonstrated that two amino acids, Val27 and Trp31, were essential for the co-factor activity, a finding reminiscent of the two critical residues in the HCV NS4A coco-factor, Ile25 and Ile29. A model for the GBV-B NS3 protease domain and NS4A cofactor complex revealed that GBV-B might have developed a similar structural strategy in the activation and regulation of its NS3 protease activity. Finally, a chimeric HCV/GBV-B bifunctional NS3, consisting of an N-terminal HCV protease domain and a C-terminal GBV-B RNA helicase domain, was engineered. Both enzymatic activities were retained by the chimeric protein, which could lead to the development of a chimeric GBV-B virus that depends on HCV protease function.
GB virus B (GBV-B) is a single-stranded (ss) positive-sense RNA virus associated with GB agent hepatitis (47, 60). It infects tamarins (Saguinusspecies) and causes acute hepatitis in naive animals (13). Phylogenetic tree analysis and the ge-nome organization of GBV-B suggest that this virus belongs to
theFlaviviridaefamily which at present consists of three
gen-era:Flavivirus,Pestivirus, andHepacivirus(47, 60). Like other
members of this family (27, 52), the genome of GBV-B en-codes a single large polyprotein of approximately 2,860 amino acids. The polyprotein is likely processed into several struc-tural (C, E1, E2, and p7) and nonstrucstruc-tural (NS2, NS3, NS4A, NS4B, NS5A, and NS5B) proteins by either host- or virus-encoded proteases (47). Among all animal viruses, GBV-B shares closest nucleotide homology with hepatitis C virus (HCV) (47, 60). Like HCV, GBV-B is hepatotropic and causes liver disease in susceptible primates, which makes it a candi-date as a separate member in the genusHepacivirusaccording to a recent suggestion by the International Committee on Viral Taxonomy (54). In addition, GBV-B shares a similar IRES structure and function (53), as well as polyprotein organization (47, 48), with HCV.
HCV is the leading etiological agent of non-A non-B hepa-titis (1, 12). More than 170 million people worldwide are in-fected by HCV. About 80% of patients with acute HCV infec-tion will progress to chronic hepatitis; 20% of these will develop cirrhosis, and 1 to 5% of these will develop hepa-tocellular carcinoma (67). A recently completed population-based survey revealed that in the United States alone, the overall prevalence of anti-HCV was 1.8%, which corresponds to an estimated 3.9 million individuals infected by HCV na-tionwide. More than 74% of these seropositive individuals test positive for serum HCV RNA, indicating that at least 2.7 million persons are chronically infected (2). Current therapies with alpha interferon (IFN-␣) alone and the combination of IFN-␣and Ribavirin (Rebetron; Schering-Plough Corp., Ken-ilworth, N.J.) have been shown to be effective in no more than 41% of the patients with chronic HCV infection (44, 51). Vaccine development has been hampered by a high degree of antigenic variation and the lack of protection against reinfec-tion, even with the same inoculum (16, 30, 59, 69). Develop-ment of small molecule inhibitors directed against specific viral targets has thus become the focus of HCV research. Extensive characterization of the HCV NS3 serine protease-RNA heli-case (3, 14, 22, 24, 26, 29, 31, 62) and NS5B RNA-dependent RNA polymerase (5, 17, 40, 70) has assisted in assay develop-ment and inhibitor identification against HCV replication. Ma-jor advances in the determination of crystal structures for NS3
* Corresponding author. Mailing address: Antiviral Therapy, K-15-4/4945, Schering-Plough Research Institute, 2015 Galloping Hill Rd., Kenilworth, NJ 07033-0539. Phone: (908) 3152. Fax: (908) 740-3918. E-mail: [email protected].
4291
on November 9, 2019 by guest
http://jvi.asm.org/
protease-RNA helicase, as well as NS5B polymerase, have be-gun to delineate important features relevant to the develop-ment of potent and specific anti-HCV inhibitors (32, 33, 38, 42, 43, 71, 74).
The recent development of infectious molecular clones of HCV (4, 25, 34, 72, 73) has led to the establishment of a cell-based RNA replication system using an antibiotic-selected subgenomic RNA replicon (41). This may prove to be valuable for the in vitro evaluation of inhibitors targeting HCV repli-cation. Unfortunately, the search for better and more specific antiviral agents will be complicated by the lack of a readily available animal model for hepatitis C other than the chim-panzee, which is associated with limited availability and ex-tremely high cost. This poses an almost insurmountable obsta-cle for in vivo testing of anti-HCV inhibitors before clinical trials in humans. The need for a small surrogate animal model is urgent. The GBV-B–tamarin model may provide an effective in vivo system reminiscent of the use of the woodchuck hepa-titis model, a strategy that has been successfully developed for anti-HBV drug development.
However, GBV-B is relatively new and uncharacterized. Limited studies of the GBV-B NS3 serine protease (57), RNA helicase (77), and NS5B RNA-dependent RNA polymerase (W. Zhong and Z. Hong, unpublished results) have revealed that there are many structural and enzymatic properties shared by GBV-B and HCV, further supporting the notion of GBV-B as a surrogate virus for HCV. The report by Scarselli et al., demonstrated that GBV-B NS3 protease shared similar sub-strate specificity with that of HCV and cleaved the HCV polyprotein at the correct junction sites in an HCV NS4A-independent fashion (57). Although it has not been shown that the HCV NS3 protease is capable of reciprocal processing of the GBV-B polyprotein junctions, this finding provides encour-agement for developing the GBV-B–tamarin model and for the in vivo testing of HCV protease inhibitors.
In this report, we address five key questions related to a recombinant GBV-B NS3 protein produced inE. coli. (i) Does the GBV-B NS3 protease process the GBV-B polyprotein as predicted by Scarselli et al. based on the use of the heterolo-gous HCV polyprotein substrates (56)? (ii) Does HCV NS3 protease cleave the GBV-B polyprotein at the predicted junc-tion sites? (iii) Is there an NS4A cofactor requirement by the GBV-B NS3 protease? (iv) If so, what is the minimal region of the NS4A that supports the GBV-B cofactor activity. (v) What are the critical amino acids essential for the cofactor activity? Our results support the notion that HCV and GBV-B share similar NS3 protease specificities, while retaining a virus-spe-cific NS4A cofactor requirement. A model for the GBV-B NS3 protease domain and NS4A cofactor complex is proposed that shows similar structural features in the activation and sta-bilization of the protease domain. Finally, a chimeric HCV/ GBV-B bifunctional NS3, consisting of an N-terminal HCV protease domain and a C-terminal GBV-B RNA helicase do-main, was engineered to explore the possibility of developing a chimeric GBV-B virus that depends on HCV serine protease function.
MATERIALS AND METHODS
Cloning of expression plasmids.The full-length GBV-B NS3 cDNA was iso-lated directly from infected tamarin serum as described previously (76). Extrac-tion of RNA from serum was by standard procedures. First, 0.2l of serum (GB agent pool mystrax 666, 8/93, kindly supplied by J. Bukh, National Institutes of Health) was diluted with 100l of calf fetal serum and extracted using the Trizol system (GIBCO BRL, Rockville, Md.). The pellet was dissolved in 10 mM dithiothreitol (DTT) containing 20 U of RNasin (Promega, Madison, Wis.) per ml. The selection of primers for cDNA synthesis and PCR amplification was based on published sequences (60). Reverse transcription-PCR was performed
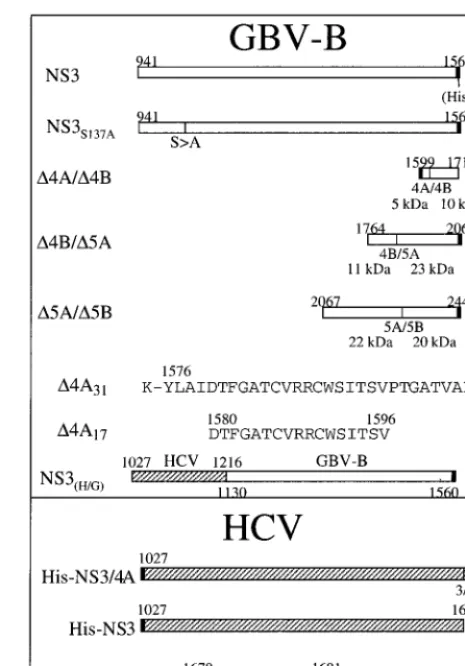
using the Superscript reverse transcriptase (GIBCO BRL) and the Advantage cDNA polymerase mix (Clontech, Palo Alto, Calif.). Four subgenomic cDNA fragments were amplified covering the entire published genome. DNA fragments containing the GBV-B cleavage sites were isolated by PCR from cDNA clones of GBV-B. The primers used to amplify the NS4A/4B junction were 5⬘-ATATGG ATCCGGTGCTACTGTCGCCCCAGTG-3⬘ and 5⬘-ATATAAGCTTCACT TGGACGCAATTGCGCCTCC-3⬘. The resulting PCR fragment was directly cloned into pET-28a (Novagen, Madison, Wis.) between theBamHI andHindIII sites. The primers for the GBV-B NS4B/5A cleavage site were 5⬘-AAATGGC TAGCGGTGAGTGGCCCACTATGGA-3⬘and 5⬘-ATATGGATCCCATGCG CACACCAGGTGTGTG-3⬘. The PCR amplified fragment was cloned into pET/ NS5B⌬CT-His (17) by replacing the NS5B coding region with that of the NS4B/5A substrate between theNheI andBamHI sites. The GBV-B NS5A/5B was cloned similarly as that of NS4B/5A with the primers 5⬘-AAATGGCTAG CCAACTTAATTTGCGTGATGCAC-3⬘and 5⬘-ATATGGATCCCATCTTCT CAACACATCTCATTTC-3⬘. The GBV-B coding regions shown in Fig. 1 were numbered according to amino acid positions published previously (47, 60). Plas-mids pNBNae, pJB1003, and pTS102 encoding the HCV NS3 cleavage sites NS4A/4B, NS4B/5A, and NS5A/5B, respectively, were cloned as described by Butkiewicz et al. (10). A chimeric HCV/GBV-B NS3 gene was constructed by joining two PCR fragments: one consisted of the HCV-1a (H) NS3 protease coding region (amino acids [aa] 1 to 190); the other consisted of the GBV-B RNA helicase coding region (aa 190 to 620). The resulting chimeric cDNA was cloned into pET/NS3-His by replacing the full-length GBV-B NS3 between the
[image:2.612.314.547.71.404.2]NheI andBamHI sites. The full-length HCV NS3 was isolated by PCR from pBRTM/HCV(1–3011) (kindly provided by Charles Rice, Washington Univer-sity, St. Louis, Mo.) and cloned into theBamHI site in pQE40 (Qiagen, Valencia, Calif.). A His tag as well as an enterokinase cleavage site (MRGSHHHHHHGS DYKDDDDKA) was inserted at the beginning of the NS3 gene to facilitate purification and removal of the His tag. All plasmids were sequenced, and their coding regions (shown in Fig. 1) were verified with an ABI Prism 377 DNA sequencer with XL upgraded gels (PE Applied Biosystems, Foster City, Calif.).
FIG. 1. Schematic illustration of various plasmid constructs and cofactor peptides used in this study. The numbers above or below each construct or peptide indicate the amino acid positions corresponding to the published full-length polyprotein for each virus. The predicted molecular masses for each substrate and its cleavage products were indicated under each substrate plasmid.
on November 9, 2019 by guest
http://jvi.asm.org/
Expression and purification.The full-length GBV-B NS3, HCV/GBV-B chi-meric NS3, and HCV NS3 DNAs were freshly transformed into appropriate
Escherichia colistrains [JM109(DE3) for GBV-B NS3 and HCV/GBV-B NS3; M15(pREP4) for HCV NS3]. Protein production was induced at 24°C for 4 h by 0.2 mM isopropyl--D-thiogalactoside (IPTG) in Luria-Bertani media. Each pro-tein (GBV-B NS3-His, HCV/GBV-B NS3-His or HCV His-NS3) was purified similarly on a nickel-chelated (Ni-nitriloacetic acid) affinity column, followed by a gel filtration column (Superdex 200; Amersham Pharmacia Biotech, Piscat-away, N.J.) (76). The purity of each protein was greater than 95%. The final protein concentration was determined by Bradford protein assay (Bio-Rad Lab-oratories, Melville, N.Y.) according to the manufacturer’s instructions. The pro-tein was stored in small aliquots at⫺70°C in the presence of 10% glycerol and 5 mM DTT.
In vitro translation of GBV-B and HCV substrates.Plasmids pNBNae, pJB1003, and pTS102, encoding the HCV NS4A/4B, NS4B/5A, and NS5A/5B cleavage sites, respectively, were linearized as described by Butkiewicz et al. (10). Plasmids containing the GBV-B NS4A/4B, NS4B/5A, and NS5A/5B cleavage sites were all linearized withXhoI. All plasmids were transcribed in vitro with T7 RNA polymerase. The in vitro-transcribed RNAs were translated in rabbit re-ticulocyte lysates (Promega) in the presence of [35S]methionine (Amersham Pharmacia Biotech) at 30°C for 1 h according to the supplier’s recommendation. All translation reactions were terminated by adding DNase-free RNase (Boehr-inger Mannheim, Indianapolis, Ind.) and cycloheximide for 15 min at 30°C.
trans-cleavage translation assays.Standard translation assays for GBV-B NS3 protease were performed in a 20-l reaction volume, initiated by adding 2l of 35S-labeled translated substrate to purified protease in a final reaction mix containing 50 mM morpholine propanesulfonic acid (MOPS; pH 7.5), 50 mM NaCl, 0.1% lauryl maltoside, 10% glycerol, and 1 mM DTT. HCV protease was assayed similarly in a final mix containing 50 mM MOPS (pH 7.5), 300 mM NaCl, 0.1% NP-40, 10% glycerol, and 1 mM DTT. Reactions were incubated as indi-cated in the figure legends, from 1 to 2 h at 30°C, and then terminated by adding an equal volume of 2⫻Laemmli sample buffer followed by boiling for 3 min. Cleavage products were separated by sodium dodecyl sulfate (SDS)–15% poly-acrylamide gel electrophoresis (PAGE), detected by a PhosphorImager, and quantified by the ImageQuant software (both from Molecular Dynamics, Sunny-vale, Calif.).
Preparation of substrates for RNA helicase assay.RNA helicase substrates containing two complementary RNA strands were annealed and gel purified. Both strands were separately transcribed in vitro using the bacteriophage SP6 or T7 RNA polymerase (Promega) according to the manufacturer’s instructions. The annealed double-stranded RNA (dsRNA) substrates were end labeled using [␥-33P]ATP and T4 polynucleotide kinase (Amersham Pharmacia Biotech). The standard substrate (5⬘-3⬘) was prepared as follows. PvuII-digested plasmid pGEM-1 was transcribed with SP6 RNA polymerase to generate a 98-base RNA strand.XbaI-digested plasmid pSP65 was transcribed with SP6 RNA polymerase to generate a 38-base strand. The two RNA strands, containing a 29-base com-plementary region, were annealed and purified according to protocols published previously by other groups (31, 37, 65).
RNA helicase assay.The standard RNA helicase assay was carried out in a 20-l reaction volume containing various concentrations of NS3 protein as indi-cated. A total of 50 fmol of labeled dsRNA substrates was added to the reaction buffer containing 50 mM Tris-HCl (pH 7), 1 mM MgCl2, 2 mM ATP, 2 mM DTT, 0.1 mg of bovine serum albumin per ml, and 4 U of RNasin RNase inhibitor (Promega). The reaction mixture was incubated at 37°C for 1 h, and the reaction was terminated by the addition of 5l of the stop buffer (100 mM HEPES, pH 8; 20 mM EDTA; 0.1% NP-40; 30% glycerol; 0.3% bromophenol blue). The RNA products were electrophoresed on a 4 to 20% polyacrylamide-TBE gel (Novex, San Diego, Calif.). The gel was dried, and33P-labeled release strands were detected by autoradiography and quantified with a PhosphorImager.
RESULTS
A previous report (56) described the protease activity of GBV-B NS3 in which a catalytic domain was produced from
E. coli. The study concluded that the GBV-B NS3 protease was
able to recognize the authentic cleavage sites in the HCV polyprotein and to process the HCV polyprotein at the correct junctions. This finding was supported by a computer model prediction that the GBV-B protease possesses a similar sub-strate recognition site (the S1 pocket) for a small P1 residue (Cys) next to the scissile bond (49). The amino acid that de-fines the S1 pocket specificity (Phe154) is conserved between the HCV and GBV-B NS3 molecules (57). However, the re-ported results were surprising in that the GBV-B NS3 protease did not seem to require a cofactor for activity, a hallmark of flavivirus-like viruses (6, 14, 15, 39, 63, 64, 66). This lack of cofactor dependency could be due to the fact that only a truncated catalytic domain was used in the characterization of
the protease activity which might be different from that of the full-length NS3 as demonstrated with HCV (20). Hence, we elected to further characterize the GBV-B protease activity using a full-length GBV-B NS3 molecule produced inE. colias previously described (76).
Cleavage of HCV polyprotein junctions by the full-length GBV-B NS3 depends on the presence of a cofactor.We initially studied the role of GBV-B NS4A in GBV-B-mediatedtrans
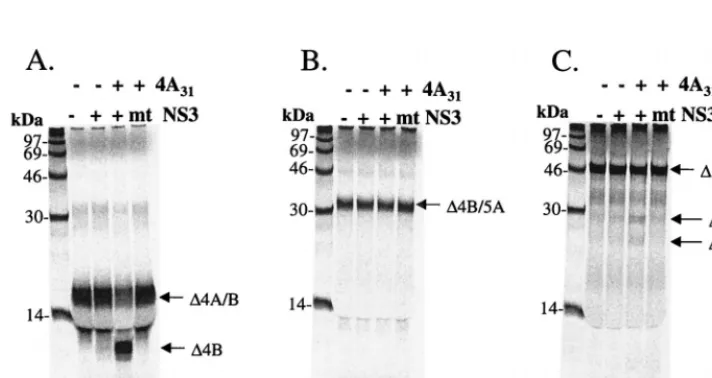
cleavages by using in vitro-translated polyprotein substrates containing the NS4A/4B, NS4B/5A, and NS5A/5B cleavage sites of the HCV polyprotein. We arbitrarily chose a 31-aa peptide (aa 16 to 46) from the central region of GBV-B NS4A (⌬4A31) to test whether the protease activity of the full-length GBV-B NS3 required an NS4A cofactor for optimal activity, as is the case with HCV. The in vitro-translated substrates were incubated as described previously (10) with the full-length GBV-B NS3 protease in the presence or absence of the⌬4A31 peptide. The results shown in Fig. 2 demonstrated that there was little processing of the HCV substrates at the NS4A/4B, NS4B/5A, and NS5A/5B junction sites in the absence of the NS4A peptide. As a control for appropriate polyprotein pro-cessing in this experiment, the full-length HCV NS3/4A de-scribed by Sali et al. (55) was used to produce the similar cleavage products as size markers (Fig. 2, lanes 2). However, processing of these substrates was increased and readily de-tectable with the addition of the NS4A peptide (compare Fig. 2, lanes 4 and 5), suggesting that, like HCV, GBV-B has also acquired a cofactor function in the central region of NS4A.
GBV-B NS3 protease processes its own polyprotein at the predicted sites. Several plasmids encoding the predicted GBV-B substrate cleavage sites at NS4A/4B, NS4B/5A, and NS5A/5B were constructed (Fig. 1) in order to evaluate ho-mologous processing by GBV-B NS3. Details of the cloning and sequences of these substrate-encoding plasmids are given in Materials and Methods. In vitrotranscleavage assays were established with these substrates similarly to those described previously (10). In these experiments, a mutant GBV-B NS3 protease in which the predicted active site serine 137 was replaced by an alanine (S137A) served as a negative control for the protease activity. The results are shown in Fig. 3. Cleavage at the NS4A/NS4B junction by the wild-type GBV-B NS3 was readily detectable only in the presence of NS4A cofactor (Fig. 3A, lane 3), producing an NS4B product with a predicted molecular mass of 10 kDa. Similarly, processing at the NS5A-NS5B junction was also NS4A cofactor dependent, producing appropriately sized NS5A and NS5B products of 22 and 20 kDa, respectively (Fig. 3C). As expected, no processing was observed for the NS3S137Amutant protease with either sub-strate (lanes 4 in Fig. 3A and C). For an unknown reason, the wild-type GBV-B NS3 did not cleave the NS4B/5A substrate, either with or without the NS4A cofactor (Fig. 3B, lanes 2 and 3). The full-length NS3 of GBV-B consisted of a consensus sequence when compared to 12 independently isolated NS3 cDNA clones. It is also identical to the published sequences (47, 60) as well as to that of the infectious clone (9). In the report by Scarselli et al. the cDNA coding for the catalytic domain of NS3 was synthesized based on the published se-quences. Thus, our full-length GBV NS3 consists of a protease domain that is identical to the one characterized by Scarselli et al. The only difference is the presence of the RNA helicase domain and a C-terminal His tag in our construct. The lack of activity was not due to the presence of the His tag, because either replacing the His tag at the N terminus of NS3 or removal of it by thrombin cleavage failed to improve the pro-tease activity (data not shown). Alternatively, the in vitro-translated GBV-B NS4B/5A substrate may be somehow
on November 9, 2019 by guest
http://jvi.asm.org/
tive in a way that prevents proper cleavage by the protease. Further investigation of this issue is under way. Given the poor cleavage efficiency at this site in HCV, requiring 100- to 1,000-fold more enzyme than that required to cleave the NS4A/4B and NS5A/5B junctions (10, 61), it is not surprising that the cleavage at GBV-B NS4B/5A may be too inefficient to be detectable.
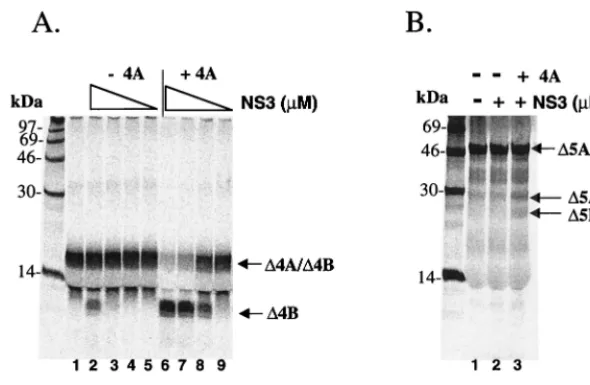
NS4A cofactor-dependent cleavage of GBV-B substrates by HCV NS3 protease.Having shown the NS4A cofactor-depen-dent GBV-B NS3 protease activity on HCV, as well as the GBV-B substrates, it was important to determine whether the HCV NS3 protease could reciprocally process the heterolo-gous GBV-B polyprotein cleavage sites. In these studies, a full-length HCV NS3 was expressed and purified to ⬎95% homogeneity fromE. coli. Its activity for proteolytic processing was tested in the presence or absence of a 13-aa cofactor
[image:4.612.95.508.72.260.2]peptide from HCV NS4A (aa 22 to 34,⌬4A13) which has been shown to be sufficient for cofactor activation of this NS3 pro-tease (10, 39, 58, 64). The results (Fig. 4A) showed a dose-dependent and NS4A cofactor-enhanced cleavage activity at the NS4A/4B junction site (lanes 6 to 9). In the absence of NS4A cofactor, some processing occurred with the highest concentration of HCV protease tested at 2.5M (Fig. 4A, lane 2). Cleavage of the GBV-B NS5A/5B site was inefficient and required 10 M of HCV NS3 protease as well as the HCV NS4A cofactor (Fig. 4B, lanes 2 and 3). Again, no processing was observed at the predicted NS4B/5A site (data not shown). To map the exact cleavage site, several synthetic peptides con-taining the predicted cleavage sites were generated. High-pres-sure liquid chromatography and mass spectrometric analyses of the cleavage products confirmed the prediction of Cys as the FIG. 2. Cofactor-dependenttranscleavage activity of GBV-B NS3 protease on HCV substrates.35S-labeled in vitro-translated substrates from HCV cleavage sites were translated as described in Materials and Methods: NS4A/NS4B (⌬4A/⌬4B) (A), NS4B/NS5A (⌬4B/⌬5A) (B), and NS5A/NS5B (⌬5A/⌬5B) (C). Labeled substrates and purified GBV-B protease were incubated in the presence or absence of 20M GBV-B NS4A peptide (aa 16 to 46,⌬4A31) for 1.5 h at 30°C. Final concentrations of GBV-B protease were 250 nM for NS4A/4B and 5M for NS4B/5A and NS5A/5B. In parallel, cleavages of HCV substrates by purified HCV NS3/4A are shown in each panel (lane 2). Samples were analyzed on an SDS–15% PAGE gel and detected by phosphorimaging scan.
FIG. 3. Cofactor-dependenttranscleavage activity of GBV-B NS3 protease on GBV-B substrates.35S-labeled in vitro-translated substrates from GBV-B cleavage sites were mixed with 2M purified protease in the presence or absence of 20M GBV-B NS4A peptide (⌬4A31):⌬4A/⌬4B (A),⌬4B/⌬5A (B), and⌬5A/⌬5B (C). A mutant (NS3S137A) GBV-B protease was also tested (lane 4 of each panel). Proteins were separated and analyzed as described in the legend to Fig. 2.
on November 9, 2019 by guest
http://jvi.asm.org/
[image:4.612.125.481.512.701.2]P1 residue at the NS4A/4B junction (data not shown) as de-scribed previously (57).
Mapping the minimal cofactor domain within GBV-B NS4A.
The results described above demonstrated that the GBV-B cofactor activity resides in the central region of NS4A from aa 16 to 46. To determine the minimum cofactor region required by GBV-B NS3 protease, we synthesized a series of truncated peptides derived from this central region of GBV-B NS4A. These synthetic peptides were tested for their ability to activate GBV-B NS3 protease activity at the NS4A/4B junction site. As summarized in Fig. 5, the C-terminal half (an 18-mer peptide
[image:5.612.156.451.72.264.2]corresponding to aa 29 to 46) of the central 31-aa peptide did not possess detectable cofactor activity since it failed to en-hance the protease activity. On the other hand, a 17-mer pep-tide representing aa 20 to 36 of NS4A retained cofactor activ-ity. Further progressive truncations from the N terminus of this peptide produced a 15-mer peptide (aa 22 to 36), which rep-resented the minimum region that could efficiently activate the protease in thesetranscleavage assays. Removal of additional residues from either terminus severely reduced this cofactor activity. It is interesting to note that this minimum cofactor region in GBV-B NS4A (aa 22 to 36) overlaps with that of FIG. 4. Cofactor-dependenttranscleavage activity of HCV NS3 protease on GBV-B substrates. (A)transcleavage reaction using GBV-B NS4A/4B as the substrate. Purified HCV full-length NS3 protease was mixed with translated substrate in the presence or absence of 100M HCV NS4A cofactor peptide (⌬4A13). Decreasing concentrations of protease were tested in this assay (indicated by a triangular descending slope: 2.5, 0.25, 0.025, and 0.0025M). (B)transcleavage reaction using GBV-B NS 5A/5B as the substrate. HCV NS3 protease at 10M was tested in the presence or absence of 100M NS4A cofactor peptide (⌬4A13).
FIG. 5. Mapping the minimum region within GBV-B NS4A required for the cofactor activity. Sequences of the entire HCV and GBV-B NS4A proteins are aligned at the top with the minimum cofactor region for HCV NS4A underlined. Vertical lines represent identity while colons represent similarity between the two viruses within the NS4A region. Dashes represent a gap created to maximize the homology alignment. To compare the cofactor activity,35S-labeled GBV-B NS4A/4B substrate
(⌬4A/⌬4B) was incubated with 1.5M GBV-B protease in the presence of various GBV-B NS4A peptides as shown in the figure. Cleavage products were then analyzed and detected by phosphorimaging and quantified by ImageQuant software (Molecular Dynamics). The cofactor activity was graded by comparing the enhancement of the protease activity to that of the background (cleavage activity in the absence of NS4A cofactor and scored as follows:⫹⫹⫹, 11- to 20-fold;⫹⫹, 5- to 10-fold;⫹, 2.5- to 5-fold;⫾, 1.5- to 2-fold; and⫺,ⱕ1.5-fold of the background activity. Amino acids critical for cofactor activation of the NS3 protease are shown in boldface, larger type; the minimum GBV-B NS4A cofactor region is also underlined.
on November 9, 2019 by guest
http://jvi.asm.org/
HCV which has been mapped to the central region (aa 22 to 33) of HCV NS4A (10, 39, 58, 64) (Fig. 5).
NS4A cofactor peptide activity is virus specific.The exper-iments described above demonstrated that the full-length GBV-B NS3 protease has the ability totranscleave the homol-ogous GBV-B substrates as well as the corresponding heterol-ogous HCV substrates. Conversely, the HCV full-length NS3 protease can cleave the heterologous GBV-B substrates in addition to the HCV substrates. Since this cleavage activity was cofactor dependent for both proteases, it was of interest to determine whether or not the required cofactor activity was virus specific. For this purpose, the NS4A/4B substrates from both GBV-B and HCV were chosen to evaluate cofactor spec-ificity. Each NS3 protease was tested with its homologous sub-strate or the heterologous subsub-strate in the presence of either the HCV NS4A cofactor peptide (aa 22 to 34,⌬4A13) or the GBV-B NS4A cofactor peptide (aa 20 to 36, ⌬4A17). The results (shown in Fig. 6) demonstrated that the cofactor re-quirement was specific for each viral protease. Each protease was activated only by the NS4A cofactor from the same virus (for HCV, compare lanes 3 and 10 with lanes 4 and 11; for GBV-B, compare lanes 6 and 13 with lanes 7 and 14). This finding is consistent with the lack of sequence homology be-tween the NS4A cofactor regions of GBV-B and HCV.
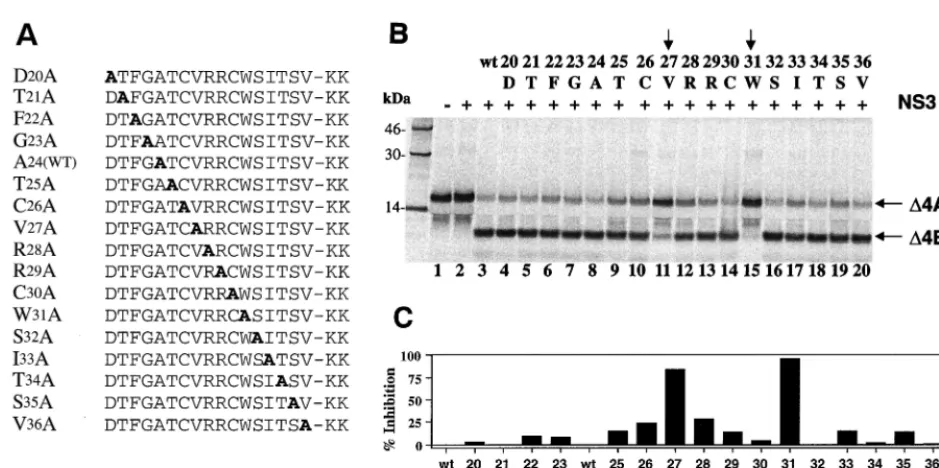
Probing essential amino acid requirements for GBV-B NS4A cofactor activity.To investigate how GBV-B NS4A co-factor activates the protease activity and what the critical de-terminants of this cofactor activity might be, a series of alanine substitutions (alanine scan) were designed using the 17-mer cofactor peptide (⌬4A17) as a template (Fig. 7A). Two lysine residues (KK) were added to the C terminus of each peptide to improve solubility. Each mutant peptide was tested for its cofactor activity at a concentration of 200M to ensure the maximum activation of the GBV-B protease on a GBV-B substrate containing the NS4A/4B cleavage site. As shown in Fig. 7B, two amino acids were clearly identified as the critical determinants of the cofactor activity: Trp31 (W31, lane 15) and Val27 (V27, lane 11). When compared to the activity of wild-type cofactor peptide (see lane 3, and also lane 8), the alanine
substitution at W31almost completely abolished the cofactor-dependent cleavage, inhibiting the product formation by 96% as determined by using a PhosphorImager (Fig. 7C). Similarly, replacing V27with alanine was also detrimental to the cofactor activity, inhibiting the cleavage by 86% (Fig. 7C). These results reveal a strikingly similar pattern of NS4A activation of NS3 protease between the two viruses, in that V27 and W31 of GBV-B mirror the two critical amino acids (I25and I29) defin-ing the HCV cofactor activity (10, 58) (Fig. 5). It appears that the hydrophobicity of the critical cofactor amino acids, as well as the spacing between these two residues, is important for the activation of NS3 proteases. Other amino acid residues in the cofactor regions may play a role in defining the viral specificity of cofactor function.
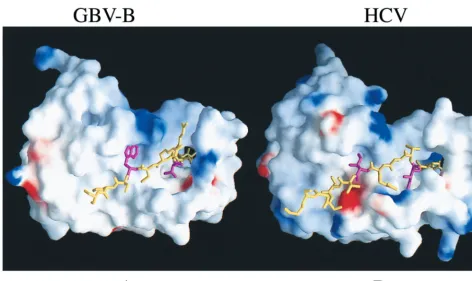
Computer model for GBV-B NS3/4A interaction.To further understand how GBV-B cofactor peptide might interact with and enhance the NS3 protease activity, the HCV NS3 protease structure was used as a template for the modeling of GBV-B protease structure using the program QUANTA (Molecular Simulations, Inc., San Diego, Calif.). The primary sequences of the NS3 protease domain between HCV and GBV-B share approximately 30% identity, whereas the full-length NS3 pro-teins share 40% identity. The secondary structure elements of the GBV-B protease model were transferred from the existing HCV NS3 protease crystal structures (33, 43, 71). Side chain atom coordinates of HCV were adopted to model and position those in the corresponding GBV-B protease. Those unresolved side chains were built by using the program CHARMm (Mo-lecular Simulations), and the newly modeled molecule was relaxed to yield a smooth backbone with reasonable packing by molecular dynamics.
Figure 8B shows the NS3/4A interaction of HCV. The two critical residues (I25and I29, in magenta) which are buried in two well-defined hydrophobic pockets in the protease core are highlighted. The I29 side chain occupies the hydrophobic pocket located between the two-barrel subdomains and may rigidify the relative position of the subdomains that are con-nected by a single flexible loop. I29may also pivot the insertion of the N-terminal half of NS4A cofactor into the N-terminal FIG. 6. NS4A cofactor activity is virus specific. HCV or GBV-B NS3 protease was tested in the absence of NS4A peptide or in the presence of either HCV NS4A (⌬4A13) or GBV-B NS4A (⌬4A17) at 100M. The assay was performed using the NS4A/4B substrates from either HCV (A) or GBV-B (B). HCV NS3 was tested at 50 nM for both substrates; GBV-B protease was tested at 250 nM for HCV substrate and at 2M for GBV-B substrate.
on November 9, 2019 by guest
http://jvi.asm.org/
domain of NS3 protease. This is consistent with the observa-tion that I29is the most critical determinant of the NS3/4A activation (10). The hydrophobic side chain of I25, another key residue, packs against two adjacent  strands (A1 and D1), which may cause the D1-E1 loop to shift from its position in NS3 upon NS4A complex formation. However, other mecha-nisms are possible, such as the one proposed by Love et al. (43), who favored the rearrangement of salt links among charged amino acids as the cause of the D1-E1 loop shift.
The modeled GBV-B complex structure reveals two strik-ingly similar hydrophobic pockets for docking the NS4A co-factor peptide and stringent complementarity between the pro-tease and its cofactor (Fig. 8A). This is consistent with the mutation analysis described above (Fig. 7) that demonstrated the two critical amino acid determinants within the cofactor. The W31 side chain (corresponding to that of I29 in HCV) occupies a somewhat larger hydrophobic pocket between the -barrel subdomains than that present in HCV. The hydro-phobic side chain of V27(corresponding to that of I25in HCV) packs similarly against the A1 strand and the D1-E1 loop. The two Arg residues (R28 and R29) are not important for the NS4A cofactor activity (Fig. 7), which is consistent with the model in that they are solvent exposed. This computer mod-eling result suggests a general strategy in the NS4A activation of NS3 protease activity, with virus-specific variation.
A chimeric NS3 that retains both HCV protease and GBV-B RNA helicase functions.Our recent studies of the GBV-B NS3 RNA helicase demonstrated that it has enzymatic properties similar to that of the HCV enzyme (76). In the preceding experiments, we compared the NS3 protease activities of HCV and GBV-B. Several other reports described the interdomain relationship between the protease and RNA helicase by com-paring the domain-derived activities with those of the full-length NS3, revealing relatively independent enzymatic func-tions (20, 21, 28, 55). This suggests that, although physically
linked, the protease and RNA helicase domains of NS3 may be functionally separated. The recent determination of the struc-ture of the full-length NS3 of HCV revealed two well-sepa-rated structural domains of protease and RNA helicase linked together through a single-stranded peptide tether (75). The full-length NS3/4A structure reveals characteristics similar to those observed in the single domain structures (32, 33, 42, 43, 71, 74). The protease and helicase catalytic centers are segre-gated in the bifunctional enzyme. The P-side of the substrate recognition site is occupied by the molecule’s own carboxy terminus, which provides a snapshot of the structure aftercis
proteolytic processing at the NS3/4A junction (75). A more “open” conformation can be adopted with minimum interdo-main interaction in solution to allow subsequent trans cleav-ages (75). Based on the high degree of homology (40% iden-tity), the predicted full-length GBV-B NS3 structure will be very similar to that of HCV. Both the protease domain and the helicase domain connected through a flexible strand are be-lieved to adopt relatively independent functional conforma-tions optimal for each activity. Several studies concluded that the full-length HCV NS3 protein had activities comparable to those of the individual catalytic domains (20, 21, 28, 55).
[image:7.612.65.535.66.300.2]This unique interdomain structural relationship (75) prompt-ed us to conduct a molecular “domain transplant” in which the GBV-B NS3 protease domain was replaced by the correspond-ing HCV NS3 protease domain (Fig. 1). This chimeric NS3 molecule thus consists of an N-terminal HCV protease domain (aa 1 to 190) and a C-terminal RNA helicase domain of GBV-B (aa 190 to 620) with the fusion located within the peptide tether separating the two major domains. This chi-meric protein was expressed and purified from E. coli. The solubility of the chimeric protein was comparable to that of the native GBV-B NS3, indicating that the “xenografted” HCV protease domain was well “received” without affecting the proper folding of the protein, as expected from the full-length FIG. 7. Cofactor activity of alanine-substituted GBV-B NS4A peptides (⌬4A17). (A) Sequence of alanine-substituted peptides used in this study, with the alanine residue shown in boldface. The A24 wild type [A24(WT)] is the same as the wild-type peptide. (B) Cofactor activity tested in thetranscleavage assay. A 200M concentration of each peptide was used in each reaction containing the GBV-B NS4A/4B substrate and 1.5M GBV-B NS3 protease. Cleavage products were analyzed similarly as described in the previous figures. The alanine-substituted residue and the corresponding amino acid position (number) in NS4A are shown above each lane. Vertical arrows identify the amino acids critical for cofactor activity. (C) Quantitative analysis of the cofactor activities (shown in panel B) expressed as the percent inhibition of wild-type cofactor activity.
on November 9, 2019 by guest
http://jvi.asm.org/
NS3 structure. Both the protease and the RNA helicase activ-ities were subsequently characterized. The results (Fig. 9A) demonstrated that the chimeric NS3 retained protease activity capable of processing both GBV-B and HCV substrates
[image:8.612.67.539.68.349.2](con-taining the NS4A/4B cleavage site). This protease activity re-quired only the HCV NS4A cofactor peptide (⌬4A13) (lane 5) and not the GBV-B NS4A cofactor peptide (⌬4A17) (lanes 7 and 8). As a comparison, the full-length HCV NS3 protease FIG. 8. Surface representation of GBV-B and HCV protease complexed with the corresponding 4A cofactors (shown as sticks). (A) GBV-B NS3/4A model. (B) HCV NS3/4A model. The electrostatic potential distribution is color coded: blue, positive; red, negative; white, neutral. The N-terminal 11 aa were removed to expose the detail interaction between NS4A and NS3 protease. The two critical Trp31 and Val27 (shown as magenta sticks in contrast to the rest of 4A colored yellow) reveal their location and complementary with the corresponding hydrophobic pockets, which is strikingly similar to that of HCV NS3/4A complex structure in panel B. The two essential residues Ile29 and Ile25, in HCV NS4A are also colored in magenta.
FIG. 9. HCV protease and GBV-B RNA helicase activities of the chimeric NS3 protein. (A) Protease activity. The GBV-B NS4A/4B (⌬4A/4B) substrate was mixed with the chimeric HCV/GBV-B NS3 at 2M (lanes 3, 5, and 7) or at 0.07M (lanes 4, 6, and 8) in the absence NS4A cofactor or the presence of either HCV NS4A cofactor (⌬4A13) or GBV-B NS4A cofactor (⌬4A17). The final concentrations of the cofactor peptides were 100M. The cleavage products were analyzed as described previously. (B) RNA helicase activity. The RNA helicase activity of the chimeric NS3 was compared to that of the native GBV-B NS3. The⌬symbol indicates that the sample was boiled before loading onto the gel. Increasing amounts (2, 5, and 10 pmol) of each protein were tested for the dsRNA unwinding activity. The released ssRNAs were separated and analyzed as described in Materials and Methods. The ascending triangular slopes indicate the increasing concentration of the NS3 proteins. The “⫺” symbol indicates the no-enzyme controls.
on November 9, 2019 by guest
http://jvi.asm.org/
[image:8.612.93.515.473.668.2]was used to demonstrate the HCV NS4A cofactor-dependent cleavage products (lane 9). Comparison of the RNA helicase activities also revealed that the chimeric NS3 retained the RNA helicase activity similar to that of native GBV-B NS3 (Fig. 9B). Thus, the chimeric NS3 acquired the HCV protease activity and retained the native GBV-B RNA helicase activity.
DISCUSSION
Virally encoded proteases play an essential role in viral replication. Inhibitors of viral proteases have been developed to inhibit viral replication. Several human immunodeficiency virus (HIV) protease inhibitors that effectively suppress the viral production and slow disease progression are approved for the treatment of HIV infection in combination with other antiviral components (46, 50). Like the HIV protease, the HCV NS3 serine protease has been considered an important target for the development of anti-HCV inhibitors. Unfortu-nately, small and relatively inexpensive animal models that support HCV replication are not available at the present time. Efforts to establish transgenic mouse models or xenografted SCID-hu mouse models have not yielded any reliable or con-vincing animal models that are suitable for anti-HCV drug development (8, 36, 45). The only animal model permissive for HCV infection in vivo is the chimpanzee, which is prohibitively expensive for routine drug discovery efforts. The recent dis-covery of GBV-B, a hepatotropic virus closely related to HCV, provides hope for a surrogate virus to evaluate anti-HCV in-hibitors in a much less expensive small animal model.
Several HCV NS3 protease-dependent chimeric viruses have been created, providing necessary cell-based antiviral as-says to evaluate the efficacy of potential inhibitors against the HCV protease (11, 18, 19, 23). Although these chimeric viruses may alleviate the pressing need for in vitro testing of cell permeation and antiviral efficacy of candidate inhibitors of HCV protease, these artificial systems in the most part fail to duplicate the polyprotein processing events occurring naturally during HCV infection. In addition, proper animal models have not been developed for these viruses. A chimeric HCV/GBV-B virus would provide an alternative solution that better mimics an HCV infection in a small primate model. The primary findings in this report further strengthen the hypothesis that the GBV-B–tamarin model is biologically relevant to the study of HCV and may serve as a good surrogate system for HCV infection (57). In addition, our comparative characterization of GBV-B NS3 protease and identification of a virus-specific GBV-B NS4A cofactor provide insights for the future devel-opment of a viable chimeric GBV-B/HCV virus that can be used in the tamarin model. In this improved version of the GBV-B–tamarin model, a built-in HCV NS3 protease domain as well as an HCV NS4A cofactor region would replace the resident GBV-B protease and NS4A cofactor, making the chi-meric virus dependent upon the respective HCV functions.
Guided by a predicted computer model of the full-length GBV-B NS3 molecule as well as the full-length HCV NS3 structure, precise protease domain swapping was accom-plished. The resulting chimeric HCV/GBV-B NS3 retained both protease and RNA helicase activities comparable to the unaltered or native functionality. The mapping of the GBV-B NS4A cofactor region provides necessary information for splic-ing the HCV NS4A cofactor into the GBV-B genome in lieu of its own cofactor. A unique sequence of 265 bases was recently identified at the 3⬘ end of the GBV-B genome (56; D. V. Sangar and S. M. Lemon, unpublished results), reminiscent of the 3⬘X element found at the 3⬘end of HCV genome (35). This will allow the construction of full-length molecular clones of
GBV-B (9) as well as chimeric HCV/GBV-B viruses. Since the HCV and GBV-B proteases share similar substrate specificity and can cross-process each other’s polyprotein, it is likely that an HCV NS3 protease-dependent GBV-B would be capable of proper proteolytic processing and producing mature viral pro-teins that would be competent for viral replication. The iden-tification of the virus-specific NS4A cofactor requirement (Fig. 6) suggests that a concomitant chimeric engineering of the NS4A domain would be necessary to achieve efficient heterol-ogous polyprotein processing by the chimeric viruses. Such chimeric viruses would be valuable for both in vitro and in vivo testing of anti-HCV inhibitors.
The complete mechanistic pathway of NS4A activation of the NS3 protease is not fully understood. The crystal structures of HCV NS3 protease and NS4A cofactor complexes (33, 43, 71) have shown that two major conformational changes upon NS4A complexation occur: one involves the packing and or-dering of the N-terminal 30 aa; the other causes a shift of the D1-E1 loop in the N-terminal -barrel subdomain (43, 71). These conformational changes help to confine the catalytic triad in the active site to a proper orientation optimal for catalysis. Here, we propose a general mechanism for the acti-vation of NS3 protease based on the structural and mutational analyses as well as on comparative studies of other related viruses. The two critical hydrophobic amino acids separated by three noncritical amino acids (⌽x3⌽, where ⌽ represents a bulky hydrophobic amino acid and x represents any amino acid) form a protease activation motif. This is further substan-tiated by the presence of this motif in the NS4A cofactors of HCV, GB viruses, and pestiviruses, as well as in the NS2B cofactor region of flaviviruses (Fig. 10) (6, 7, 66). Based on the comparison between the structures of NS3 alone and NS3/4A complex (33, 42), the hydrophobic pocket corresponding to the second⌽(I29in HCV NS4A) remains unchanged before and after NS4A complexation, while the pocket for the first⌽(I25 in HCV NS4A) undergoes significant conformational change. We thus hypothesize that the second hydrophobic amino acid of the “⌽x3⌽” motif makes contact with the protease. It plays an essential role by occupying the hydrophobic pocket between the two-barrel subdomains and rigidifying the relative posi-tion of the subdomains which are connected by a single loop. This is followed by the insertion of the N-terminal half of NS4A cofactor into the N-terminal domain of NS3 protease, allowing the first hydrophobic amino acid of the activation motif to be inserted into a second hydrophobic pocket adjacent to two beta-strands (A1 and D1). This insertion may directly cause the D1-E1 loop shift upon cofactor complexation and stiffen the alignment of the catalytic triad in the active site. Another mechanism was proposed by Love et al. (43), who
FIG. 10. Cofactor activation motif for the flavivirus-like NS3 serine protease. Cofactor regions from various members of theFlaviviridaefamily are compared. The minimum cofactor region in HCV is underlined. A common feature involv-ing two bulky hydrophobic amino acids (in boldface type symbolized by a “⌽”) is proposed to form the activation motif,⌽xxx⌽, where “x” is any amino acid.
on November 9, 2019 by guest
http://jvi.asm.org/
favored the rearrangement of salt links among charged amino acids as the cause of D1-E1 loop shift. However, this cannot explain the results from the mutational analysis (10, 58) and the apparent lack of sequence conservation of the charged amino acids (Fig. 5 and 10). A recent study on HGV/GBV-C also revealed a similar cofactor requirement which was mapped to the central region of NS4A (6). With the alternating hydro-phobic pattern, the HGV NS4A cofactor enhanced the NS3 protease activity. Interestingly, the HGV NS4A has been shown to be a weak cofactor of HCV NS3 protease, highlight-ing the high degree of functional conservation in related vi-ruses (68). Despite the strict virus-specific cofactor require-ment, a general strategy becomes apparent in the activation of NS3 protease by its NS4A cofactor.
ACKNOWLEDGMENTS
We thank Gregory Reyes for support and Patricia Weber for en-couragement and critical reading of the manuscript. We also thank Yanhui Liu for mass spectrometry analysis and Angela Skelton and Eric Ferrari for assistance in molecular cloning and protein purifica-tion.
This work was supported in part by a grant from the National Institute of Allergy and Infectious Diseases (U19-AI40035).
REFERENCES
1.Alter, H. J., R. H. Purcell, J. W. Shih, J. C. Melpolder, M. Houghton, Q.-L. Choo, and G. Kuo.1989. Detection of antibody to hepatitis C virus in prospectively followed transfusion recipients with acute and chronic non-A non-B hepatitis. N. Engl. J. Med.321:1494–1500.
2.Alter, M. J., D. Kruszon-Moran, O. V. Nainan, G. M. McQuillan, F. Gao, L. A. Moyer, R. A. Kaslow, and H. S. Margolis.1999. The prevalence of hepatitis C virus infection in the United States, 1988 through 1994. N. Engl. J. Med.341:556–562.
3.Bartenschlager, R., L. Ahlborn-Laake, J. Mous, and H. Jacobsen.1993. Nonstructural protein 3 of the hepatitis C virus encodes a serine-type pro-teinase required for cleavage at the NS3/4 and NS4/5 junctions. J. Virol.67: 3835–3844.
4.Beard, M. R., G. Abell, M. Honda, A. Carroll, M. Gartland, B. Clarke, K. Suzuki, R. Lanford, D. V. Sangar, and S. M. Lemon.1999. An infectious molecular clone of a Japanese genotype 1b hepatitis C virus. Hepatology30: 316–624.
5.Behrens, S.-E., L. Tomei, and R. De Francesco.1996. Identification and properties of the RNA-dependent RNA polymerase of hepatitis C virus. EMBO J.15:12–22.
6.Belyaev, A. S., S. Chong, A. Novikov, A. Kongpachith, F. R. Masiarz, M. Lim, and J. P. Kim.1998. Hepatitis G virus encodes protease activities which can effect processing of the virus putative nonstructural proteins. J. Virol.72: 868–872.
7.Brinkworth, R. I., D. P. Fairlie, D. Leung, and P. R. Young.1999. Homology model of the dengue 2 virus NS3 protease: putative interactions with both substrate and NS2B cofactor. J. Gen. Virol.80:1167–1177.
8.Bronowicki, J. P., M. A. Loriot, V. Thiers, Y. Grignon, A. L. Zignego, and C. Brechot.1998. Hepatitis C virus persistence in human hematopoietic cells injected into SCID mice. Hepatology28:211–218.
9.Bukh, J., C. L. Apgar, and M. Yanagi.1999. Toward a surrogate model for hepatitis C virus: an infectious molecular clone of the GB virus-B hepatitis agent. Virology262:470–478.
10. Butkiewicz, N. J., M. Wendel, R. Zhang, R. Jubin, J. Pichardo, E. B. Smith, A. M. Hart, R. Ingram, J. Durkin, P. W. Mui, M. G. Murray, L. Ra-manathan, and B. Dasmahapatra.1996. Enhancement of hepatitis C virus NS3 proteinase activity by association with NS4A-specific synthetic peptides: identification of sequence and critical residues of NS4A for the cofactor activity. Virology225:328–338.
11. Cho, Y. G., H. S. Moon, and Y. C. Sung.1997. Construction of hepatitis C-SIN virus recombinants with replicative dependency on hepatitis C virus serine protease activity. J. Virol. Methods65:201–207.
12. Choo, Q.-L., G. Kuo, A. J. Weiner, L. R. Overby, D. W. Bradley, and M. Houghton.1989. Isolation of a cDNA clone derived from a blood-born non-A, non-B viral hepatitis genome. Science244:359–364.
13. Deinhardt, F., A. W. Holmes, R. B. Capps, and H. Popper.1967. Studies on the transmission of human viral hepatitis to marmoset monkeys. I. Trans-mission of disease, serial passages, and description of liver lesions. J. Exp. Med.125:673–688.
14. Failla, C., L. Tomei, and R. De Francesco.1994. both NS3 and NS4A are required for proteolytic processing of hepatitis C virus nonstructural pro-teins. J. Virol.68:3753–3760.
15. Falgout, B., M. Pethel, Y.-M. Zhang, and C.-J. Lai.1991. Both nonstructural proteins NS2B and NS3 are required for the proteolytic processing of dengue virus nonstructural proteins. J. Virol.65:2467–2475.
16. Farci, P., H. J. Alter, S. Govindarajan, D. C. Wong, R. Engle, R. R. Les-niewski, I. K. Mushahwar, S. M. Desai, R. H. Miller, N. Ogata, and R. H. Purcell.1992. Lack of protective immunity against reinfection with hepatitis C virus. Science258:135–140.
17. Ferrari, E., J. Wright-Minogue, J. W. S. Fang, B. M. Baroudy, J. Y. N. Lau, and Z. Hong. 1999. Characterization of soluble hepatitis C virus RNA-dependent RNA polymerase expressed inEscherichia coli. J. Virol.73:1649– 1654.
18. Filocamo, G., L. Pacini, and G. Migliaccio.1997. Chimeric Sindbis viruses dependent on the NS3 protease of hepatitis C virus. J. Virol.71:1417–1427. 19. Filocamo, G., L. Pacini, C. Nardi, L. Bartholomew, M. Scaturro, P. Delmas-tro, A. Tramontano, R. De Francesco, and G. Migliaccio.1999. Selection of functional variants of the NS3-NS4A protease of hepatitis C virus by using chimeric sindbis viruses. J. Virol.73:561–575.
20. Gallinari, P., D. Brennan, C. Nardi, M. Brunetti, L. Tomei, C. Steinkuhler, and R. De Francesco.1998. Multiple enzymatic activities associated with recombinant NS3 protein of hepatitis C virus. J. Virol.72:6758–6769. 21. Gallinari, P., C. Paolini, D. Brennan, C. Nardi, C. Steinkuhler, and R. De
Francesco.1999. Modulation of hepatitis C virus NS3 protease and helicase activities through the interaction with NS4A. Biochemistry38:5620–5632. 22. Grakoui, A., D. W. McCourt, C. Wychowski, S. M. Feinstone, and C. M. Rice.
1993. Characterization of the hepatitis C virus-encoded serine proteinase: determination of the proteinase-dependent polyprotein cleavage sites. J. Vi-rol.67:2832–2843.
23. Hahm, B., S. H. Back, T. G. Lee, E. Wimmer, and S. K. Jang.1996. Gen-eration of a novel poliovirus with a requirement of hepatitis C virus protease NS3 activity. Virology226:318–326.
24. Hijikata, M., H. Mizushima, T. Akagi, S. Mori, N. Kakiuchi, N. Kato, T. Tanaka, K. Kimura, and K. Shimotohno. 1993. Two distinct proteinase activities required for the processing of a putative nonstructural precursor protein of hepatitis C virus. J. Virol.67:4665–4675.
25. Hong, Z., M. Beaudet-Miller, R. E. Lanford, B. Guerra, J. Wright-Minogue, A. Skelton, B. M. Baroudy, G. R. Reyes, and J. Y. N. Lau.1999. Generation of transmissible hepatitis C virions from a molecular clone in chimpanzees. Virology256:36–44.
26. Hong, Z., E. Ferrari, J. Wright-Minogue, R. Chase, C. Risano, G. Seelig, C.-G. Lee, and A. D. Kwong.1996. Enzymatic characterization of hepatitis C virus NS3/4A complexes expressed in mammalian cells using the herpes simplex virus amplicon system. J. Virol.70:4261–4268.
27. Houghton, M.1996. Hepatitis C viruses, p. 1035–1058.InB. N. Fields, D. M. Knipe, and P. M. Howley (ed.), Virology, 3rd ed. Raven Press, New York, N.Y.
28. Howe, A. Y., R. Chase, S. S. Taremi, C. Risano, B. Beyer, B. Malcolm, and J. Y. Lau.1999. A novel recombinant single-chain hepatitis C virus NS3-NS4A protein with improved helicase activity. Protein Sci.8:1332–1341. 29. Jin, L., and D. L. Peterson.1995. Expression, isolation, and characterization
of the hepatitis C virus ATPase/RNA helicase. Arch. Biochem. Biophys.323: 47–53.
30. Kao, J. H., P. J. Chen, J. T. Wang, P. M. Yang, M. Y. Lai, T. H. Wang, and D. S. Chen.1996. Superinfection by homotypic virus in hepatitis C virus carriers: studies on patients with post-transfusion hepatitis. J. Med. Virol.50: 303–308.
31. Kim, D. W., Y. Gwack, J. H. Han, and J. Choe.1995. C-terminal domain of the hepatitis C virus NS3 protein contains an RNA helicase activity. Bio-chem. Biophys. Res. Commun.215:160–166.
32. Kim, J. L., K. A. Morgenstern, J. P. Griffith, M. D. Dwyer, J. A. Thomson, M. A. Murcko, C. Lin, and P. R. Caron.1998. Hepatitis C virus NS3 RNA helicase domain with a bound oligonucleotide: the crystal structure provides insights into the mode of unwinding. Structure6:89–100.
33. Kim, J. L., K. A. Morgenstern, C. Lin, T. Fox, M. D. Dwyer, J. A. Landro, S. P. Chambers, W. Markland, C. A. Lepre, E. T. O’Malley, S. L. Harbeson, C. M. Rice, M. A. Murcko, P. R. Caron, and J. A. Thomson.1996. Crystal structure of the hepatitis C virus NS3 protease domain complexed with a synthetic NS4A cofactor peptide. Cell87:343–355.
34. Kolykhalov, A. A., E. V. Agapov, K. J. Blight, K. Mihalik, S. M. Feinstone, and C. M. Rice.1997. Transmission of hepatitis C by intrahepatic inoculation with transcribed RNA. Science277:570–574.
35. Kolykhalov, A. A., S. M. Feinstone, and C. M. Rice.1996. Identification of a highly conserved sequence element at the 3⬘terminus of hepatitis C virus genome RNA. J. Virol.70:3363–3371.
36. Laconi, E., R. Oren, D. K. Mukhopadhyay, E. Hurston, S. Laconi, P. Pani, M. D. Dabeva, and D. A. Shafritz.1998. Long-term, near-total liver replace-ment by transplantation of isolated hepatocytes in rats treated with retrors-ine. Am. J. Pathol.153:319–329.
37. Lee, C.-G., and J. Hurwitz.1992. A new RNA helicase isolated from HeLa cells that catalytically translocates in the 3⬘to 5⬘direction. J. Biol. Chem. 267:4398–4407.
38. Lesburg, C. A., M. B. Cable, E. Ferrari, Z. Hong, A. F. Mannarino, and P. C. Weber.1999. Crystal structure of the RNA-dependent RNA polymerase
on November 9, 2019 by guest
http://jvi.asm.org/
from hepatitis C virus reveals a fully encircled active site. Nat. Struct. Biol. 6:937–943.
39.Lin, C., J. A. Thomson, and C. M. Rice.1995. A central region in the hepatitis C virus NS4A protein allows formation of an active NS3-NS4A serine proteinase complex in vivo and in vitro. J. Virol.69:4373–4380. 40.Lohmann, V., F. Korner, U. Herian, and R. Bartenschlager.1997.
Biochem-ical properties of hepatitis C virus NS5B RNA-dependent RNA polymerase and identification of amino acid sequence motifs essential for enzymatic activity. J. Virol.71:8416–8428.
41.Lohmann, V., F. Korner, J.-O. Koch, U. Herian, L. Theilmann, and R. Bartenschlager.1999. Replication of subgenomic hepatitis C virus RNAs in a hepatoma cell line. Science285:110–113.
42.Love, R. A., H. E. Parge, J. A. Wickersham, Z. Hostomsky, N. Habuka, E. W. Moomaw, T. Adachi, and Z. Hostomska.1996. The crystal structure of hepatitis C virus NS3 proteinase reveals a trypsin-like fold and a structural zinc binding site. Cell87:331–342.
43.Love, R. A., H. E. Parge, J. A. Wickersham, Z. Hostomsky, N. Habuka, E. W. Moomaw, T. Adachi, S. Margosiak, E. Dagostino, and Z. Hostomska.1998. The conformation of hepatitis C virus NS3 proteinase with and without NS4A: a structural basis for the activation of the enzyme by its cofactor. Clin. Diagn. Virol.10:151–156.
44.Marcellin, P., N. Boyer, A. Gervais, M. Martinot, M. Pouteau, C. Castelnau, A. Kilani, J. Areias, A. Auperin, J. P. Benhamou, C. Degott, and S. Erlinger. 1997. Long-term histologic improvement and loss of detectable intrahepatic HCV RNA in patients with chronic hepatitis C and sustained response to interferon-alpha therapy. Ann. Intern. Med.127:875–881.
45.Matsuda, J., M. Suzuki, C. Nozaki, N. Shinya, K. Tashiro, K. Mizuno, Y. Uchinuno, and K. Yamamura.1998. Transgenic mouse expressing a full-length hepatitis C virus cDNA. Jpn. J. Cancer Res.89:150–158.
46.Molla, A., G. R. Granneman, E. Sun, and D. J. Kempf.1998. Recent devel-opments in HIV protease inhibitor therapy. Antivir. Res.39:1–23. 47. Muerhoff, A. S., T. P. Leary, J. N. Simons, T. J. Pilot-Matias, G. J. Dawson,
J. C. Erker, M. L. Chalmers, G. G. Schlauder, S. M. Desai, and I. K. Mushahwar.1995. Genomic organization of GB viruses A and B: two new members of theFlaviviridaeassociated with GB agent hepatitis. J. Virol.69: 5621–5630.
48. Ohba, K., M. Mizokami, J. Y. Lau, E. Orito, K. Ikeo, and T. Gojobori.1996. Evolutionary relationship of hepatitis C, pesti-, flavi-, plantviruses, and newly discovered GB hepatitis agents. FEBS Lett.378:232–234.
49. Pizzi, E., A. Tramontano, L. Tomei, N. La Monica, C. Failla, M. Sardana, T. Wood, and R. De Francesco.1994. Molecular model of the specificity pocket of the hepatitis C virus protease: implications for substrate recognition. Proc. Natl. Acad. Sci. USA91:888–892.
50. Rana, K. Z., and M. N. Dudley.1999. Human immunodeficiency virus pro-tease inhibitors. Pharmacotherapy19:35–59.
51. Reichard, O., G. Norkrans, A. Fryden, J. H. Braconier, A. Sonnerborg, O. Weiland, and T. S. S. Group.1998. Randomised, double-blind, placebo-controlled trial of interferon alpha-2b with and without ribavirin for chronic hepatitis C. Lancet351:83–87.
52. Rice, C. M.1996. Flaviviridae: the viruses and their replication, p. 931–960.
InB. N. Fields, D. M. Knipe, and P. M. Howley (ed.), Virology, 3rd ed. Raven Press, New York, N.Y.
53. Rijnbrand, R., G. Abell, and S. M. Lemon.2000. Mutational analysis of the GB virus B internal ribosome entry site. J. Virol.74:773–783.
54. Robertson, B., G. Myers, C. Howard, T. Brettin, J. Bukh, B. Gaschen, T. Gojobori, G. Maertens, M. Mizokami, O. Nainan, S. Netesov, K. Nishioka, T. Shin-i, P. Simmonds, D. Smith, L. Stuyver, and A. Weiner.1998. Classi-fication, nomenclature, and database development for hepatitis C virus (HCV) and related viruses: proposals for standardization. Arch. Virol.143: 2493–2503.
55. Sali, D. L., R. Ingram, M. Wendel, D. Gupta, C. McNemar, A. Tsarbopoulos, J. W. Chen, Z. Hong, R. Chase, C. Risano, R. Zhang, N. Yao, A. D. Kwong, L. Ramanathan, H. V. Le, and P. C. Weber.1998. Serine protease of hepatitis C virus expressed in insect cells as the NS3/4A complex. Biochem.37: 3392–3401.
56. Sbardellati, A., E. Scarselli, L. Tomei, A. S. Kekule, and C. Traboni.1999. Identification of a novel sequence at the 3⬘end of the GB virus B genome. J. Virol.73:10546–10550.
57. Scarselli, E., A. Urbani, A. Sbardellati, L. Tomei, R. De Francesco, and C.
Traboni.1997. GB virus B and hepatitis C virus NS3 serine proteases share substrate specificity. J. Virol.71:4985–4989.
58. Shimizu, Y., K. Yamaji, Y. Masuho, T. Yokota, H. Inoue, K. Sudo, S. Satoh, and K. Shimotohno.1996. Identification of the sequence on NS4A required for enhanced cleavage of the NS5A/5B site by hepatitis C virus NS3 protease. J. Virol.70:127–132.
59. Shimizu, Y. K., M. Hijikata, A. Iwamoto, H. J. Alter, R. H. Purcell, and H. Yoshikura.1994. Neutralizing antibodies against hepatitis C virus and the emergence of neutralization escape mutant viruses. J. Virol.68:1494–1500. 60. Simons, J. N., T. J. Pilot-Matias, T. P. Leary, G. J. Dawson, S. M. Desai, G. G. Schlauder, A. S. Muerhoff, J. C. Erker, S. L. Buijk, M. L. Chalmers, C. L. Van Sant, and I. K. Mushahwar.1995. Identification of two flavivirus-like genomes in the GB hepatitis agent. Proc. Natl. Acad. Sci. USA92: 3401–3405.
61. Steinkuhler, C., A. Urbani, L. Tomei, G. Biasiol, M. Sardana, E. Bianchi, A. Pessi, and R. De Francesco.1996. Activity of purified hepatitis C virus protease NS3 on peptide substrates. J. Virol.70:6694–6700.
62. Suzich, J. A., J. K. Tamura, F. Palmer-Hill, P. Warrener, A. Grakoui, C. M. Rice, S. M. Feinstone, and M. S. Collett.1993. Hepatitis C virus NS3 protein polynucleotide-stimulated nucleoside triphosphatase and comparison with the related pestivirus and flavivirus enzymes. J. Virol.67:6152–6158. 63. Tanji, Y., M. Hijikata, S. Satoh, T. Kaneko, and K. Shimotohno.1995.
Hepatitis C virus-encoded nonstructural protein NS4A has versatile func-tions in viral protein processing. J. Virol.69:1575–1581.
64. Tomei, L., C. Failla, R. L. Vitale, E. Bianchi, and R. De Francesco.1996. A central hydrophobic domain of the hepatitis C virus NS4A protein is neces-sary and sufficient for the activation of the NS3 protease. J. Gen. Virol.77: 1065–1070.
65. Warrener, P., and M. Collett.1995. Pestivirus NS3 (p80) protein possesses RNA helicase activity. J. Virol.69:1720–1726.
66. Wiskerchen, M., and M. S. Collett.1991. Pestivirus gene expression: protein p80 of bovine viral diarrhea virus is a serine proteinase involved in polypro-tein processing. Virology184:341–350.
67. World Health Organization.1996. Hepatitis C. Seroprevalence of hepatitis C virus (HCV) in a population sample. Weekly Epidemiol. Rec.71:346–349. 68. Wright-Minogue, J., N. Yao, R. Zhang, N. J. Butkiewiez, B. M. Baroudy, J. Y. N. Lau, and Z. Hong.Cross-genotypic interaction between hepatitis C virus NS3 protease domains and NS4A cofactors. J. Hepatol., in press. 69. Wyatt, C. A., L. Andrus, B. Brotman, F. Huang, D.-H. Lee, and A. M. Prince.
1998. Immunity in chimpanzees chronically infected with hepatitis C virus: role of minor quasispecies in reinfection. J. Virol.72:1725–1730. 70. Yamashita, T., S. Kaneko, Y. Shirota, W. Qin, T. Nomura, K. Kobayashi,
and S. Murakami.1998. RNA-dependent RNA polymerase activity of the soluble recombinant hepatitis C virus NS5B protein truncated at the C-terminal region. J. Biol. Chem.273:15479–15486.
71. Yan, Y., Y. Li, S. Munshi, V. Sardana, J. L. Cole, M. Sardana, C. Steinkuehler, L. Tomei, R. De Francesco, L. C. Kuo, and Z. Chen.1998. Complex of NS3 protease and NS4A peptide of BK strain hepatitis C virus: a 2.2 Å resolution structure in a hexagonal crystal form. Protein Sci.7: 837–847.
72. Yanagi, M., R. H. Purcell, S. U. Emerson, and J. Bukh.1997. Transcripts from a single full-length cDNA clone of hepatitis C virus are infectious when directly transfected into the liver of a chimpanzee. Proc. Natl. Acad. Sci. USA94:8738–8743.
73. Yanagi, M., M. St. Claire, M. Shapiro, S. U. Emerson, R. H. Purcell, and J. Bukh.1998. Transcripts of a chimeric cDNA clone of hepatitis C virus genotype 1b are infectious in vivo. Virology244:161–172.
74. Yao, N., T. Hesson, M. Cable, Z. Hong, A. D. Kwong, H. V. Le, and P. C. Weber.1997. Structure of the hepatitis C virus RNA helicase domain. Nat. Struct. Biol.4:463–467.
75. Yao, N., P. Reichert, S. Taremi, W. W. Prosise, and P. C. Weber.1999. Molecular views of viral polyprotein processing revealed by the crystal struc-ture of the hepatitis C virus bifunctional protease-helicase. Strucstruc-ture 7: 1353–1363.
76. Zhong, W., P. Ingravallo, J. Wright-Minogue, A. Skelton, A. S. Uss, R. Chase, N. Yao, J. Y. N. Lau, and Z. Hong.1999. Nucleoside triphosphatase and RNA helicase activities associated with GB virus B nonstructural protein 3. Virology261:216–226.