Increase the Processivity of Hepatitis C Virus NS5B Polymerase
Activity
In Vitro
Nagraj Mani,aAlexander Yuzhakov,aOlga Yuzhakov,aJoyce T. Coll,bJim Black,bKumkum Saxena,bJohn R. Fulghum,b Judith A. Lippke,bB. Govinda Rao,c*Rene Rijnbrand,aAnn D. Kwonga*
Infectious Diseases Department, Vertex Pharmaceuticals Incorporated, Boston, Massachusetts, USAa; Protein Sciences Department, Vertex Pharmaceuticals Incorporated, Boston, Massachusetts, USAb; Computational Sciences Department, Vertex Pharmaceuticals Incorporated, Boston, Massachusetts, USAc
ABSTRACT
The precise role(s) and topological organization of different factors in the hepatitis C virus (HCV) RNA replication complex are
not well understood. In order to elucidate the role of viral and host proteins in HCV replication, we have developed a novel
in
vitro
replication system that utilizes a rolling-circle RNA template. Under close-to-physiological salt conditions, HCV NS5B
⌬
21,
an RNA-dependent RNA polymerase, has poor affinity for the RNA template. Human replication protein A (RPA) and HCV
NS5A recruit NS5B
⌬
21 to the template. Subsequently, NS3 is recruited to the replication complex by NS5B
⌬
21, resulting in RNA
synthesis stimulation by helicase. Both RPA and NS5A
(S25-C447), but not NS5A
(S25-K215), enabled the NS5B
⌬
21-NS3 helicase
com-plex to be stably associated with the template and synthesize RNA product in a highly processive manner
in vitro
. This new
in
vitro
HCV replication system is a useful tool that may facilitate the study of other replication factors and aid in the discovery of
novel inhibitors of HCV replication.
IMPORTANCE
The molecular mechanism of hepatitis C virus (HCV) replication is not fully understood, but viral and host proteins collaborate
in this process. Using a rolling-circle RNA template, we have reconstituted an
in vitro
HCV replication system that allows us to
interrogate the role of viral and host proteins in HCV replication and delineate the molecular interactions. We showed that HCV
NS5A
(S25-C447)and cellular replication protein A (RPA) functionally cooperate as a processivity factor to stimulate HCV
replica-tion by HCV NS5B
⌬
21 polymerase and NS3 helicase. This system paves the way to test other proteins and may be used as an
as-say for discovery of HCV inhibitors.
H
epatitis C virus (HCV) is a blood-borne pathogen that infects
an estimated 130 million to 200 million people worldwide
and constitutes a global health problem (
1
). A majority of people
exposed to HCV (
⬃
85%) go on to develop chronic hepatitis,
which can result in liver cirrhosis, liver failure, or hepatocellular
carcinoma (
2
).
HCV is a positive-stranded RNA virus belonging to the
Flavi-viridae
family. It has an
⬃
9.6-kb genome which encodes a single
polyprotein that is co- and posttranslationally processed by
cellu-lar and viral proteases to give rise to structural proteins C, E1, and
E2 and nonstructural (NS) proteins P7, NS2, NS3, NS4A, NS4B,
NS5A, and NS5B, which are involved in polyprotein processing,
genome replication, or virion packaging (
3
). HCV replication is
carried out by a membrane-associated ribonucleoprotein
com-plex which is comprised of HCV nonstructural proteins and host
factors (
4
,
5
). While the functions of some of the HCV proteins,
such as NS3 protease, NS3 helicase, and NS5B RNA-dependent
RNA polymerase (RdRp), are well understood, the functions of
others, such as NS4B and NS5A, are less clear (
3
). In addition, it is
unknown how the various viral and host proteins assemble into a
replication complex and participate in the replication process (
6
).
NS3 is a bifunctional protein with an N-terminal serine
pro-tease and a C-terminal NTPase/helicase domain. NS4A is a
54-amino-acid (aa) cofactor that is essential for optimal proteolytic
activity and forms a tight complex with NS3 to form NS3-4A. The
proteolytic activity of NS3-4A is required to process cleavage sites
between NS3 and NS4A, NS4A and NS4B, NS4B and NS5A, and
NS5A and NS5B in the HCV polyprotein to release components of
the HCV replicase (
5
,
7
,
8
). The structure of the NS3 protease
domain alone and in complex with the NS4A cofactor has been
solved and reveals that the central region of NS4A is buried deeply
in the core of NS3, forming a tight noncovalent complex with the
NS3 protease catalytic domain, and assists in organizing the active
site of the enzyme (
9
,
10
).
The NS3 helicase activity is essential for HCV replication in
cultured cells, and it can unwind RNA/RNA, RNA/DNA, and
DNA/DNA substrates in a 3
=
-to-5
=
direction (
11–16
). The
un-winding activity of the NS3 helicase domain has been shown to be
Received11 June 2014 Accepted3 October 2014
Accepted manuscript posted online15 October 2014
CitationMani N, Yuzhakov A, Yuzhakov O, Coll JT, Black J, Saxena K, Fulghum JR, Lippke JA, Rao BG, Rijnbrand R, Kwong AD. 2015. Nonstructural protein 5A (NS5A) and human replication protein A increase the processivity of hepatitis C virus NS5B polymerase activityin vitro. J Virol 89:165–180.doi:10.1128/JVI.01677-14.
Editor:M. S. Diamond
Address correspondence to Nagraj Mani, [email protected].
* Present address: B. Govinda Rao, Biogen Idec, Cambridge, Massachusetts, USA; Ann D. Kwong, InnovaTID Pharmaceuticals, Cambridge, Massachusetts, USA.
N.M. and A.Y. contributed equally to this article.
Copyright © 2015, American Society for Microbiology. All Rights Reserved.
doi:10.1128/JVI.01677-14
on November 7, 2019 by guest
http://jvi.asm.org/
exquisitely sensitive to salt concentrations. The helicase activity
was reduced to 10% in the presence of 50 mM NaCl, and no
activity was detected at 150 mM NaCl (
17
). In other studies,
max-imal activity was obtained in the absence of potassium ions (
11
,
15
). Similarly, strand displacement by the full-length NS3 was
maximal in the absence of sodium ions (
18
). The NS3 helicase
sensitivity to inhibition by salt is unusual; most DNA and RNA
helicases are stimulated by salt. For example, the optimal salt
con-centration for Rep52 DNA helicase is 50 to100 mM NaCl, and that
for RNA helicase A is 50 to 100 mM KCl (
19
,
20
).
The structures of the NS3 helicase/NTPase domain alone and
in complex with an oligonucleotide have been published
previ-ously (
21–27
). In a crystal structure of a single-chain fusion of
NS4A with full-length NS3, the helicase domain is separated from
the protease domain by a flexible polypeptide linker (
28
). A
com-parison of the unwinding activity of the single-chain NS4A-NS3
fusion with that of the helicase domain or full-length NS3 revealed
that the protease domain and the core peptide of NS4A are
impor-tant for optimal HCV helicase activity (
29–32
). A

-strand located
at the C terminus of NS3 that interacts with the NS3 protease
domain serves as a toggle in inducing a large conformational
change in NS3 that presumably allows NS3 to switch between its
protease and helicase activities (
33
).
The NS4A protein cofactor of the protease domain has been
shown to promote RNA-coupled ATP hydrolysis by the NS3
he-licase in NS3-4A (
34
,
35
). There have been various reports that the
HCV helicase is modulated by the NS5B polymerase and that the
effect differs depending on whether the helicase domain or
full-length NS3 is used (
32
). Other investigators have demonstrated a
direct interaction between NS3 and NS5B that is primarily
medi-ated through the protease domain of NS3 (
36
).
NS5B is an RdRp whose structure adopts a unique shape in the
polymerase family due to extensive interactions between the
fin-ger and thumb subdomains that encircle the active site of the
enzyme (
37–39
). NS5B appears to lack specificity, has poor
affin-ity for HCV RNA, and can copy heterologous nonviral RNA or
DNA templates (
40–46
). Compared to other well-studied
RNA-and DNA-dependent polymerases, NS5B has very poor catalytic
activity
in vitro
on primed single-stranded templates (
46
,
47
). This
lack of specificity for HCV RNA and the poor catalytic activity of
NS5B
in vitro
may reflect the possibility that additional viral and
host proteins are required for specific protein-protein and
pro-tein-RNA interactions that are essential for initiation and
elonga-tion of HCV RNA synthesis (
48–53
).
NS5A is an
⬃
450-amino-acid, membrane-associated zinc
metalloprotein that is essential for viral replication and virion
pro-duction and may serve as a molecular switch between these two
aspects of the HCV life cycle (
3
,
5
,
54
). The protein is
phosphor-ylated and organized into three domains (
5
,
54–59
). The structure
of full-length NS5A has not yet been solved; however, three
differ-ent crystal structures of N-terminal domain I of NS5A have been
published (
60–62
). The structure published by Tellinghuisen et al.
(
60
) shows domain I as a homodimer with a positively charged
cleft that can serve as a potential RNA binding groove. In contrast,
the cleft is absent in the second published structure (
61
), and the
proposed RNA binding surfaces of monomers that formed the
cleft in the first structure are exposed to the solvent and face away
from each other. In the most recent crystal structure of NS5A
domain I, two new dimeric forms of this domain were observed
(
62
). The biological significance of these configurations of NS5A
dimeric structure is unknown. While multiple functions have
been ascribed to NS5A, its precise roles in HCV replication have
not been delineated (
5
).
The two aims of this study were to (i) create an
in vitro
HCV
replication assay that utilized both HCV NS5B RdRp and NS3
helicase and (ii) use the system to elucidate the roles of other viral
and host proteins that might be involved in HCV RNA replication.
We describe the development of a novel
in vitro
HCV replication
system that utilizes a rolling-circle RNA template and provide
experimental evidence supporting a role for HCV NS5A and
hu-man replication protein A (RPA) in HCV replication. RPA is a
single-stranded binding (SSB) protein comprised of a
three-sub-unit protein complex (70 kDa, 32 kDa, and 14 kDa) that has
mul-tiple essential activities in eukaryotic DNA replication and
signal-ing pathways (
63–65
). NS5B
⌬
21 alone has no activity on the
rolling-circle template and requires RPA, truncated NS5A
(S25-K215)containing domain 1 alone, or NS5A
(S25-C447)for RNA synthesis
to occur. The addition of NS3 helicase increases the rate of RNA
synthesis. Under close-to-physiological salt conditions, RPA and
almost full-length NS5A
(S25-C447)or truncated NS5A
(S25-K215)form protein-protein interactions with NS5B
⌬
21 and recruit the
polymerase to the RNA template, thus stimulating RNA synthesis.
However, the presence of both RPA and NS5A
(S25-C447), but not
truncated NS5A
(S25-K215), was required for the replication
com-plex containing NS5B
⌬
21 and NS3 helicase to stably interact with
the rolling-circle RNA template in a highly processive manner.
Thus, NS5A
(S25-C447)and human RPA show functional
cooper-ativity and behave as a processivity factor for HCV RNA
poly-merase.
MATERIALS AND METHODS
Materials.Labeled nucleotides, [35S]methionine (1,000 Ci/mmol), and
[32P]UTP (specific activity, 5,000 cpm/pmol) were obtained from
PerkinElmer (Waltham, MA); unlabeled nucleotides and T4 gene 32 pro-tein (gp32) were obtained from Amersham Pharmacia Biotech (GE Healthcare Biosciences, Pittsburgh, PA). T7 RNA polymerase and RNasin were obtained from Promega (Madison, WI). M13mp18 single-stranded DNA (ssDNA) and restriction enzymes were obtained from New England BioLabs (Ipswich, MA). Unless mentioned otherwise, all other chemicals were obtained from Sigma-Aldrich (St. Louis, MO).
Expression and purification of HCV genotype 1a NS3 helicase do-main.The HCV NS3 RNA helicase domain (S1207 to T1657 of the HCV polyprotein sequence; GenBank accession no.ABV46164.2) was cloned into pET-BS(⫹). The resulting plasmid, pET-BS(⫹)/HCV/NS3 helicase, contains a six-histidine tag fused to the N terminus of the NS3 helicase domain to facilitate protein purification.Escherichia coliBL21(DE3) cells, freshly transformed with the pET-BS(⫹)/HCV/NS3 helicase plasmid, were grown at 30°C in Luria broth (LB) supplemented with 50g/ml of carbenicillin. When an optical density at 600 nm (OD600) of 1.0 was
reached, the cells were induced for 3 h at 30°C by the addition of isopro-pyl--D-thiogalactopyranoside (IPTG) to a final concentration of 0.8 mM. After induction, the cells were harvested and stored frozen at⫺70°C until purification.
Cell paste containing the cloned NS3 helicase domain (NS3h) was lysed in buffer A (50 mM HEPES [pH 8.0], 200 mM NaCl, 10% glycerol, 2.5 mM-mercaptoethanol [-ME]) containing 0.1% beta-octogluco-side and 0.1 mM phenylmethylsulfonyl fluoride (PMSF). The lysate was treated with DNase to eliminate DNA bound to the helicase and centri-fuged at 54,000⫻gto remove debris. The protein was batch adsorbed overnight to Talon resin (Clontech Laboratories, Mountain View, CA). The resin was washed to baseline with buffer A to remove detergent and eluted with buffer A containing 50 mM imidazole adjusted to pH 7.5. The protein was precipitated from the pool by the addition of solid (NH2)SO4
on November 7, 2019 by guest
http://jvi.asm.org/
to 45% (2 M) for 30 min on ice. The precipitate was harvested by centrif-ugation at 54,000⫻g. The pellet was resolubilized in buffer B (50 mM HEPES [pH 8.0], 100 mM NaCl, 10% glycerol, 2 mM dithiothreitol [DTT]) and run over a Sephacryl S-100 column (GE Healthcare Biosci-ences) in buffer B. The eluted peak was treated with thrombin (2 U/mg for 2 h at room temperature [RT]) to remove the histidine tag. The NaCl was diluted to 30 mM and the helicase protein was purified to homogeneity by exchange on a Mono Q ion-exchange column (GE Healthcare Biosci-ences). The protein was eluted at approximately 150 mM NaCl.
Expression and purification of HCV genotype 1a NS3-4A (protease-helicase domain and NS4A).The region encoding the 631-amino-acid NS3 full-length protein sequence (A1027 to T1657 of HCV genotype 1a polyprotein sequence; GenBank accession no.ABV46164.2) with the 54-amino-acid NS4A (S1658 to C1711) at its C terminus was cloned into the baculovirus transfer vector pVL1392 (Life Technologies, Grand Island, NY). The resulting NS3-4A (695-aa) plasmid construct containing a six-histidine tag fused to the N terminus of the NS3 protease domain was transfected into Sf9 cells to generate plaque-purified high-titer baculovi-rus stock, and recombinant NS3-4A was expressed for purification as described previously (66).
Cell paste containing the expressed NS3-4A protein was resuspended in 5 volumes of buffer A (50 mM Na2HPO4[pH 8.0], 10% glycerol, 300
mM NaCl, 5 mM-mercaptoethanol, 0.2 mM PMSF, 2.5g/ml of leu-peptin, 1.0g/ml of E64, and 2.0g/ml of pepstatin). The suspension was passed once through a microfluidizer (Microfluidics Corporation, New-ton, MA), and the lysate was centrifuged at 100,000⫻gfor 30 min at 4°C. The pellet was resuspended in buffer B (buffer A containing 0.5% n-do-decyl--D-maltoside [Affymetrix, Santa Clara, CA]; 2.5 ml/g of paste),
Dounce homogenized, and mixed at 4°C for 3 h before centrifugation at 100,000⫻gfor 30 min at 4°C. The imidazole concentration of the super-natant fraction was adjusted to 10 mM and batch adsorbed by mixing overnight with nickel-nitrilotriacetic acid (NTA) agarose metal affinity resin (Qiagen, Valencia, CA; 1 ml of resin/5 mg of expected NS3-4A protein). The resin was poured into a gravity flow column (Kontes FlexColumn; 2.5 cm by 10 cm) and washed with 10 volumes of buffer C (buffer A containing 0.1%n-dodecyl--D-maltoside and 10 mM imida-zole). NS3-4A was eluted on ice in 3 to 4 column volumes of buffer D (buffer C containing 300 mM imidazole), and fractions were evaluated for the presence of NS3-4A by SDS-PAGE. Fractions containing NS3-4A were pooled and stored at⫺70°C until the next purification step.
The pooled NS3-4A protein-containing fraction from nickel affinity chromatography was purified further on a Superdex 200 26/60 column (GE Healthcare Biosciences) that was equilibrated with buffer D (20 mM HEPES [pH 8.0], 10% glycerol, 300 mM NaCl, 10 mM -mercaptoetha-nol, and 0.05%n-dodecyl--D-maltoside). Column fractions were ana-lyzed by SDS-PAGE, and NS3-4A-containing fractions were pooled and stored at⫺70°C.
Expression and purification of HCV genotype 1b C-terminally trun-cated NS5A(S25-K215).The N-terminal domain I of NS5A (S25-K215; HCV 1b; GenBank accession no.KC124831.1) lacking the N-terminal amphipathic helix (24 amino acids) was cloned into pET30b (EMD Mil-lipore, Billerica, MA) with a C-terminal polyhistidine tag and enteroki-nase cleavage site and expressed in 6 liters of BL21(DE3) cells. An over-night seed culture grown in LB medium containing 30 g/ml of kanamycin was used to inoculate 6 1-liter cultures. The cells were grown at 37°C shaking at 250 rpm to an OD600of 0.55 in brain heart infusion
medium (BD Biosciences, Franklin Lakes, NJ) and induced with 1 mM IPTG for 5 h at 25°C. Cells were harvested by centrifugation at 7,000⫻g for 10 min, and the pellets were stored at⫺80°C. All purification steps, except for the polyhistidine tag cleavage, were performed on ice or at 4°C. Cell paste from 6 liters of culture producing recombinant NS5A(S25-K215)
was resuspended in lysis buffer (50 mM HEPES containing 10% glycerol, 250 mM NaCl, 5 mM imidazole-HCl, 5 mM-ME, 20l/liter of Benzo-nase nuclease [EMD Millipore], 2g/liter of leupeptin, 1g/liter of pep-statin [Roche Diagnostics Corp., Indianapolis, IN], 20M
diisopropyl-fluoro-phosphate [DFP; pH 7.9; Sigma-Aldrich, St. Louis, MO]) at a ratio of 10 ml per g of cell paste. The suspension was passed two times through a microfluidizer (Microfluidics Corporation, Newton MA), and the lysate was centrifuged at 54,000⫻gfor 1 h. The supernatant fraction was batch adsorbed by mixing overnight with 5 ml of His-Select nickel affinity gel (Sigma-Aldrich). The resin containing bound NS5A(S25-K215)was packed in a column, washed with wash buffer (50 mM HEPES containing 10% glycerol, 250 mM NaCl, 5 mM imidazole-HCl, and 5 mM-ME [pH 7.9]), and eluted with 4 column volumes of elution buffer (wash buffer containing 250 mM imidazole-HCl [pH 8.0]). The eluate was collected into 75 ml of buffer A (20 mM HEPES containing 10% glycerol and 2 mM DTT [pH 8.0]) and loaded onto a 5-ml HiTrap Q HP column (GE Health-care Biosciences) that had been preequilibrated in buffer B (buffer A con-taining 50 mM NaCl). The HiTrap Q column was washed with buffer B and eluted with a 40-column-volume linear gradient from 50 mM NaCl to 400 mM NaCl. Fractions containing NS5A(S25-K215)were pooled,
concen-trated using a centrifugal concentrator (VS2002; Sartorius Stedim Bio-tech, Bohemia, NY), and incubated overnight at room temperature with recombinant enterokinase (New England BioLabs, Ipswich, MA) to re-move the polyhistidine tag. The cleaved protein was further purified on a 5-ml HiTrap Q HP column followed by a Superdex 75 column (GE Healthcare Biosciences) that had been preequilibrated in buffer C (20 mM HEPES containing 250 mM NaCl and 2 mM DTT [pH 8.0]). The peak monodisperse fractions were pooled, concentrated to 1.15 mg/ml, and stored at⫺80°C.
Expression and purification of HCV genotype 1b NS5A(S25-C447). NS5A(S25-C447)(HCV 1b; GenBank accession no.KC124831.1)
contain-ing a deletion of the N-terminal amphipathic helix was cloned in pET30b (EMD Millipore) containing a C-terminal polyhistidine tag and an en-terokinase cleavage site and expressed at the 1-liter scale in 2.5 liters of ultrayield baffled flasks (Thomson Instrument Company, Oceanside, CA), via autoinduction, using Overnight Express instant TB medium (EMD Millipore) containing 30g/ml of kanamycin. Cultures were in-oculated with 10 ml of an overnight LB medium seed and grown at 37°C for 4 h with shaking at 250 rpm; the temperature was then dropped to 15°C for an additional 48 h. Cells were harvested via centrifugation (5,000⫻g) for 15 min, and the cell paste was frozen at⫺80°C until purification.
All purification steps were performed on ice or at 4°C. Cell paste from 1 liter of culture producing recombinant NS5A(S25-C447)was resuspended in lysis buffer (50 mM potassium phosphate containing 10 mM Tris-HCl, 5% glycerol, 500 mM NaCl, 0.25% Tween 20, 5 mM imidazole-HCl, 5 mM-ME, 20l/liter of Benzonase nuclease, 2g/liter of leupeptin, 1
g/liter of pepstatin, and 20M DFP [pH 8.0]) at a ratio of 10 ml per g of cell paste. The resuspension was lysed by sonication, and the lysate was centrifuged at 43,000⫻gfor 1 h. The supernatant fraction was batch incubated overnight with 4 ml of His-Select nickel affinity gel (Sigma-Aldrich, St. Louis, MO) with mixing. The resin containing the bound NS5A(S25-C447)was packed into a column, washed with 20 column
vol-umes of wash buffer (50 mM potassium phosphate containing 10 mM Tris-HCl, 5% glycerol, 500 mM NaCl, 0.25% Tween 20, 5 mM imidazole-HCl, and 5 mM-ME), and eluted with 4 column volumes of wash buffer containing 300 mM imidazole-HCl. The eluate was immediately loaded onto a HiPrep 26/10 desalting column (GE Healthcare Biosciences) that had been preequilibrated in buffer B (20 mM HEPES containing 10% glycerol and 200 mM NaCl). Fractions containing NS5A(S25-C447)were
pooled and concentrated in a centrifugal concentrator and loaded onto a Superdex 200 10/300 GL column (GE Healthcare Biosciences). The peak monodisperse fractions were collected and stored at⫺80°C.
Cloning, expression, and purification of HCV genotype 1b NS5B⌬21 polymerase.NS5B polymerase (aa 2420 to 2989 of the HCV polyprotein) from hepatitis C virus subtype 1b (GenBank accession no.
AAK08509) containing a hexahistidine tag at the N terminus and lacking the C-terminal 21-amino-acid amphipathic helix was cloned into a pET21B vector and transformed intoE. coliBL21(DE3) cells using a stan-dard protocol. Freshly transformed cells were grown at 37°C in brain heart
on November 7, 2019 by guest
http://jvi.asm.org/
infusion medium (Difco Laboratories, Detroit, MI) supplemented with 100g/ml of ampicillin. Cells were grown at 37°C up to an optical density of 0.7 at 600 nm, and expression was induced at 28°C with 0.2 mM IPTG. Cells were harvested via centrifugation 4 h postinduction and flash frozen at⫺80°C prior to purification.
Frozen cell pellets (⬃30 g) were thawed in 10 volumes of buffer A (40 mM potassium phosphate buffer [pH 7.4], 200 mM NaCl, 10% [vol/vol] glycerol, 3 mM-ME, and 5 mM imidazole) containing 50M DFP, 1
g/ml of E-64 protease inhibitor (Sigma-Aldrich), 1g/ml of leupeptin, and 10g/ml of pepstatin (Roche Diagnostics Corp., Indianapolis, IN) and lysed in a microfluidizer (Microfluidics, Newton, MA). The lysate was centrifuged at 54,000⫻gfor 45 min, and the supernatant was incubated with 0.5 ml of nickel metal affinity resin (Sigma-Aldrich) per g of cell paste overnight at 4°C. The resin was washed with 20 column volumes of buffer B (40 mM potassium phosphate buffer [pH 7.4], 500 mM NaCl, 10% [vol/vol] glycerol, 3 mM-ME, and 5 mM imidazole), followed by a wash with 5 column volumes of buffer A and elution with buffer A containing 200 mM imidazole. The fractions containing the polymerase were pooled and desalted into buffer C (25 mM HEPES [pH 7.5], 0.25 mM EDTA, 10% glycerol, and 1 mM DTT) containing 200 mM NaCl using a Sephadex G-25 16/60 column and loaded onto a 5-ml HiTrap SP Sepharose column (Thermo Fisher Scientific, Waltham, MA) that had been preequilibrated with buffer C plus 200 mM NaCl. The column was washed with 2 column volumes of buffer C containing 200 mM NaCl, followed by a steep wash gradient from 200 to 500 mM NaCl in buffer C over 2 column volumes. The polymerase was eluted with a 500 to 700 mM NaCl gradient in buffer C over 10 column volumes, concentrated by ultrafiltration using a Vivas-pin with a 30-kDa molecular mass cutoff (Thermo Fisher Scientific) and loaded onto a Superdex 200 column (60 by 2.6 cm; Thermo Fisher Scien-tific) equilibrated in buffer D (10 mM Tris-HCl [pH 7.5], 600 mM NaCl, 10% [vol/vol] glycerol, and 5 mM DTT). Fractions were pooled based on SDS-PAGE and stored at⫺80°C until further use.
Purification of RPA endogenous complex from HeLa cells.HeLa cells from a 3-liter suspension cell culture (⬃2⫻109cells) were used for
the purification procedure. The cells were collected by centrifugation for 10 min at 3,000 rpm in a Beckman JS-4.2 rotor at 4°C. The cell pellets were resuspended in 40 ml of ice-cold phosphate-buffered saline (PBS), trans-ferred into a conical 50-ml centrifugation tube, and pelleted in a Beckman CS-6R centrifuge for 10 min at 1,000 rpm at 4°C. The cell pellet was resuspended in 1.5 times the packed cell volume of ice-cold hypotonic buffer H (20 mM HEPES [pH 7.5], 5 mM KCl, 1.5 mM MgCl2, 1 mM
DTT, 1 mM PMSF, and Roche complete mini-cocktail protease inhibitors at the recommended concentrations) and centrifuged immediately for 10 min at 2,000 rpm in a Beckman CS-6R centrifuge (Beckman Coulter Inc., Brea, CA) at 4°C. The cells were resuspended in 12 ml (3 times the column volume) of ice-cold hypotonic buffer and kept on ice for 20 min, allowing them to swell. The swollen cells were transferred into a glass Dounce homogenizer and homogenized with a glass B pestle until the release of nuclei was seen under the microscope by trypan blue (Invitrogen, Carls-bad, CA) inclusion in about 80% of cells (30 to 50 up-and-down strokes). While the cell homogenate was stirred on ice, the salt concentration was adjusted to 0.2 M by dropwise additions of 5 M NaCl, and the cell homog-enate was kept on ice with occasional swirling for an additional 20 min (nuclear extraction). The cell homogenate was cleared from nuclei by centrifugation in a Beckman Avanti J-25 (Beckman Coulter Inc.) centri-fuge with a JA-25.50 rotor at 20,000 rpm (17,217⫻g) for 30 min at 4°C. The supernatants were collected and centrifuged again at 100,000⫻gin a 45Ti rotor in a Beckman Optima LE-80K ultracentrifuge for 30 min at 4°C. The supernatant containing the cell extract was dialyzed overnight against buffer A (20 mM Tris [pH 7.5], 0.1 mM EDTA, 50 mM NaCl, 0.01% NP-40, 10% glycerol, Roche complete mini-cocktail protease in-hibitors at the recommended concentrations) at a volume ratio of 1:200 at 4°C. The dialyzed cell extract was collected and precipitated by dropwise addition of saturated ammonium sulfate salt solution. The concentration of ammonium sulfate in the cell extract was adjusted to 35% of saturation
at 4°C (0.56 ml of saturated ammonium sulfate per 1 ml of cell extract), and the precipitates were pelleted by centrifugation at 10,000⫻gfor 30 min. The precipitates were resuspended in 4 ml of ice-cold buffer B (20 mM Tris [pH 7.5], 0.1 mM EDTA, 500 mM NaCl, 0.01% NP-40, 10% glycerol, and Roche complete mini-cocktail protease inhibitors at the rec-ommended concentrations) and, after filtration through a Whatman filter paper, loaded onto a 0.5-ml ssDNA-cellulose column (Sigma-Aldrich). After being washed with 20 ml of buffer B, the RPA-containing fractions were eluted with 2 ml of buffer C (20 mM Tris [pH 7.5], 0.1 mM EDTA, 2 M NaCl, 0.01% NP-40, 10% glycerol, Roche complete mini-protease in-hibitor cocktail). The eluate was dialyzed overnight against 4 liters of buffer A at 4°C. The next day, the sample was loaded onto a Q-Sepharose column (HiTrap Q HP; GE Healthcare Biosciences) equilibrated with buffer A. After washing with 5 column volumes of equilibration buffer, the bound proteins were eluted with a linear 50 to 400 mM NaCl gradient in 25 ml. The fractions of gradient were collected in 0.5-ml volumes. RPA came off the column at 125 to 150 mM NaCl. The presence of RPA in the gradient fractions was determined by SDS-PAGE and Western blotting with specific antibody against the largest p70 subunit of RPA (H-7; Santa Cruz Biotechnology Inc., Dallas, TX). The fractions containing RPA were combined, concentrated in protein concentrators with a molecular mass cutoff of 20 kDa (Pierce; Thermo Fisher Scientific) up to 0.25g per ml, dialyzed against buffer D (20 mM Tris [pH 7.5], 0.1 mM EDTA, 150 mM NaCl, 20% glycerol), aliquoted in 100-l volumes, and stored at⫺80°C. The purity of isolated RPA, as estimated by silver staining of SDS-PAGE gel, was around 70%.
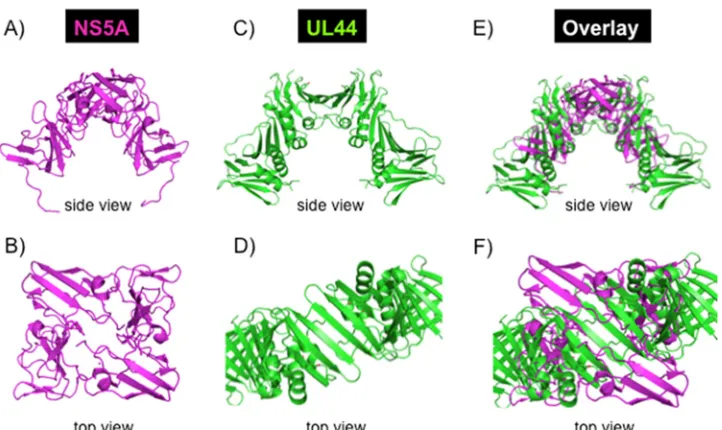
Structural comparison of HCV NS5A and CMV UL44.The dimeric structure of HCV NS5A domain I (PDB code 1ZH1) was overlaid with the dimeric structure of the cytomegalovirus (CMV) DNA polymerase sub-unit UL44 (PDB code 1T6L), which has a C clamp shape and is known to act as a processivity-promoting factor in CMV replication. The PyMOL Molecular Graphics System (Schrödinger, LLC) was used for overlaying the two structures. As there are no similarities in amino acid sequence or secondary-structure elements of the two structures, the 3-dimensional (C clamp) shape of the two structures was used to guide the manual overlay of the two structures.
Preparation of RNA rolling-circle template.Briefly, two RNA oligo-nucleotides (74 oligo-nucleotides [nt]and 106 oligo-nucleotides) were synthesized with T7 RNA polymerase on a parental DNA template primed with a T7 promoter primer. The 74-nt RNA oligonucleotide was hybridized to a bridging DNA oligonucleotide and ligated with DNA ligase. The double-stranded RNA rolling-circle template was constructed by hybridization of the RNA circle and the 106-nucleotide RNA oligonucleotide. Four DNA oligonucleotides were synthesized to generate the rolling-circle template forin vitrotranscription reactions: DNA1 oligonucleotide, GAGTGGTA TAGTGGAGTGAAGTGAGGTGAAGGGTTGATGGTGAATATTGGG GAGGGTGGAGGTTATGGTGTCTCCCTATAGTGAGTCGTATTA; DNA2 oligonucleotide, CTCCCCAATATTCACCATCAACCCTTCACC TCACTTCACTCCACTATACCACTCGGGAGACACCATAACCTCCA CATAAAAAAAAAAAAAAAAAAAAAAAAATCTCCCTATAGTGAGT CGTATTA; T7 promoter oligonucleotide, TAATACGACTCACTATAG GGAGA; and bridge oligonucleotide, CCCTCTGTGGTAGAGTGGTAT AGT. Specifically, 20g of DNA1 or DNA2 was incubated in 20l of annealing buffer (20 mM Tris-HCl [pH 7.5] and 100 mM NaCl) in the presence of 200g of the T7 promoter oligonucleotide for 1 min at 90°C and then cooled slowly to 24°C. Twelve micrograms of each annealed oligonucleotide was used to prepare short RNA transcripts using the Am-bion MEGAshortscript transcription kit (Life Technologies, Grand Is-land, NY). After incubation for 3 h at 37°C, the reaction mixtures were incubated with DNase for 15 min at 37°C, cooled to 4°C, extracted with phenol-chloroform, and precipitated with ethanol as per standard proto-cols. RNA oligonucleotide 1 (the transcript of DNA1; 5.16 nmol) and bridge DNA oligonucleotide (25.8 nmol) were incubated in 60l of buffer A for 1 min at 65°C and then cooled slowly to 16°C. DNA ligase (10,000 U; New England BioLabs) was added, and incubation was continued for an
on November 7, 2019 by guest
http://jvi.asm.org/
additional 12 h at 16°C. After DNase treatment for 30 min at 37°C, circular RNA was purified by 10% urea-PAGE. A total of 270 pmol of circle RNA was mixed with 11,382 pmol of RNA oligonucleotide 2 (the transcript of DNA2) in 50l of annealing buffer and incubated for 5 min at 75°C, after which the mixture was cooled slowly to 4°C.
RNA synthesis assays using the rolling-circle template.Sixty-six nanograms (1 pmol) of rolling-circle RNA was incubated with 2 pmol (126 ng) (or as indicated) of NS5B⌬21, 4 pmol (200 ng) of NS3h (when present), and 16.7 pmol (2g) of RPA (when present) in 25l of repli-cation buffer (20 mM Tris-HCl [pH 7.5], 8 mM MgCl2, 2 mM ATP, 250 M [each] CTP, GTP, and UTP, 0.1 mM EDTA, 5 mM DTT, 4% glycerol, 40g/ml of bovine serum albumin [BSA], 1g/ml of RNasin, [32P]UTP
[5,000 cpm/pmol], and 150 mM NaCl) for 2 h at 30°C. At different time points, aliquots of 4l were removed and analyzed as described below.
RNA synthesis assays using a poly(A)/oligo(dT) template.A 2.5-g quantity of poly(A)/oligo(dT) (Life Technologies, Grand Island, NY) was incubated in 50l of replication buffer (described above) at 30°C. At different time points, aliquots of 4l were removed, 50g of carrier yeast tRNA (Sigma-Aldrich, St. Louis, MO) was added, and the mixtures were precipitated in 1 ml of 30% trichloroacetic acid (TCA) and spotted onto Whatman GF/F filters (Thermo Fisher Scientific); the filters were washed with 10% TCA followed by 70% ethanol, dried, and counted in a liquid scintillation counter.
Interaction of35S-labeled proteins with RPA bound to ssDNA.35
S-labeled proteins were prepared using a commercially available transcrip-tion/translation kit (TnT Quick Coupled Transcription/Translation Sys-tem; Promega, Madison, WI) using DNA templates encoding proteins under the control of a T7 promoter as per the manufacturer’s instructions. Reactions were performed in 150l containing 800 fmol of M13mp18 ssDNA, 50g of RPA (when present) or 75g of gp32 (when present), and 25 fmol of35S-labeled protein in column buffer (20 mM Tris-HCl
[pH 7.5], 8 mM MgCl2, 5 mM DTT, 5% glycerol, 50g/ml of BSA)
containing 150 mM NaCl. Reaction mixtures were incubated for 10 min at 30°C and then applied to 5-ml agarose BioGel A15 columns (Bio-Rad Laboratories, Hercules, CA) equilibrated with column buffer at 4°C. Frac-tions of 200l were collected, and 150-l aliquots were TCA precipitated and analyzed for35S-protein using liquid scintillation counter.
SPR.Immobilization of NS5B⌬21 (5,900 resonance units [RU]) and NS3h (3,900 RU) was performed on Biacore CM dextran matrix-coated sensor chip CM5 (GE Healthcare Biosciences). After each analysis, the surface was regenerated by a 15-min wash with 100 mM ethanolamine without decreasing the capacity of the immobilized proteins. Surface plas-mon resonance (SPR) analysis was performed by passing 35l of RPA, NS3h, or NS5B⌬21 (40 nM protein solution) over a chip containing im-mobilized protein. All proteins were dialyzed against SPR buffer (10 mM HEPES-NaOH [pH 7.4], 150 mM NaCl, 3.4 mM EDTA, and 0.005% Tween 20) to remove buffer-related artifacts. All proteins were tested for interaction with a sensor chip containing no protein.
Gel filtration analysis of35S-NS3h and NS5B⌬21.The mixtures con-tained 0.3 mg of NS5B⌬21 and 600 fmol of35S-NS3h or35S-NS3h alone in
a volume of 200l of column buffer (20 mM Tris-HCl [pH 7.5], 8 mM MgCl2, 5 mM DTT, 5% glycerol, 50g/ml of BSA) containing 200 mM
NaCl. Proteins were incubated at 30°C for 10 min before injection of the mixture into an HR 10/30 Superose 12 column equilibrated with column buffer containing 200 mM NaCl. Fractions of 200l were collected, and 5-l aliquots were analyzed for35S-protein using liquid scintillation
counting.
Processivity competition experiment.A standard rolling-circle reac-tion with 66 ng (1 pmol) of the rolling-circle RNA template, 2 pmol (126 ng) of NS5B⌬21, 4 pmol (200 ng) of NS3h, 16.7 pmol (2g) of RPA (when present), and 3 pmol of NS5A (when present) was used. Protein mixtures were premixed in the replication buffer (20 mM Tris-HCl [pH 7.5], 8 mM MgCl2, 2 mM ATP, 250M GTP, 0.1 mM EDTA, 5 mM DTT,
4% glycerol, 40g/ml of BSA, 1g/ml of RNasin [Promega, Madison, WI], and 150 mM NaCl) and incubated on ice for 30 min before addition
of the rolling-circle RNA template, [32P]UTP (250M), and CTP (250 M). Reaction mixtures were warmed up to 30°C for 5 min, and 5g of poly(A)/oligo(U) was added. The mixtures were incubated for an addi-tional 2 min, and synthesis was initiated by addition of GTP and [32P]UTP
(5,000 cpm/pmol). At the desired time points, the reactions were stopped with 0.5 M EDTA and 10g of yeast tRNA. After phenol-chloroform extractions and ethanol precipitation, the reaction mixtures were diluted in 1⫻NorthernMax formaldehyde loading dye (Life Technologies, Grand Island, NY), electrophoresed on a formaldehyde-0.5% agarose gel, and autoradiographed.
RESULTS
Purification of HCV proteins and human RPA.
In order to set up
an
in vitro
HCV replication system, NS3-4A, the NS3 helicase
domain, NS5A
(S25-K215), NS5A
(S25-C447), and NS5B
⌬
21
polymer-ase of HCV were cloned and expressed as described in Materials
and Methods. Native human RPA was purified from HeLa cell
extracts as described in Materials and Methods. The protein
prep-arations were visualized by SDS-PAGE (
Fig. 1A
), and their
iden-tities were confirmed by Western blot analyses (data not shown).
Design of a rolling-circle RNA template.
Rolling-circle
sys-tems are well established for the T4 and
E. coli in vitro
DNA
rep-lication systems (
67
,
68
). In these systems, a helicase unwinds the
DNA duplex at the fork of the template and the replicative DNA
polymerase is in constant contact with the helicase as it
incorpo-rates nucleotides at the 3
=
end of the annealed strand. The product
of the synthesis is a continuously growing,
high-molecular-weight, 5
=
DNA tail. Previous studies have shown that NS5B can
utilize both RNA and DNA templates (
40–46
). The NS3 helicase
can unwind RNA/RNA, RNA/DNA, and DNA/DNA substrates in
a 3
=
-to-5
=
direction (
11
,
13–15
). To incorporate both the RNA
polymerase activity of NS5B and the RNA helicase activity of NS3
into one assay, we prepared a rolling-circle RNA template as
de-scribed in Materials and Methods. In our system and in contrast to
the
E. coli
and the T4 DNA replication systems described above,
the product of synthesis on a rolling-circle RNA template by
RNA-dependent RNA polymerase will result in a continuously growing,
high-molecular-weight 5
=
RNA tail (
Fig. 1B
).
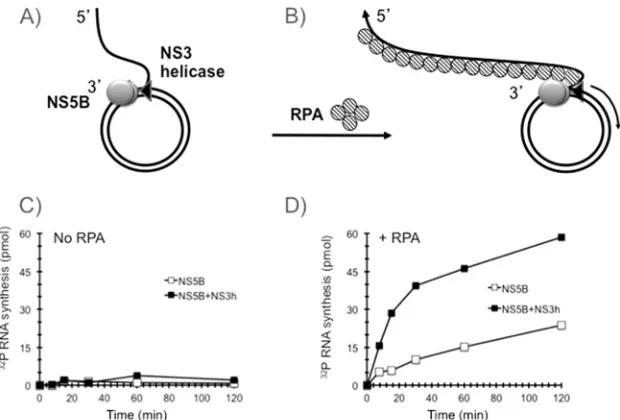
RNA synthesis by NS5B
⌬
21 on a rolling-circle RNA template
requires RPA and is stimulated by NS3 helicase.
In order to test
whether NS5B
⌬
21 can synthesize RNA on a rolling-circle RNA
template under close-to-physiological salt conditions, we added 2
pmol of NS5B
⌬
21 and 4 pmol of NS3 helicase domain
individu-ally and in combination to the rolling-circle template and initiated
synthesis by adding NTP in the presence of 100 mM NaCl. These
reactions were carried out in both the absence and presence of 2
g of human RPA (schematically shown in
Fig. 2A
and
B
,
respec-tively). In the absence of RPA, no significant RNA synthesis was
observed with NS5B
⌬
21 alone or in combination with the NS3
helicase domain (
Fig. 2C
). In contrast, the addition of RPA
stim-ulated RNA synthesis by NS5B
⌬
21 (
Fig. 2D
). The addition of NS3
helicase domain significantly increased RNA synthesis, from 15 to
50 pmol of RNA product per hour per pmol of the rolling-circle
RNA template (
Fig. 2D
). The continuous synthesis of RNA
prod-uct by HCV NS5B
⌬
21 and the NS5B
⌬
21-NS3 helicase domain in
the presence of RPA did not reach a plateau after 2 h, suggesting that
RNA synthesis was robust and stable and that the accumulation of
product did not inhibit the reaction. The exquisite sensitivity of the
unwinding activity of the HCV NS3 helicase domain to salt
inhibi-tion has been a conundrum because the helicase has to be active at
physiological salt conditions during infection. Using a
on November 7, 2019 by guest
http://jvi.asm.org/
circle RNA template, we observed significant stimulation of RNA
synthesis by the NS3 helicase domain in the presence of NS5B
⌬
21
and RPA in the presence of 100 mM NaCl.
NS5B
⌬
21 has poor activity on a multiply primed linear
tem-plate, and activity is not stimulated by the addition of RPA, T4
gp32, or NS3 helicase.
One way in which RPA might stimulate
NS5B RNA synthesis is by melting secondary structures through
its interaction with single-stranded RNA. If this hypothesis is true,
any SSB-like protein that interacts with RNA and prevents
sec-ondary-structure formation should be able to substitute for RPA
in the rolling-circle reaction. Another SSB protein that fits these
requirements is the bacteriophage T4 protein gp32. T4 gp32 is a
component of the T4 DNA replicase and binds RNA with an
af-finity similar to that of RPA (
69
,
70
). First we demonstrated that
gp32 and RPA were able to bind a
32P-labeled RNA
oligonucleo-tide comparably in a gel mobility shift assay (
Fig. 3A
). Next we
tested the ability of both RNA-binding SSB proteins to support
HCV RNA synthesis on a multiply primed poly(A)/oligo(dT)
lin-ear template. It is important to note that a single rolling-circle
template has only one 3
=
end, whereas the poly(A)/oligo(dT)
tem-plate contains multiple oligo(dT) primers hybridized to a single
poly(A) molecule. NS5B
⌬
21 had very poor activity (
⬍
1 pmol of
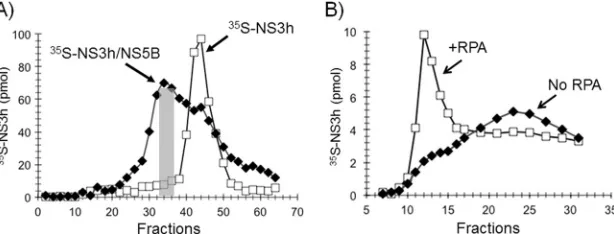
FIG 1Setting up anin vitroHCV rolling-circle replication system. (A) Purification of HCV proteins and RPA. HCV proteins NS3-4A, NS3 helicase, NS5A(S25-C447), NS5A(S25-K215), NS5B⌬21 polymerase and the native human RPA were purified as described in Materials and Methods and analyzed using SDS-polyacrylamide gel electrophoresis and Coomassie blue staining. The arrowheads indicate the bands corresponding to individual proteins or protein subunits in the case of NS3-4A and RPA. (B) Design of the rolling-circle RNA template. Two RNA oligonucleotides (74 nt and 106 nt) were synthesized with T7 RNA polymerase on the primed DNA templates with T7 promoter sequence primers as described in Materials and Methods. The 74-nt RNA oligonucleotide was hybridized with the DNA oligonucleotide “bridge” and ligated with DNA ligase to create a RNA circle. The rolling-circle RNA template was constructed by hybridization of the RNA circle and the 106-nt RNA oligonucleotide to complete the formation of the rolling-circle RNA template.
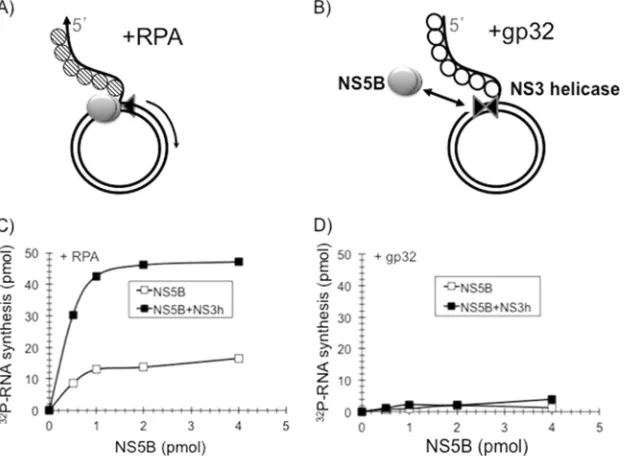
FIG 2RPA is required for RNA rolling-circle synthesis. (A and B) Schematic representations of the templates that were used in the reactions. (C and D) NS5B⌬21 (open squares) or NS5B⌬21-NS3h (filled squares) were assembled onto a rolling-circle template without RPA (C) or in the presence of RPA (D). Synthesis was initiated by adding NTP, aliquots were withdrawn at the indicated time points, and [32P]UMP incorporation was determined as described in Materials and Methods.
on November 7, 2019 by guest
http://jvi.asm.org/
[image:6.585.137.447.472.682.2]RNA product in 2 h) on the linear template (
Fig. 3B
, top) that was
not increased by the addition of RPA (
Fig. 3B
, middle) or T4 gp32
(
Fig. 3B
, bottom).
NS5B
⌬
21 RNA synthesis on a rolling-circle template is
stim-ulated by RPA but not by T4 gp32.
Next we compared the
effi-ciencies of RNA synthesis using a rolling-circle RNA template
coated either with RPA (schematically shown in
Fig. 4A
) or with
gp32 (schematically shown in
Fig. 4B
) in the presence of
increas-ing concentrations of NS5B
⌬
21 and a constant amount of the NS3
helicase domain. As shown in
Fig. 4C
, RPA but not T4 gp32 (
Fig.
4D
) was able to support RNA synthesis by NS5B
⌬
21 on the
roll-ing-circle template, while in the absence of NS5B, no detectable
RNA synthesis was observed. The inability of another RNA
bind-ing SSB (T4 gp32) to substitute for RPA suggests that the
stimu-lation of NS5B
⌬
21 activity by RPA is not likely to be mediated
through the nonspecific melting of the RNA secondary structure.
Based on these data, we hypothesized that RPA facilitates the
binding of NS5B to the rolling-circle template through specific
protein-protein contacts, increasing the initiation rate of RNA
synthesis by NS5B.
RPA forms a stable complex with NS5B
⌬
21 and ties it to the
template.
In order to test whether RPA ties NS5B
⌬
21 to the
tem-FIG 3RPA and gp32 interact with RNA. (A)32P-RNA gel shift assay.32P-RNA was preincubated with RPA or gp32 and loaded onto a 10% agarose gel. The two proteins interact with the RNA with similar affinities, producing a supershifted band. (B1) NS5B⌬21 synthesis on a poly(A)/oligo(dT) template: The reaction was started by the addition of NTP, and the incorporation [32P]UTP into the synthesized product was measured in samples obtained at the indicated time points. (B2) NS5B⌬21 synthesis in the presence of RPA on the poly(A)/oligo(dT) template. Conditions were as described above. (B3) NS5B⌬21 synthesis in the presence of gp32 on the poly(A)/oligo(dT) template. Conditions were as described above.
FIG 4RPA is specifically required for rolling-circle RNA synthesis. The rolling-circle RNA template (1 picomole) was coated with RPA as shown schematically in panel A or gp32 as shown schematically in panel B. (C and D) The templates were tested for the ability to support RNA synthesis in the presence of NS5B⌬21 (open squares) or NS5B⌬21-NS3h (filled squares) for 60 min with increasing amounts of NS5B as described in Materials and Methods. Robust synthesis of RNA was observed only in the presence of RPA (C) and not in the presence of gp32 (D).
on November 7, 2019 by guest
http://jvi.asm.org/
[image:7.585.135.452.64.241.2] [image:7.585.138.454.455.683.2]plate, the NS3 helicase domain and NS5B
⌬
21 were individually
labeled with [
35S]methionine using an
in vitro
translation
sys-tem and purified by gel filtration chromatography.
35S-labeled
NS5B
⌬
21 and the NS3 helicase domain were incubated with
ss-DNA alone or coated with RPA or T4 gp32 and assayed for stable
association by gel filtration chromatography (
Fig. 5
). Complexes
of proteins bound to ssDNA were excluded from the column in
the void volume (fractions 10 to 15), whereas unbound proteins
were eluted with the included column volume (fractions 16 to 32).
Thus, the gel filtration column separates proteins bound to DNA
from proteins that are not bound to DNA. As shown in
Fig. 5
,
35
S-NS5B
⌬
21 stably bound to RPA-coated ssDNA (
Fig. 5A
) but
not to gp32-coated DNA (
Fig. 5B
) or to ssDNA alone (
Fig. 5C
)
under physiological salt conditions (150 mM NaCl). As shown in
Fig. 5
, under the same conditions, the
35S-labeled NS3 helicase
domain did not stably interact with either ssDNA coated with RPA
(
Fig. 5D
), gp32 (
Fig. 5E
), or ssDNA alone (
Fig. 5F
). These studies
support the hypothesis that RPA ties NS5B
⌬
21 to the template
and suggest that there may be specific protein-protein interactions
between NS5B
⌬
21 and RPA.
NS5B
⌬
21 interacts directly with RPA.
In order to further
in-vestigate whether NS5B
⌬
21 forms protein-protein interactions
with RPA in the absence of ssDNA, we performed surface plasmon
resonance (SPR) analysis. In the SPR technique, one
macromole-cule is immobilized on a sensor chip and a solution containing
another molecule is passed over the chip. If a stable interaction is
formed between the two macromolecules, the resultant increase in
mass on the sensor chip is detected as an increase in surface
plas-mon resonance that can be measured in response or resonance
units (RU). NS5B
⌬
21 (
Fig. 6A
) or the NS3 helicase domain
(
Fig. 6B
) was immobilized on a chip and a solution of RPA or gp32
was passed over the chip. These results showed that RPA, but not
gp32, can stably interact directly with NS5B
⌬
21. In contrast,
nei-ther RPA nor gp32 could stably interact with the NS3 helicase
domain.
The NS3 helicase domain interacts directly with NS5B
⌬
21
and forms a stable complex.
Our studies have shown that
al-though the NS3 helicase domain stimulates the polymerase
activ-ity of NS5B
⌬
21 in the presence of RPA on a rolling-circle
tem-plate, it does not interact with ssDNA alone or with ssDNA coated
with RPA in the presence of 150 mM NaCl. In order to investigate
how NS3 helicase is recruited to the replication complex, we used
SPR to examine interactions between NS5B
⌬
21 and the NS3
he-licase domain. We found that the NS3 hehe-licase domain and
NS5B
⌬
21 strongly interacted with each other, independent of
which partner was immobilized on the chip, with a
Kd
(dissocia-tion constant) below 4.8
⫻
10
⫺9M (
Fig. 6C
and
D
).
The NS3 helicase domain and NS5B
⌬
21 form a stable
com-plex that can bind ssDNA.
In order to investigate whether the
NS3 helicase domain and NS5B
⌬
21 can form a stable complex
that is capable of binding ssDNA stably, we incubated
35S-labeled
NS3 helicase domain with NS5B
⌬
21 and isolated the complex on
a gel filtration column. The
35S-NS3 helicase domain
NS3h-NS5B
⌬
21 complex was eluted from the column in the void
vol-ume earlier than the
35S-NS3 helicase domain alone, which was
eluted in the included volume, indicating that NS3 helicase
do-main and NS5B
⌬
21 form a stable protein complex (
Fig. 7A
).
Frac-tions 32 to 36 (
Fig. 7A
, gray bar) containing the
35S-NS3h–
NS5B
⌬
21 complex were pooled and analyzed for the ability of the
complex to interact with ssDNA alone or ssDNA coated with RPA
FIG 5RPA ties NS5B⌬21 to the template.35S-labeled NS5B⌬21 and NS3h, individually synthesized in transcription/translation reactionsin vitroin the presence of [35S]methionine, were incubated with circular ssM13mp18 DNA coated with RPA (A and D) or gp32 (B and E) or alone (C and F) and run on a BioGel A100 5-ml gel filtration column to resolve protein bound to DNA (fractions 10 to 15) from free protein (fractions 16 to 30).35S-NS5B⌬21 stably interacted with ssDNA coated with RPA but not with ssDNA alone or ssDNA coated with T4 gp32.35S-NS3h, on the other hand, did not show a stable interaction with ssDNA alone or when coated with RPA or T4 gp32.
on November 7, 2019 by guest
http://jvi.asm.org/
[image:8.585.134.449.66.321.2](
Fig. 7B
). As expected, the
35S-NS3h–NS5B
⌬
21 complex was able
to stably interact with ssDNA only when it was coated with RPA
and not with ssDNA alone or ssDNA coated with T4 gp32 (data
not shown).
NS5A stimulates NS5B
⌬
21 RNA synthesis on a rolling-circle
template.
In order to investigate the role of HCV NS5A, a
trun-cated NS5A
(S25-K215)containing domain I and almost full-length
NS5A
(S25-C447), both lacking the N-terminal 24-amino-acid
am-phipathic domain, were preincubated with NS5B
⌬
21 and
incu-bated with the rolling-circle RNA template and NS3h in the
pres-ence or abspres-ence of RPA (
Fig. 8
). NS5B
⌬
21 alone exhibited no
activity, but both NS5A
(S25-C447)(
Fig. 8A
) and domain
I-contain-ing truncated NS5A
(S25 to K215)(
Fig. 8B
) were able to stimulate
NS5B
⌬
21 RNA synthesis similar to that observed with RPA (
Fig.
8A
). It was previously reported that preincubation of NS5B and
NS5A, prior to template addition and initiation of reaction, was
needed to observe stimulation of RNA synthesis by NS5B (
71
). We
observed a similar preincubation requirement using the rolling-circle
RNA template (data not shown). Addition of NS5A
(S25-C447), but not
the truncated NS5A
(S25-K215)containing domain I, to RPA
re-sulted in an additive increase in RNA synthesis on the
rolling-circle RNA template (
Fig. 8A
and
B
). Two minor contaminants,
E.
coli
GroEL and DnaK, that copurified with NS5A
(S25-C447)did not
stimulate RNA synthesis (data not shown). It is interesting that
there was no difference in stimulation by the helicase domain
alone compared with NS3-4A containing both the protease and
helicase domains and NS4A (
Fig. 8C
).
RPA and NS5A
(S25-C447)confer high processivity on the
NS5B
⌬
21 replication complex on a rolling-circle template.
In
order to test whether the combination of NS5A
(S25-C447)and RPA
ties NS5B
⌬
21 more tightly to the rolling-circle template than
NS5A
(S25-C447)or RPA alone, a classical template competition
ex-periment was performed. Nonprocessive replication complexes
continuously fall off the template and have to reinitiate. Highly
processive replication complexes tend to stay on the template,
resulting in high-molecular-weight product synthesis. HCV
rep-lication assays were carried out in the presence or absence of a
large molar excess of a poly(A)/oligo(U) template which acts as a
sponge and sequesters the replication complex when it falls off the
rolling-circle template, thus preventing the reinitiation of RNA
synthesis. Radiolabeled RNA products resulting from the addition
of either RPA or NS5A
(S25-C447)or RPA-NS5A
(S25-C447)to reaction
mixtures containing NS5B
⌬
21 polymerase and the NS3 helicase
domain were analyzed by polyacrylamide gel electrophoresis and
FIG 6NS5B⌬21 directly interacts with RPA and NS3. Panels A through D show surface plasmon resonance analysis of interactions between NS5B⌬21, NS3h, RPA, and T4 gp32. NS5B⌬21 (A) or NS3h (B) was immobilized to the sensor chip, and solutions of either RPA or T4 gp32 were passed over the chip. NS5B⌬21 directly interacted with RPA but not with T4 gp32, whereas NS3h showed no interaction with either RPA or T4 gp32. Panels C and D show NS3h passed over immobilized NS5B⌬21 (C) and NS5B⌬21 passed over immobilized NS3h (D). In both configurations, NS5B⌬21 showed direct interaction with NS3h.
FIG 7Preformed NS3h-NS5B⌬21 complex is a stable functional complex. (A)35S-NS3h–NS5B⌬21 complex formation and isolation. Superose 12 gel filtration chromatography profiles of35S-NS3h alone (open squares) and with preincubation with cold NS5B⌬21 (filled diamonds). The gray box shows fractions 32 to 38 of35S-NS3h–NS5B⌬21 complex, which were pooled and examined for interaction. (B)35S-NS3h–NS5B⌬21 complex interaction with ssDNA (filled diamonds) and with ssDNA coated with RPA (open squares) on a BioGel A100 gel filtration column.
on November 7, 2019 by guest
http://jvi.asm.org/
[image:9.585.137.451.65.234.2] [image:9.585.139.446.566.683.2]autoradiography. RNA synthesis from the rolling-circle template
in the presence of RPA (
Fig. 9A
) or NS5A
(S25-C447)(
Fig. 9B
) was
completely abolished in the presence of the competing poly(A)/
oligo(U) template. These data suggest that the addition of either
RPA or NS5A
(S25-C447)separately to the NS5B
⌬
21-NS3 helicase
domain complex on a rolling-circle template results in an
in-creased rate of synthesis of RNA but not in an inin-creased length of
RNA synthesis or processivity (the replicase falls off the template
and gets trapped by the excess poly(A)/oligo(U) templates). In
contrast, when NS5A
(S25-C447)and RPA were both present in the
reaction, robust synthesis of a high-molecular-weight RNA
prod-uct occurred from the rolling-circle template even in the presence
of the poly(A)/oligo(U) template (
Fig. 9C
). These data suggest
that NS5A
(S25-C447)and RPA functionally cooperate in imparting
high processivity to the HCV replicase containing NS5B
⌬
21 and
the NS3 helicase domain on a rolling-circle RNA template.
NS5A
(S25-C447)forms a stable complex with NS5B
⌬
21 but not
with the NS3 helicase domain.
As described above, an interaction
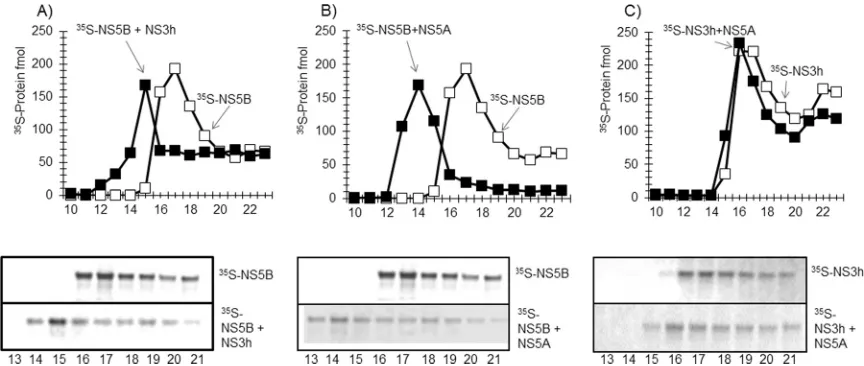
FIG 8NS5A stimulates NS5B⌬21 RNA synthesis on a rolling-circle template. (A) NS5A(S25-C447)and RPA stimulate RNA synthesis by NS5B⌬21 and show additive stimulatory effects in combination. (B) Domain I of NS5A(S25-K215)does not stimulate RNA synthesis by NS5B⌬21 in the presence of RPA. (C) The native NS3-4A complex comprising the 631-amino-acid NS3 protein containing the protease and the helicase domain complexed with the full-length 54-amino-acid NS4A protein stimulated RNA synthesis by NS5B⌬21 polymerase on a rolling-circle RNA template that was comparable to that of the NS3 helicase (NS3h) domain both in the presence and absence of NS5A(S25-C447)and human RPA.
FIG 9RPA and NS5A cooperate to form RNA polymerase processivity factor. The radiolabeled RNA products resulting fromin vitroreactions carried out as described in Materials and Methods were analyzed by polyacrylamide gel electrophoresis and autoradiographed. (A) RNA synthesis by NS5B⌬21-NS3h from the rolling-circle template in the presence of RPA without and with poly(A)/oligo(U). (B) RNA synthesis by NS5B⌬21-NS3h from the rolling-circle template in the presence of NS5A(S25-C447)without and with poly(A)/oligo(U). (C) RNA synthesis by NS5B⌬21-NS3h from the rolling-circle template in the presence of RPA and NS5A(S25-C447), without and with poly(A)/oligo(U).
on November 7, 2019 by guest
http://jvi.asm.org/
[image:10.585.48.544.65.223.2] [image:10.585.94.493.417.673.2]between NS3 helicase and NS5B
⌬
21 was previously demonstrated
using SPR (
Fig. 6C
and
D
). In order to examine the interactions
between NS5A
(S25-C447)with NS5B
⌬
21 and the NS3 helicase
do-main, gel filtration chromatography experiments were performed.
Consistent with previous results (
Fig. 6
and
7
), incubation of
35S-NS5B
⌬
21 with NS3 helicase resulted in the formation of a
higher-molecular-weight complex that was eluted from the gel filtration
column faster than
35S-NS5B
⌬
21 alone (
Fig. 10A
). Similarly,
in-cubation of
35S-NS5B
⌬
21 with NS5A also resulted in the
forma-tion of a higher-molecular-weight complex, indicating that NS5B
and NS5A interact with each other (
Fig. 10B
). Interestingly, the
elution profile of
35S-NS3 helicase domain plus NS5A
(S25-C447)
was
similar to that of
35S-NS3 helicase domain alone, indicating that
these two proteins do not directly interact with each other (
Fig.
10C
).
DISCUSSION
Development of an HCV
in vitro
replication system.
The goal of
this study was to create an
in vitro
HCV replication assay to study
viral and cellular proteins that constitute the HCV replication
complex and influence the activity and processivity of HCV
poly-merase on an RNA template. All replication systems utilize a
com-plex of different proteins that are required to deliver certain core
functions required for RNA or DNA synthesis. In the case of viral
replication, the functions can come from the virus or the host.
Based on a comparison of different viral
in vitro
replication
sys-tems, we hypothesized that a minimum of six replication
func-tions are required: (i) an origin recognition protein, (ii) a
poly-merase, (iii) a priming mechanism, (iv) a single-stranded binding
protein, (v) an unwinding protein, and (vi) one or more
proces-sivity factors. Based on our assumption that the polymerase and
helicase must form part of a replication complex, we set the
min-imum criteria for our
in vitro
replication system to be one where
polymerase activity is stimulated by the presence of NS3 helicase.
We could readily identify the RNA-dependent RNA polymerase
for NS5B and the helicase for the C-terminal region of NS3. Using
a rolling-circle RNA template and these two purified proteins as
our starting points, we looked for other proteins of the HCV
rep-lication complex, such as processivity factors.
HCV replication has been shown to be dependent on human
proteins or factors (
72
). We hypothesized that at least one of those
host factors could be the human single-stranded binding (SSB)
protein also known as replication protein A (RPA) (
63
). In the
in
vitro
replication system, we showed that RPA strongly stimulated
RNA synthesis by NS5B
⌬
21 on a rolling-circle template in the
presence of NS3 helicase. We believe that this is the first example
of a viral RNA polymerase that uses human RPA as a processivity
factor. Finally, we investigated the mechanism of action of NS5A
in the
in vitro
replication system, which revealed that NS5A
(S25-C447)and RPA interact with NS5B polymerase, forming a highly
pro-cessive complex that includes NS3 helicase on a rolling-circle RNA
template, resulting in the synthesis of high-molecular-weight
RNA product. These results help to extend our understanding
into at least one of the roles of NS5A, a complex multifunctional
protein, in the HCV replication complex.
Multiple roles for RPA and other SSBs in human and viral
DNA replication
in vitro
.
The requirement of RPA for human
and viral DNA replication has been well established (
63
).
Al-though not much is known about the function of RPA in RNA
metabolism, it has been shown that direct interactions between
RPA and the RdRp QDE-1 are essential for RNA synthesis (
73
). T4
gp32, the SSB protein of bacteriophage T4, plays important roles
in phage DNA replication, repair, and recombination, facilitates
its activity at the replication fork through binding to ssDNA,
me-diates specific protein-protein interaction with the major
compo-nents of the DNA replication complex, including DNA
polymer-ase (T4 gp43) activity, and recruits primpolymer-ase-helicpolymer-ase (T4 gp59)
assembly to the primosome complex T4 gp61-41 (
74–77
).
Role of SSB protein in leading- and lagging-strand DNA
syn-thesis
in vitro
.
In previous studies, rolling-circle templates have
been used in DNA replication experiments to elucidate the
re-quirements for coordinated leading- and lagging-strand
replica-tion. In the
E. coli
and T7 bacteriophage rolling-circle DNA
repli-cation systems, the presence of
E. coli
SSB protein or the T7 SSB
FIG 10Protein-protein interaction between NS5B⌬21, NS5A(S25-C447), and NS3h. NS5B⌬21 and NS3h form a complex (A), NS5B⌬21 and NS5A(S25-C447)form a stable protein complex (B), and NS3h and NS5A(S25-C447)do not interact directly (C).
35S-labeled proteins were incubated with the high excess of “cold” protein to form a stable protein complex. Proteins were separated on a Superose 12 gel filtration column, and fractions were collected and analyzed using a scintillation counter and by polyacrylamide gel electrophoresis followed by autoradiography.
on November 7, 2019 by guest
http://jvi.asm.org/
[image:11.585.76.508.66.249.2]gp2.5, respectively, is not required for leading-strand synthesis but
is essential for lagging-strand synthesis, in which SSB protein plays
a role in protein-protein switching (
67
,
78
,
79
). The simian virus
40 (SV40) proliferating cell nuclear antigen (PCNA)-dependent
dipolymerase replication system and the eukaryotic human DNA
replication system follow the same rules whereby RPA is
specifi-cally required for protein-protein switching on the lagging strand
through direct interactions with components of the replication
complex but is not essential for synthesis of the leading strand
because it can be replaced with
E. coli
SSB protein in the primer
extension reaction (
80
,
81
).
Role of RPA in the HCV rolling-circle replication system.
On
a multiprimed template [poly(A)/oligo(dT)], NS5B
⌬
21 alone was
able to synthesize a small amount of product (0.7 pmol) in 2 h and
RPA did not show any stimulatory effects on RNA synthesis (
Fig.
3B
). In contrast, on a rolling-circle RNA template, RPA was
abso-lutely required for the activity of NS5B
⌬
21 (
Fig. 2
). The inability
of gp32, which has SSB protein-like properties, to substitute for
RPA (
Fig. 4
), despite its high affinity to bind RNA (
Fig. 3
), suggests
that RPA plays an additional role other than melting RNA
second-ary structures. RPA is known to be a nuclear factor for host and
viral DNA replication, although RPA is available in the cytoplasm,
where HCV replicates (
63
). Competition studies using ssDNA
sat-urated with RPA in the presence of competitor RNA have shown
that the
K
dof RPA for RNA in the presence of ssDNA is 10
⫺6to
10
⫺7M. Although the affinity of RPA for ssDNA is 3 orders of
magnitude stronger than for RNA, there is no ssDNA in the
cyto-plasm to compete with HCV RNA for RPA (
63–65
).
Role of HCV helicase in the HCV rolling-circle replication
system.
The addition of NS3 helicase to HCV polymerase and
RPA on a rolling-circle template increased RNA synthesis from
⬃
7.0 to
⬃
50 pmol in 2 h. Other investigators have demonstrated
various effects of the protease domain on the helicase domain
alone, in the full-length NS3 (631 amino acids containing the
HCV protease and helicase domains), or in the NS3-4A complex
(
29
). In contrast, we observed that NS3-4A had activity in
rolling-circle replication similar to that of the NS3 helicase domain alone
(
Fig. 8
), suggesting that the protease domain does not participate
in the interaction between helicase and NS5B.
HCV helicase activity has been shown to be extremely sensitive
to NaCl concentrations approaching physiological levels (
82
).
This led to a hypothesis that NS3 helicase is associated with other
HCV nonstructural proteins or host factors as part of a replication
complex
in vivo
, and these additional factors are needed to ensure
the functionality of HCV helicase and efficient replication of HCV
in cells (
16
). Consistent with this hypothesis, the stimulatory
ac-tivity of the helicase domain or NS3-4A in the replication complex
on a rolling-circle template is significantly less sensitive to salt and
HCV helicase is able to form a stable complex with HCV
polymer-ase under physiological salt conditions. Taken together, these data
suggest that the presence of NS5B and RPA stabilizes helicase
ac-tivity under physiological salt conditions either through an
in-duced conformation of the helicase or by excluding salt from the
active site of the helicase.
Role of NS5A in the HCV rolling-circle replication system.
Most viral RdRps are not processive enzymes and can switch
tem-plates or jump off the template during RNA synthesis (
83
).
Previ-ous studies have shown that HCV NS5B alone is not a processive
enzyme
in vitro
(
40–46
). NS5A has been shown to interact with
RNA (
84
) and NS5B (
85
) and to play a role in HCV replication
(
86
) In this study, NS5A
(S25-C447)and RPA formed a functional
cooperative interaction which enabled RNA synthesis by
NS5B
⌬
21 to be highly processive on a rolling-circle RNA
tem-plate.
NS5A has been predicted to comprise three domains, and
while the crystal structure of domain 1 has been solved, the
struc-tures of domains 2 and 3 are unknown; nuclear magnetic
reso-nance (NMR) studies suggest that they are intrinsically
disor-dered, at least in experimentally manipulated proteins (
87–90
).
Interestingly, in our studies, functional cooperativity between
NS5A and RPA was observed with the almost full-length
NS5A
(S25-C447)but not with a truncated NS5A
(S25-K215)compris-ing solely domain 1, suggestcompris-ing a role for the other domains of
native NS5A in HCV replication (
Fig. 8
). It has been shown
pre-viously that domain 3 plays a role in virus assembly and can be
deleted without affecting RNA replication (
54
,
58
,
91
). Our results
that show that NS5A
(S25-C447)and NS5B
⌬
21 form a stable
com-plex are consistent with previous findings regarding RNA and
NS5B interaction domains in NS5A (
54
,
55
,
85
,
92
,
93
).
Protein-protein interactions within the replicase.
The
inter-actions between replicative DNA polymerases and helicases are
known from studies in the T7 and
E. coli
DNA replication systems
(
67
,
78
). We have shown that NS5B
⌬
21 binds poorly to nucleic
acid and to a rolling-circle template by itself, and previous studies
have also shown that NS3 helicase binds poorly to an ssDNA
tem-plate at physiological salt levels (
⬃
150 mM) (
82
). Our studies
suggest that human RPA is required to bring NS5B
⌬
21 to the
template through specific NS5B
⌬
21-RPA interactions (
Fig. 5
) and
that NS3 helicase, in turn, is brought to the template through
specific NS3 helicase-NS5B
⌬
21 interactions (
Fig. 6
and
7
). The
interaction between the NS3 helicase domain and NS5B was
pre-dicted from
in vivo
studies, and it was shown that the NS3 helicase
domain modulates template recognition by NS5B and increases
the amount of product synthesized (
94
,
95
). Similar results were
observed with NS3-4A, consistent with their distinct roles in HCV life
cycle. Thus, the interactions of the NS3 helicase domain with NS5B
and between NS5B and RPA are crucial for formation of the
replica-tion complex and might represent new antiviral drug targets.
NS5A and RPA functionally cooperate to tie the replication
complex to the template.
Our biochemical data demonstrate that
functional cooperativity exists between NS5A
(S25-C447)and RPA in
tying the replication complex to the template and promoting RNA
synthesis by NS5B
⌬
21 on a rolling-circle RNA template (
Fig. 9
).
While RPA and NS5A
(S25-C447)proteins can independently
stim-ulate RNA synthesis by NS5B
⌬
21 polymerase in the presence of
NS3 helicase, both of these replication complexes were easily
com-peted away from the rolling-circle RNA template by the addition
of a large molar excess of poly(A)/oligo(U). In contrast, in the
presence of both NS5A
(S25-C447)and RPA, the replication complex
was highly processive and could not be competed off the
rolling-circle template by a lar
![FIG 5 RPA ties NS5B�21 to the template. 35S-labeled NS5B�21 and NS3h, individually synthesized in transcription/translation reactions in vitro in the presenceof [35S]methionine, were incubated with circular ssM13mp18 DNA coated with RPA (A and D) or gp32 (](https://thumb-us.123doks.com/thumbv2/123dok_us/143719.20863/8.585.134.449.66.321/individually-synthesized-transcription-translation-reactions-presenceof-methionine-incubated.webp)