JOURNAL OFVIROLOGY, Mar. 1990, p. 1063-1069 0022-538X/90/031063-07$02.00/0
Copyright C 1990, American Society for Microbiology
cis
Rescue of
a
Mutated
Reverse Transcriptase Gene of Human
Hepatitis
B
Virus by
Creation of
an
Internal ATG
SUSANTA ROYCHOUDHURYANDCHIAHO SHIH*
Departmentof Biochemistry andBiophysics, School of Medicine, University of Pennsylvania, Philadelphia, Pennsylvania 19104-6059
Received 28 August 1989/Accepted 27 November 1989
Using mutational analysis, we have investigatedthe translation strategy ofthe reverse transcriptasegene (pol) of humanhepatitisB virus. It has been proposed thatthispolgeneproductissynthesizedas acore-pol
fusion protein fromapolycistronic mRNA template via ribosomalframeshifting, a mechanism oftenseen in
retroelements. Ourdata indicate that creation ofa novel ATG initiation codonnear the original ATG can
compensate for alethal missensemutation atthe first ATG position of thepolopen reading frame. Genetic
analysishasrigorously ruledoutthe possibilities offrameshifting, non-ATG initiation,orRNAediting. These
resultsare discussed in the context ofa 5'-endentry model versus anovel model of direct internalentry of
ribosomes.
Reversetranscriptase (pot)wasindependently discovered
in retroviruses almost 2 decades ago by Baltimore (1) and
TeminandMizutani (39). Itwas demonstrated bySummers
andMason that DNA animal viruses suchasduck hepatitis
B virus (duck HBV) could also replicate viareverse
tran-scription (38). The ubiquity ofreversetranscriptase hasalso
been supported by the recent discovery ofthis enzyme in
procaryotes (11, 21). In many cases, thepol open reading frame (ORF) is overlapped by and out of frame to a 5'
upstream nucleocapsid ORF within the same polycistronic
mRNA(e.g., for HBV,see Fig. 1). The biosynthesis ofthis enzyme inretroviruses involves the formation ofagag-pol
fusionprotein byeither ribosomal frameshifting (fs) (12) or
readthroughofanamberstopcodon between the
nucleocap-sid (gag) andpolgenes (44). For human HBV, it has been proposed that the synthesis of thepolgeneproduct from a
3.5-kilobase(kb) polycistronicmRNA(Fig. 1)alsoinvolves
the formation ofanucleocapsid-pol (designatedc-pot)fusion
proteinvia fs(43).
Identification andcharacterization ofthepolgeneproduct
with anti-pol antibodies in hepatocytes by active HBV replication (2, 22) have proven difficult because of either poor immunogenicity or smallamounts of thepol protein.
One hypothesis concerning HBVpol gene expression via
ribosomal fs has been examined by genetic analysis in the
duck HBV system (4, 33). Using a trans-rescue genetic
approach, a pol-defective mutant has been successfully complemented bycotransfection withacore-defective non-sensemutant,indicating thatcoreandpolgeneproductscan
besynthesized separately (4, 33). Here,wereporta
comple-mentarycis-rescue approachtoinvestigatepolgene expres-sionin the human HBVsystem. Our resultsprovide further convincing evidence against the requirement of the forma-tion ofacore-pol fusion protein in HBV via ribosomal fs.
MATERIALS AND METHODS
Site-directed mutagenesis. The procedurefor site-directed
mutagenesiswas adapted fromthatofKunkel(18). Plasmid RG6 containsafull-length HBVmonomerDNA in the minus
orientation in theEcoRI site ofvectorM13mpl8. A
uracil-containing single-stranded DNAtemplate of RG6 was pre-pared by infectingRG6bacteriophageinagrowingculture of
* Correspondingauthor.
CJ236 in the presenceof uridine (0.25 ,ug/ml)and
chloram-phenicol (30 ,ug/ml).Theoligonucleotides used for
construc-tion of the HBVmutants areshown below.
Nucleotide no. of Olignucleotidesequence
mutantposition
5' *
2306 GAC CAC TAA ATG CCC CTATC
2310 GAC CAC CAA ACG CCC CTATC
*
2471 TGG ACT CAT TAG GTG GGGAA
305 CGT GTG TCT TAG CCA AAATT
*
1314 AGC AGG TCT TGA GCA AACAT
2299 CTC CAG CTT ATG GAC CACCA
2342 CGG AGA CTA ATG TTG TTAGA
Approximately100to200ngofoligonucleotidewas phos-phorylatedwith T4polynucleotidekinase and then annealed to 1 ,ug ofuracil-containing, single-stranded RG6 template
DNA. The resulting hybrid DNA was extended with T4
DNApolymeraseandligatedwith T4 DNAligase.Asample
of the ligation mixture (about 100 ng ofDNA) was
trans-formed into MV1304 Escherichiacolicells and thenplatedin
thepresenceof MV1304. Mutant HBV cloneswerescreened
for the mutant sequence by the Sanger dideoxy technique
(32) (Sequenase obtained from U.S. BiochemicalCo.).
Construction of MC-3vector. A pSV2ANeo-HBV
mono-mervectorwasconstructedasdescribedpreviously (14, 35).
ThisvectorcontainstwoEcoRI sitesflankingthefull-length
3.2-kb HBV DNA fragments (ayw subtype ofHBV). The
transcription of HBV pre-corepromoterin this vector is in
thesamedirectionasthatfor theneomycinresistancegene.
Inordertodimerize the HBVgenomeefficiently,the
down-stream EcoRI site of the pSV2ANeo-HBV monomer was eliminated, resulting inapSV2ANeo-HBVmonomerwitha 1063
Vol.64, No. 3
on November 10, 2019 by guest
http://jvi.asm.org/
C
S
P or (Pol)
Rl
HBV
Rl RI
HBV
0 1 3
0/3182
J- pSV2
3182 3.5 kb mRNA 2.5 kb mRNA
N,'%> -> 2.1 kbmRNA
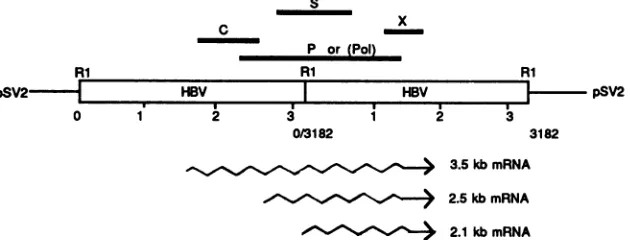
FIG. 1. Transcriptionmap ofHBV drawnwith a DNAtemplate in ahead-to-tail tandem dimerconfiguration, which is likeacircular
permutationofacovalentlyclosedcircular form.The fourdifferent ORFsare shownbysolidbars. The2.5-kbmRNAencodesthepre-surface
antigen (pre-Sl), andthe 2.1-kb mRNAencodesanotherpre-surface antigen (pre-S2) and surface antigen (S). The polymerasegene(po)is
inthe+1frame withrespect to theoverlappingnucleocapsidgene(C),and the X ORF encodes aputative transactivator (forareview, see
references7,23, and 41).
single upstream EcoRI site. This vector was designated MC-3. Briefly, the pSV2ANeo-HBV monomer (35) was
linearized by EcoRI partial digestion. The 9.5-kb DNA fragment of this linearized pSV2ANeo-HBV monomer was
electroeluted froma1.2%agarosegel. The EcoRI end of this eluted fragment was eliminated by Klenow repair. After blunt-end religation, the ligated DNA mixture was trans-formed into E. coli. Loss of the downstream or upstream EcoRI site was differentiated by cleavage with an AccI or AccI-plus-EcoRI double digest.
Dimerization of HBV DNA. The construction of a wild-type HBV dimer expression vector has been described elsewhere(35). For thedimerization ofmutantHBVDNAs, the aforementioned MC-3 plasmid was used. The 3.1-kb
mutant HBV DNA fragment was isolated from the phage
DNA of the M13mpl8-HBV mutant by complete digestion with EcoRI and was electroeluted from agarose gel. This purifiedmutantHBV DNAfragmentwasligatedtotheMC-3 vectorwhichhad beenlinearized by EcoRI and dephospho-rylated by alkaline phosphatase. A sample of the ligation mixture was transformed intoE. coli MV1304. Head-to-tail tandem dimerswere screened byXhoI orAccIdigestionof miniprep DNA. For theconstructionof double mutants, the uracil template ofmutant 2310 was used. Double mutants were also confirmed by the Sanger dideoxy sequencing method(32).
Cell culture and transfection. The human hepatoma cell line HepG2 was maintained in 10% fetal bovine serum in DulbeccomodifiedEaglemediumat37°Cinthe presence of 5.5% CO2. The calcium phosphate transfection procedure was followed as detailed elsewhere (35). Briefly, 7 x
105
cells per 10-cm dish were transfected with 5 ,ug of either wild-typedimer or mutantdimerDNAplus 30 ,ugofhuman genomic DNA as carrier. A control dish transfected with
HBVmonomerDNAwasalwaysincluded. Donor DNAwas
removed 4 h aftertransfection,andcells were fed with fresh Dulbecco modified Eagle mediumcontaining 10% fetal
bo-vine serum.
Analysisof HBVexpression. Five days after transfection, medium was removed and saved for further analysis (see below).Cells from one 10-cm dish (6 x
106
cells) were lysedto prepare intracellular, extrachromosomal HBV DNA by theHirtmethod (10). Total cellular RNA was prepared from cellsfromanother 10-cmdishby the method ofChirgwinet al. (5) (see Fig. 3b), and cells from a third 10-cm dish were used to prepare intracellular core particles for an endoge-nous-polymerase assay (15, 38; seebelow).
Core particle-associated endogenous-polymerase activity.
Theprocedure for preparation of intracellular coreparticles wasadapted from the method of Summers andMason(38). Briefly, cells from a 10-cm dish (6 x 106cells) were lysed in 0.5 ml of chilled extraction buffer(20 mM Tris hydrochloride [pH 7.4], 7 mM MgSO4, 50 mM NaCl, 0.1%
P-mercaptoeth-anol, 100 ,ug of bovine serum albumin per ml, 0.25 M sucrose) with a Dounce homogenizer. The homogenates werethen spun in aMicrofuge (Beckman Instruments,Inc.) at 4°C for 30min to remove cellular debris and nuclei. The clearhomogenates were loadedona sucrosegradient (5 ml of 15 to 30% sucrose [wt/vol] in extraction buffer). The gradients were run at 32,000 rpm for 6 h at 4°C in a Beckman SW50.1 rotor. Fractions (200
RIu)
were collected from the bottom of the gradient, and core particle fractions were pooled. The particles from pooled fractions werepelletedby spinning them at 35,000 rpm for14 h at 4°C ina Beckman SW50.1 rotor. The pellets were suspended in 100 ,ul of extraction buffer, and 40-pd samples were used for the polymerase assay as described elsewhere (35).Preparation of extracellularHBV DNA.Extracellular DNA
was prepared from 72-h-conditioned medium from three 10-cm dishes (total, 30 ml) collected 5 days after transfec-tion. The mediumwasprecleared byspinningitat 35,000 x
gfor 30 minat 4°C. Particles from the precleared medium werepelleted byspinningthem at 25,000 rpmin a Beckman
SW28rotorfor 16 hat4°C. Thepelletsweresuspended in 0.5
ml of 10 mM Tris hydrochloride-10 mM EDTA-0.6%
so-dium dodecylsulfate buffer(pH 7.5) anddigested with 400 ,ugofproteinase K per ml for 3 hat37°C. After phenol and chloroform extractions, the DNAs were precipitated and redissolved in 30 ,ulof TE (10 mM Trishydrochloride,1mM EDTAbuffer[pH7.5]) andanalyzedon a1% agarosegelby the Southernprocedure (37).
RESULTS
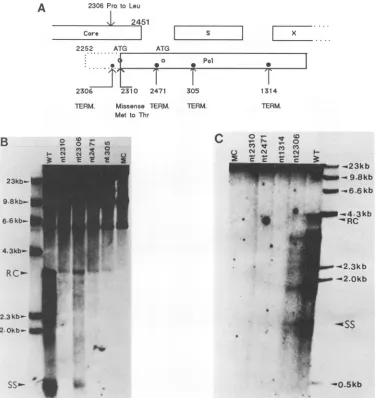
Mutant 2310. A point mutation was introduced into the
first ATG codon of theHBVpolORFatnucleotide (nt)2310, converting methionine to threonine while keeping the core
proteinunaltered(Fig. 2A). Upon tandemdimerizationand transfection of thismutantinto the humanhepatomaHepG2 cell line, the functional activities of mutant 2310 were
assayedin parallel with those of the wild-type HBVdimer control asdescribed in the legend ofFig. 2. Neither HBV-specific DNA replicative
intermediates,
as measured by Southern blot analysis, nor polymerase activity could be detected.These results with thismutantsuggestthatribosomalfs is
pSV2 L--
T
on November 10, 2019 by guest
http://jvi.asm.org/
[image:2.612.152.467.74.194.2]EXPRESSION STRATEGY OF HBV REVERSE TRANSCRIPTASE 1065
2306 Pro to Leu
1o
24512252...2 .2 1ATG ATG
2306 2310 2471
TERM. Missense TERM.
Met to Thr
I
s
I
S Pol
305 TERM.
B
[image:3.612.123.499.72.470.2]23kb-9.8kb-| 6.6
kbm-4.3kb
I
RC
2.3kb
-2. 0kb'-I
SSI'-o N In
3c9) t
C4 co
~:...t
u CY N _. C4
2E. - .10
3--o4.3 kb
RC
_w2.3kb
_ -2.0kb
-0O.5kb
FIG. 2. (A) Schematicrepresentation of different mutation positions in thepolORF. The nucleotide number(HBV subtype ayw) indicates theposition of the base change. None of the mutations exceptthatatnt2306 changethe codingcapacity ofanyoverlapping ORF. For the
sake ofclarity, thisfigure isnotdrawntoscale. Symbols:0,ATG; *, stopcodon.S,X, andpolareexplained in the legendtoFig. 1. (B) Southern blotanalysis of wild-type andmutantHBV DNAreplicative intermediates. Both relaxed-circle and minus-strand, single-stranded formsarecharacteristic replicative-intermediate molecular forms of HBV thatoccurduringreversetranscription (7, 23, 41). All thebands
above the6.0-kbpositionrepresentthe residual donor DNA of the HBV dimer in thepSV2Avector.These"contaminating"bandsserve as
convenientinternal referencestoassesstransfection efficiency and the relativeamountof donor DNAappliedtoeachrecipient culture. (C) Coreparticle-associated endogenous polymerase activity of wild-type and mutant HBV. Base substitutionmutants 1314and 2471, which containatruncatedpolgene, areincludedasnegative controls. Withalonger chase,morefull-lengthopencirclescanbeseenatpositions
near4.3 kb.RC, Relaxed circle; SS, single stranded; WT, wild-type HBV. Lanes MC, MC-3vector(see Materials and Methods). notlikely to occurdownstream from the ATG codon. Iffs
does occurdownstream fromthis position, the change from methionine tothreonine atposition 2310 would notchange
the amino acid sequence of thec-pol fusion protein;
there-fore, mutant2310 should notbe lethal. Thefact thatmutant
2310 exhibits no detectable pol activity (Fig. 2B and C)
suggests that fs cannot occur downstream from position 2310.
Mutant2306. Aribosomal fsoccurringupstreamfrom the
first ATG ofpolin wild-type HBV willgenerate acore-pol
fusionproteinwithaninternalmethionineresidueat nt2310.
A missense mutation at this position might abolish the
enzymatic activityofpol completely.Torule thisout,astop
codon was introduced at position 2306, which is in frame
with andimmediately adjacenttothe first ATG codon ofpol
(Fig. 2A). If fs occurs upstream from position 2306, the
frameshifted ribosome should be arrested when it
encoun-tersthecreatedstopcodonatnt2306. Functionalanalysisof
thisnonsensemutant2306 indicated that both DNA
replica-tive intermediates and polymerase activity are clearly
de-tectable (Fig. 2). Although the levelofactivityappearedto
be significantly lower than that of the wildtype,it isclearly significantly above the background level. This difference
betweenwild-typeandmutant2306cannotbe attributedtoa
concurrent structuralalteration of thecoreprotein by muta-tion 2306, which converts a proline residue into a leucine,
since cotransfection ofmutant 2306 with otherpolmutants
producing wild-typecore protein failedto restore the poly-merase activity to the wild-type level (see Discussion).
Examination of the steady-state mRNA levels by Northern
A
Li2...
1314 TERM.
-.o23kb
I -.-a9.8kb
0 000--w6.6kb
VOL.64, 1990
I
I-0
v
on November 10, 2019 by guest
http://jvi.asm.org/
1066 ROYCHOUDHURY AND SHIH
Arg to Gly
I
Thr to Asn TAG
Core
I
A
ATG
Pol
2310/2299 2'
Ileto Met MO
N N ,.C
cN c N
310
letto Thr
B
o o 0 0
c'X CO cCo)
0N N N N4
C Cs N CS C
.23
kb-.9,8kb
6.6kb
_4.3k b
-aRC
_
_6
- 2.3k b
--42.Okb
SS
.'.'.i C
3.5kb
I.
.2.5
kb
w w w~~~~~--2.1 kb
T
2310/2342
Leuto Met
c" cn0) 0)0)
Cq
')C'Je000 0 Ui)v- I- 'r-.
cje4Ncs
Cce)clCMes
, _*0 _0 4.f
3 :4ca - r- £
_.
jJ
a
RCm v i
v.4a
[image:4.612.145.486.72.509.2]
;s-lntracelflular
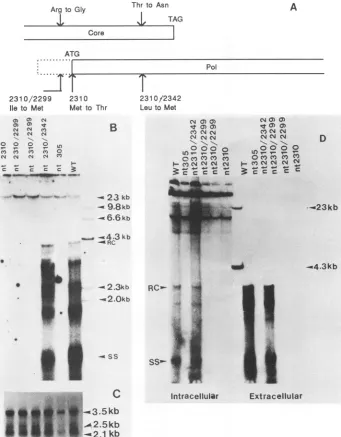
FIG. 3. (A) Schematic representation ofthe positions of newly created methionine codons in thepol ORF. The nucleotide number indicates thepositionatwhich the base has beenchanged. Theconcurrentchange of amino acid residue in thecoreprotein is indicated.(B) Coreparticle-associated endogenous polymerase activity of wildtype and2310/2342 doublemutant. Theduplicated lanes of2310/2299in panels B, C, and Daretwoindependent isolates ofmutant2310/2299. (C)Northern blot analysis of the steady-state level ofHBV-specific transcripts in HepG2 cells transiently transfected with double mutants. (D) Southern blot analysis of intracellular and extracellular HBV-specific DNAs from HepG2 cellstransiently transfected with 2310/2342 doublemutant.WT,Wild-typeHBV;RC, relaxedcircle;SS, single stranded.
(RNA) blot analysis did not reveal any differences among
wild-type and any mutant HBV (data not shown). Taken
together, these data strongly suggestthat fs doesnot occur
eitherdownstreamorupstreamfromthefirst ATG position
of pol within the c-pol overlap. Nevertheless, it is still
possible that fs occurs in the vicinity of positions 2306 to
2310. Anothersimilarpossibility is that the fs signal
neces-sary for the fs event (12) is located near this position. A
missense mutation at the first ATG may therefore have
abolished thefs signal.
One prominent band near the 4.3-kb position of the
relaxedcircle ofHBVcanalso be detected in othermutants,
includingmutant2310 (Fig. 2B).This band doesnotappear
to represent a bona fide replicative intermediate, since
neitherapositive polassayresult(Fig. 2C)northepresence
of a characteristic single-stranded replicative intermediate
(Fig. 2B) hasever been observed in mutant 2310, 2471,or 305. Infact, when less donor DNAwasused, this pseudosig-nal disappeared(e.g., see Fig. 3).
Creationof internal novel ATG codons. To better
under-stand translational initiation of the pol gene, we created
novel ATGcodons either 3 aminoacidsupstream(position 2299)or11amino acidsdownstream (position 2342) fromthe
mutatedATG codoninmutant2310 (Fig. 3A). Thesedouble
mutantswereanalyzed ina mannersimilartothatdescribed previously (Fig. 3B and D). Bothpol enzymatic assay and
C-4 0) 0)
.0') 0)
('N
(%4c
0000
C Cc C C
D
---23kb
-4.3 k b
Extracellular
J. VIROL.
,,,...
.. _on November 10, 2019 by guest
http://jvi.asm.org/
EXPRESSION STRATEGY OF HBV REVERSE TRANSCRIPTASE 1067
4.3kbm-
-o
L-(%4 C4j CY) e)) 0) 0) cn)
o C NC N. NM Ct C4 c4 cN c4
00 0 00
04 C4 ('4 N N'
4. 4' 4' 4' 4'
c e c c X
N 1 Cl YC
0
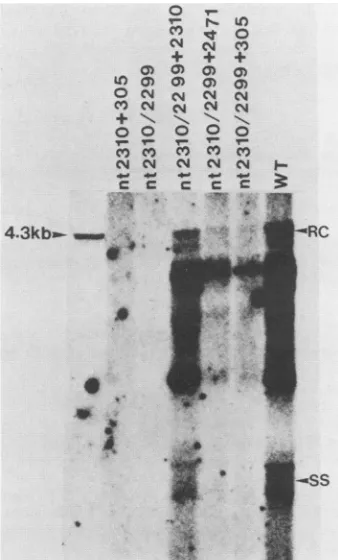
FIG. 4. Restoration of polymerase activity ofmutant 2310 by
creation ofanupstreamATG codonat nt2299after cotransfection
withpol-defective mutantsuch as 305, 2471, or 2310. Controls to
testfor recombinationwerealways included bycotransfectingtwo
differentpol-defective mutants (e.g., 305 and2310). RC, Relaxed
circle; SS, single stranded;WT, wild-typeHBV.
Southern blot analysis clearly indicated that the missense
mutant 2310 can be rescued by providing a novel
down-streamATG codonat nt2342(Fig.3B andD). In ordertobe
certain that the difference between these mutants indeed reflects a difference in translational control rather than in transcriptionalcontrol or mRNAstability, we examined the steady-state level of the 3.5-kbpol-specific mRNAin these
mutants by Northern blot analysis (Fig. 3C). Our results
indicate that these mutants do not differ from each other regarding levels of allspecies of HBV-specific mRNAs. One explanation for the failureto rescuethepolactivityindouble mutant2310/2299is a concurrent structural alteration of the coreprotein (from Arg toGlyatposition2299).Althoughthe core gene product of mutant 2310/2299 is detectable by immunoprecipitation with anti-core antibody (data not shown), it is possible that this mutated core protein is no
longerfunctional. Inanattempttocomplementthepotential defectofthecoreproteinindoublemutant2310/2299witha
functional core protein from mutant2310, wecotransfected thisdouble mutant 2310/2299with singlemutant2310. Nei-ther single mutant 2310 nordouble mutant2310/2299 alone was sufficient to produce detectable pol activity (Fig. 4). When cotransfected with both mutant 2310 and double mutant 2310/2299, cells showed wild-type levels of pol activity. Similar results (Fig. 4) were obtained when DNA replicative intermediates were assayed by Southern blot analysis (datanotshown).Thepossibilityofrescueby DNA recombination between a core-producing plasmid (mutant 2310) andapol-producingplasmid(mutant2310/2299)canbe excluded since in a control experiment, pol-truncated
mu-tant 305 did not rescue mutant2310. This result provesnot
only that rescue of mutant 2310 with an upstream ATG
codon is possible,but also that thecoreandpol
proteins
of HBVcanbesynthesized separately.DISCUSSION
Potential mechanisms of HBV pol gene expression. The
genetic data presented in this paper argue
convincingly
against ribosomal fs ornon-ATG initiation as amechanism ofpol geneexpressionin HBV. Furthermore, theystrongly
[image:5.612.96.265.76.356.2]supporttheconclusionthat the first ATG in thepolORF is essential fortranslational initiation. Several different mech-anisms can explainthis observation (Fig. 5). First, transla-tion may occur by direct internal initiation, as recently described for the picornavirus system (13, 29). A major difference between picornavirus and 3.5-kb HBV-specific mRNAsis that theformerarenaturallyuncapped,while the latterarebelievedtobecapped (19, 20).Thelikelypresence ofacapped structure at the 5' end ofthe 3.5-kb mRNAof HBVdoesnotargueagainstthepossibilityof direct internal initiation, since it has been shown that methylated capped poliovirus mRNAcanbe translated invitroat an
efficiency
FIG. 5. Potentialstrategies ofHBVpolgeneexpression. Seetextfor references.
VOL.64, 1990
.o.. .&
".II'3k.4..,
on November 10, 2019 by guest
http://jvi.asm.org/
[image:5.612.91.535.540.711.2]equaltothat of the uncappedcounterpartin a cap-indepen-dent fashion (28). As described in the legend to Fig. 2, the
diminished pol activity ofmutant2306 cannotbe explained by the concurrent structural alteration of the core gene product. Ifpol expression indeed involves direct internal initiation, diminished activity of pol inmutant2306(from CC AAATGtoCTAAATG)canbeexplained by aperturbation of unknownsignals for direct internal initiation. Second, the
pol proteinmaybeencoded byan asyetunidentified mRNA species which exposes the ATG at nt 2310 as the 5'-most proximal initiation codon. The scanning 40S ribosome sub-unit would thus commence initiation at nt 2310 when it encountered this first ATG of pol. It should be noted that this hypothetical transcript has neverbeen identified by primer extension-nuclease Si mapping (3, 6, 24, 42). Third, it has previously been proposed that ribosomes, after reaching the
stopcodon,maybe ableto scanbackwards andreinitiate at the once-bypassed, upstream ATG codon (26, 27, 40). We consider this a less likely possibility because the distance betweenthe end of thecore gene(nt 2451) and thefirstATG
of pol (nt 2310) is approximately 140 nt. This distance
appears to be too long for this type of reinitiation to take place (26, 27). Another possibility, such as leaky scanning (16, 17), is unlikely because within the 3.5-kb mRNA, there arefourorfiveATG codons 5' upstreamfrom the first ATG
ofpol at nt2310 in HBV subtype ayw. Moreover, some of theseupstreamATG codonsarein optimalsequencecontext fortranslational initiation, whiletheauthentic ATGcodon of polatnt2310 and thecreated ATG codonsat nt2299or2342 are in poor sequencecontext, with acytosine ratherthan a purineatthe -3 position (17). Neither RNA editing (36)nor
non-ATG initiation (9, 25, 31) appears tobeconsistentwith
thedatapresented in Fig. 2, 3, and 4.
Althoughourstudies of HBVpolgene translationarenot
consistent withapreviousreportof detectionofc-polfusion
proteins incertain human hepatoma tissue (43), it should be notedthat human hepatoma tissues often contain integrated andrearranged HBV DNA (34). Therefore, onelikely inter-pretation of this discrepancy isthatthereportedc-polfusion
protein is actually produced from the rearrangedHBVDNA
rather than synthesized as a natural product during the
normal lifecycle of wild-type HBV.
Our observation of genetic complementation between
core-defective and pol-defective HBV plasmids argues
against c-pol fusion (Fig. 4) and substantiates an earlier
reportonthe duck HBV system(4, 33). Asdiscussed in the legendto Fig. 2, byitself the missense mutation of thefirst ATGofpolat nt2310 doesnotprovide conclusive evidence against fs occurring downstream fromthisposition, sincethe
potential perturbation ofa +1 fssignalcannotbe excluded. Our report here of the successful rescue of the missense
mutant 2310 by creation of an internal ATG constitutes
strong support for either direct internal initiation or the
existence ofa novel species of mRNA responsible for pol
translation which may have escapeddetection.
Packaging strategyofpolgene product. Of potential
bio-logical significance are the formation of a gag-pol fusion
protein in retrovirusesandits abilitytotarget and
compart-mentalize thepolgene product in the coreparticles during
packaging. Another aspect of potential significance is the
stoichiometricregulation of thegagandgag-polproductsin
aproperratio (forareview, seereference 8). However, the
biosyntheses of the nucleocapsid and the reverse
tran-scriptaseappeartobeseparablein HBV(Fig. 3and 4). This
isalso thecasein theclosely related duckHBV (4, 33)and
even incauliflower mosaicvirus (30), anotherretroelement
in plants. The stoichiometric regulation of capsid andpol gene products and the packaging strategy of pol during morphogenesis of these viruses promise to be rewarding areasofinvestigationin the future.
ACKNOWLEDGMENTS
WethankMarioChen for theconstruction oftheMCvectorfor
dimerization, Richard Greenberg fortheRG6vectorfor
mutagene-sis and mutant 305 as anegative control, and Duanqing Pei for mutant 1314. We also thank Jonathan Israel, Steve Liebhaber, VincentRacaniello,William Mason, JohnTaylor,andcolleagues in
ourlaboratoryfor carefulreading ofthemanuscript.Wearegrateful
toMaliaMcCarthyfor herhelpinpreparation ofthemanuscript.
This work was funded by Public Health Service grants ROl
CA43835 and CA48198 from theNational Institutes of Healthto C.S.
LITERATURECITED
1. Baltimore, D. 1970. Viral RNA-dependent DNA polymerase. Nature(London)226:1209-1211.
2. Bavand, M. R.,and0.Laub. 1988. Twoproteinswithreverse
transcriptase activities associated with hepatitis B virus-like
particles.J. Virol.62:626-628.
3. Buscher, M., W. Reiser, H.Will,and H. Schaller. 1985.
Tran-scripts and the putative RNA pregenome ofduck hepatitis B
virus:implication forreversetranscription.Cell 40:717-724. 4. Chang,L.J.,P. Pryciak,D. Ganem, and H.E. Varmus.1989.
Biosynthesis ofthereversetranscriptase ofhepatitisB viruses
involves de novo translational initiationnot ribosomal
frame-shifting.Nature(London) 337:364-368.
5. Chirgwin, J. M., A. E. Przybyla,R. J. MacDonald, and W.J.
Rutter. 1979. Isolation of biochemically active RNA from sourcesenrichedin ribonuclease. Biochemistry 18:5294-5299. 6. Enders, G.H., D. Ganem,andH. Varmus. 1985. Mappingthe
major transcripts of ground squirrel hepatitis virus: the
pre-sumptive template forreversetranscriptase is terminally
redun-dant. Cell 42:297-308.
7. Ganem, D., and H. E. Varmus. 1987. The molecularbiologyof
thehepatitis Bviruses. Annu. Rev. Biochem. 56:651-693.
8. Goff,S. P.,and L.I.Lobel. 1987. Mutants of murineleukemia
viruses andretroviralreplication. Biochim.Biophys.Acta 907: 93-123.
9. Hann, S. R., M. W. King, D. L. Bentley, C. W. Anderson, and R. N. Eisenman. 1988. A non-AUG translational initiation in c-mycexon1generatesanN-terminally distinctproteinwhose
synthesisisdisruptedinBurkitt'slymphomas.Cell52:185-195.
10. Hirt, B. 1967. Selective extraction of polyoma DNA from infectedmouse cell cultures.J.Mol. Biol. 26:365-369.
11. Inouye,S.,M. Y. Hsu, S.Eagle, and M.Inouye. 1989. Reverse
transcriptase associated withthebiosynthesis ofthebranched
RNA-linked msDNA inMyxococcusxanthus. Cell 56:709-717. 12. Jacks, T.,and H. E.Varmus.1985.Expression ofRoussarcoma
virus pol gene by ribosomal frameshifting. Science
230:1237-1242.
13. Jang, S. K., M. V. Davies, R. J. Kaufman, and E. Wimmer. 1989. Initiation ofprotein synthesis by internal entry of
ribo-somesinto the 5' nontranslatedregionofencephalomyocarditis
virus RNA in vivo. J. Virol. 63:1651-1660.
14. Kadesch, T., andP. Berg. 1986. Effects of theposition of the simian virus 40 enhanceronexpression of multipletranscription
unitsinasingleplasmid. Mol. Cell. Biol. 6:2593-2601.
15. Kaplan,P.M.,R.L.Greenman, J.L.Gerin,R. H.Purcell, and
W.S.Robinson.1973. DNApolymeraseassociated with human
hepatitisBantigen.J. Virol. 12:995-1005.
16. Kozak,M. 1986. Bi-functionalmessenger RNAs ineukaryotes.
Cell 47:481-483.
17. Kozak,M.1989.Thescanningmodelfor translation:anupdate. J.Cell Biol. 108:229-241.
18. Kunkel,T.A.1985.Rapidand efficientsite-specific mutagenesis
without phenotypic selection. Proc. Natl. Acad. Sci. USA 82:488-492.
19. Lien, J.-M., C. E. Aldrich, and W. S. Mason. 1986. Evidence
on November 10, 2019 by guest
http://jvi.asm.org/
EXPRESSION STRATEGY OF HBV REVERSE TRANSCRIPTASE 1069 thata cappedoligoribonucleotide is the primer for duck
hepa-titis B virus plus-strand DNA synthesis. J. Virol.57:229-236. 20. Lien, J.-M., D. J. Petcu, C. E. Aldrich, and W. S. Mason. 1987.
Initiation and termination of duck hepatitis B virus DNA
synthesis during virus maturation. J. Virol. 61:3832-3840. 21. Lim, D., and W. K.Maas. 1989. Reverse
transcriptase-depen-dent synthesis of a covalently linked, branched DNA-RNA compound in E. coli B. Cell 56:891-904.
22. Mack, D. H., W. Bloch, N. Nath, and J. J. Sninsky. 1988. Hepatitis B virus particles contain a polypeptide encoded by the largest open reading frame: a putative reverse transcriptase. J. Virol. 62:4786-4790.
23. Mason, W. S., J. M.Taylor, and R. Hull. 1987. Retroid virus genomereplication. Adv. Virus Res. 32:35-96.
24. Moroy, T., J. Etiemble, C. Treko, P. Tiollais, and M. A. Buendia. 1985. Transcription ofwoodchuck hepatitis virus in thechronicallyinfectedliver. EMBO J. 4:1507-1514.
25. Peabody, D.S. 1987. Translation initiation at an ACG triplet in
mammaliancells. J.Biol. Chem. 262:11847-11851.
26. Peabody, D. S., and P. Berg. 1986. Termination-reinitiation occursinthe translationofmammalian cell mRNAs. Mol. Cell. Biol. 6:2695-2703.
27. Peabody, D. S., S. Subramani, and P. Berg. 1986. Effect of upstreamreading framesontranslation efficiencyinsimian virus
40recombinants. Mol.Cell. Biol.6:2704-2711.
28. Pelletier, J., G. Kaplan, V. R. Racaniello, and N. Sonenberg. 1988. Cap-independenttranslation of poliovirus mRNA is
con-ferred by sequence elements within the 5' noncoding region.
Mol.Cell.Biol. 8:1103-1112.
29. Pelletier, J., and N. Sonenberg. 1988. Internal initiation of translation ofeukaryoticmRNAdirectedby a sequencederived frompoliovirusRNA. Nature (London) 334:320-325. 30. Penswick, J., R.Hubler,and T. Hohn. 1988. Aviablemutation
incauliflower mosaic virus, aretroviruslike plant virus,
sepa-rates its capsid protein and polymerase genes. J. Virol. 62: 1460-1463.
31. Prats, H., M. Kaghad, A. C. Prats, M.Klagsburn,J. M. Lelias, P. Liauzun, P. Chalon, J. P. Tauber, F. Amalric, J. A. Smith, and D. Caput. 1989. High molecular mass forms of basic
fibroblast growth factor are initiated by alternative CUG
codons. Proc.Natl. Acad. Sci. USA86:1836-1840.
32. Sanger, F., A. R. Coulson, B. G. Barrell, A. J. H. Smith, and B.A. Roe. 1980.Cloning in single-stranded bacteriophageas an aid torapid DNA sequencing. J. Mol. Biol. 143:161-178. 33. Schlicht, H. J., G. Radziarill, and H. Schaller. 1989. Synthesis
and encapsidation of duck hepatitis B virus reverse
tran-scriptase do not require formation of core-polymerase fusion proteins. Cell 56:85-92.
34. Shih, C., K. Burke, M.-J. Chou, J. B. Zeldis, C.-S. Yang, C.-S. Lee, K. J. Isselbacher, J. R. Wands, and H. M.Goodman. 1987. Tight clustering ofhuman hepatitisB virusintegrationsites in hepatomas near atriple-stranded region. J.Virol. 61:3491-3498. 35. Shih, C., L. S. Li, S. Roychoudhury, and M. H. Ho. 1989. In vitro propagation of humanhepatitis B virus in a rat hepatoma cell line. Proc. Natl. Acad. Sci. USA86:6323-6327.
36. Simpson, L., and J. Shaw. 1989. RNAeditingand the
mitochon-drialcryptogenesofkinetoplastidprotozoa.Cell 57:355-366.
37. Southern, E. M. 1975. Detection ofspecific sequences among
DNAfragments separated by gelelectrophoresis. J. Mol. Biol.
98:503-517.
38. Summers,J., and W. S. Mason.1982.Replicationof the genome
ofahepatitis B-like virus by reversetranscription ofanRNA
intermediate. Cell29:403-415.
39. Temin, H., and S. Mizutani. 1970. RNA-dependent
DNA-polymerasein virions of Rous sarcoma virus. Nature(London)
226:1211-1213.
40. Thomas, K. R., and M. R. Capecchi. 1986. Introduction of
homologous DNA sequences into mammalian cells induces
mutationsin the cognate gene. Nature (London)324:34-38.
41. Tiollais, P., C. Pourcel, and A. Dejean. 1985. The hepatitis B virus. Nature (London) 317:489-495.
42. Wil, H., W. Reiser, T. Weimer, E. Pfaff, M. Buscher, R. Sprengel, R. Cattaneo, and H. Schaller. 1987.Replication strat-egyofhumanhepatitis Bvirus. J. Virol.61:904-911.
43. Will, H., J. Salfeld, E.Pfaff, C.Manso, L. Thelmann,and H. Schaller. 1986. Putative reversetranscriptase intermediates of human hepatitis B virusin primary livercarcinomas. Science 231:594-596.
44. Yoshinaka, Y.,I.Katoh, T. D.Copeland,and S.Oroszlan. 1985. Murineleukemia virusprotease is encodedbythegag-polgene and issynthesizedthrough suppression ofanambertermination codon. Proc. Natl. Acad. Sci. USA82:1618-1622.
VOL. 64, 1990