Heat Shock Protein 90 Ensures Efficient
Mumps Virus Replication by Assisting
with Viral Polymerase Complex
Formation
Hiroshi Katoh, Toru Kubota, Yuichiro Nakatsu, Maino Tahara, Minoru Kidokoro,
Makoto Takeda
Department of Virology III, National Institute of Infectious Diseases, Tokyo, Japan
ABSTRACT
Paramyxoviral RNAs are synthesized by a viral RNA-dependent RNA
poly-merase (RdRp) consisting of the large (L) protein and its cofactor phosphoprotein
(P protein). The L protein is a multifunctional protein that catalyzes RNA
synthe-sis, mRNA capping, and mRNA polyadenylation. Growing evidence shows that the
sta-bility of several paramyxovirus L proteins is regulated by heat shock protein 90 (Hsp90).
In this study, we demonstrated that Hsp90 activity was important for mumps virus
(MuV) replication. The Hsp90 activity was required for L-protein stability and activity
be-cause an Hsp90-specific inhibitor, 17-allylamino-17-demethoxygeldanamycin (17-AAG),
destabilized the MuV L protein and suppressed viral RNA synthesis. However, once
the L protein formed a mature polymerase complex with the P protein, Hsp90
activ-ity was no longer required for the stabilactiv-ity and activactiv-ity of the L protein. When the
Hsp90 activity was inhibited, the MuV L protein was degraded through the CHIP (C
terminus of Hsp70-interacting protein)-mediated proteasomal pathway. High
concen-trations of 17-AAG showed strong cytotoxicity to certain cell types, but combined
use of an Hsp70 inhibitor, VER155008, potentiated degradation of the L protein,
al-lowing a sufficient reduction of 17-AAG concentration to block MuV replication with
minimum cytotoxicity. Regulation of the L protein by Hsp90 and Hsp70 chaperones
was also demonstrated for another paramyxovirus, the measles virus. Collectively,
our data show that the Hsp90/Hsp70 chaperone machinery assists in the maturation
of the paramyxovirus L protein and thereby in the formation of a mature RdRp
com-plex and efficient viral replication.
IMPORTANCE
Heat shock protein 90 (Hsp90) is nearly universally required for viral
protein homeostasis. Here, we report that Hsp90 activity is required for efficient
propagation of mumps virus (MuV). Hsp90 functions in the maintenance of the
cata-lytic subunit of viral polymerase, the large (L) protein, prior to formation of a mature
polymerase complex with the polymerase cofactor of L, phosphoprotein. Hsp70
col-laborates with Hsp90 to regulate biogenesis of the MuV L protein. The functions of
these chaperones on the viral polymerase may be common among paramyxoviruses
because the L protein of measles virus is also similarly regulated. Our data provide
important insights into the molecular mechanisms of paramyxovirus polymerase
maturation as well as a basis for the development of novel antiviral drugs.
KEYWORDS
heat shock protein 90, large protein, mumps virus, polymerase
M
umps is a common childhood illness characterized by painful swelling of the
parotid glands and is often accompanied by severe complications such as
orchi-tis, aseptic meningiorchi-tis, pancreatiorchi-tis, and deafness (1). Mumps virus (MuV), which
be-longs to the genus
Rubulavirus
of the family
Paramyxoviridae
and the order
Monon-egavirales
, is the causative agent of mumps (2). The viral genome consists of a
Received11 November 2016Accepted21
December 2016
Accepted manuscript posted online4
January 2017
CitationKatoh H, Kubota T, Nakatsu Y, Tahara
M, Kidokoro M, Takeda M. 2017. Heat shock protein 90 ensures efficient mumps virus replication by assisting with viral polymerase complex formation. J Virol 91:e02220-16.
https://doi.org/10.1128/JVI.02220-16.
EditorDouglas S. Lyles, Wake Forest University
Copyright© 2017 American Society for
Microbiology.All Rights Reserved. Address correspondence to Hiroshi Katoh, [email protected].
VIRUS-CELL INTERACTIONS
crossm
March 2017 Volume 91 Issue 6 e02220-16 Journal of Virology jvi.asm.org 1
on November 7, 2019 by guest
http://jvi.asm.org/
nonsegmented, negative-strand RNA approximately 15 kb in length and encodes eight
viral proteins. The genomic RNA of mononegaviruses is encapsidated by the
nucleo-capsid (N) protein, forming the ribonucleonucleo-capsid (NC) (3). The viral RNA-dependent RNA
polymerase (RdRp) binds to NC and forms the viral ribonucleoprotein (RNP) complex,
which acts as an active template for both transcription and genome replication. The
RdRp is composed of two viral proteins, the large (L) protein and its cofactor
phos-phoprotein (P).
The L protein of mononegaviruses harbors the catalytic centers required for RNA
synthesis (4), mRNA capping (5), and polyadenylation (6). Since the L protein alone is
unable to bind efficiently to NC, the P protein positions RdRp on NC (7). Recent studies
have shown that the MuV P protein directly interacts with the N-terminal core region
of the N protein so that the L protein locates in close proximity to the NC (8, 9). In
addition, the P protein of paramyxoviruses, such as Sendai virus, human parainfluenza
virus (HPIV), measles virus (MeV), and Nipah virus (NiV), acts as a chaperone, which
prevents the L protein from aggregation and ensures the proper RdRp formation
(10–12). A similar chaperone-like function of the P protein has been reported for the N
proteins of several mononegaviruses (13, 14). The N and P proteins form a soluble N
0-P
complex that prevents the N protein from aggregation and nonspecific binding to
cellular RNAs, and this complex is used as an encapsidation substrate for the nascent
viral RNA during genome replication. In addition to these coordinated interactions of
the N, P, and L proteins, many other cellular proteins play important roles in
paramyxo-viral RNA synthesis (15–17).
Heat shock protein 90 (Hsp90) is a ubiquitous ATP-dependent molecular chaperone
and plays crucial roles in the folding, maturation, and activation of its client proteins to
maintain cellular homeostasis and survival (18). Systematic analyses have revealed that
Hsp90 associates with 60% of kinases, 7% of transcription factors, and 30% of E3
ubiquitin ligases (19). The chaperoning process of Hsp90 has been studied most
extensively for the steroid hormone nuclear receptors (20). First, Hsp40 and Hsp70
recognize the receptors (21), and then Hsp90 is recruited to the Hsp70-Hsp40-receptor
complex by the cochaperone Hsp70/Hsp90-organizing protein (HOP) (22). The
recep-tors gain hormone-binding activity when ATP binds to Hsp90, replacing the three
cochaperones (Hsp70, Hsp40, and HOP) with another cochaperone, p23 (23). If Hsp90
remains unbound to ATP, the receptors undergo proteasomal degradation. An E3
ubiquitin ligase, the C terminus of Hsp70-interacting protein (CHIP), associates with
Hsp90 or Hsp70, inducing proteasomal degradation of the client proteins of these
chaperones (24).
Many viruses also use Hsp90 for maturation and to maintain the stability of their
viral proteins (25). Hsp90 regulates different stages of the viral life cycle through its
interaction with diverse types of viral proteins, such as polymerase, capsid, and
attach-ment proteins. Hsp90 activity is also required for the capsid assembly of picornaviruses,
including poliovirus, rhinovirus, and coxsackievirus (26). Hsp90 plays a role in the
regulation of viral polymerase activity, rather than protein folding, during herpes
simplex virus (27) and hepatitis B virus infections (28, 29) and is important for proper
cleavage of the newly synthesized hepatitis C virus NS2/3 protein (30) and for RNP
assembly of influenza virus (31, 32). In the cases of mononegavirus infections, the L
protein has been identified as a client protein of Hsp90, and its stability and maturation
are dependent on the Hsp90 chaperone machinery (12, 33–36).
In the present study, the roles of Hsp90 in MuV infection were analyzed. Our data
revealed that Hsp90 activity is required for biogenesis of the MuV L protein and
formation of the functional polymerase complex.
RESULTS
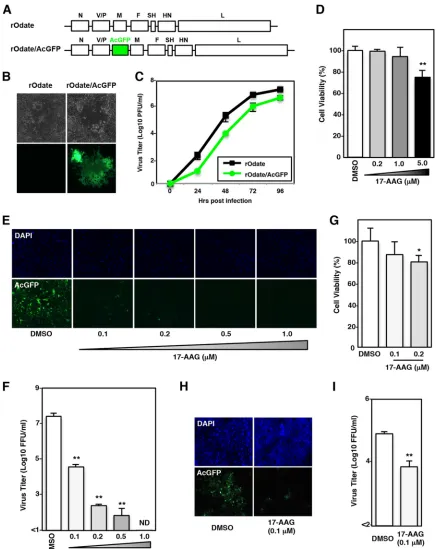
Hsp90 activity is required for MuV propagation.
For sensitive monitoring of MuV
infection in live cells, a recombinant MuV (Odate strain) expressing
Aequorea
coerule-scens
green fluorescent protein (rOdate/AcGFP) was generated. The plasmid
pMuV-Odate/AcGFP was constructed by inserting the AcGFP gene between the V/P and M
on November 7, 2019 by guest
http://jvi.asm.org/
genes in the plasmid pMuV-Odate (Fig. 1A) and used for the rescue of rOdate/AcGFP.
The expression of AcGFP was confirmed in Vero cells infected with rOdate/AcGFP (Fig.
1B). In Vero cells, rOdate/AcGFP replicated less efficiently than the parental rOdate, but
the virus titer reached as high as
⬃
10
7PFU/ml at 96 h postinfection (hpi) (Fig. 1C). To
examine whether Hsp90 activity is required for MuV propagation, an Hsp90 inhibitor,
17-allylamino-17-demethoxygeldanamycin (17-AAG), was used for analyses. First, the
toxicity of 17-AAG to Vero cells was analyzed. Cell viability was not affected significantly
by 17-AAG at concentrations of up to 1.0
M (Fig. 1D). Therefore, in further experiments
using Vero cells, 17-AAG was used at concentrations of no higher than 1.0
M. Vero
cells were infected with rOdate/AcGFP at a multiplicity of infection (MOI) of 0.01 and
incubated for 96 h at various concentrations of 17-AAG. MuV propagation estimated by
AcGFP signals (Fig. 1E) and virus titers in the culture supernatants (Fig. 1F) were
significantly reduced by 17-AAG (Fig. 1E and F). Similar results were obtained when
A549 cells were used. The viability of A549 cells was not affected significantly by
17-AAG at a concentration of 0.1
M (Fig. 1G), while MuV propagation was reduced
significantly by 17-AAG at the same concentration (Fig. 1H and I). These data indicated
that Hsp90 activity was important for MuV propagation in cultured cells.
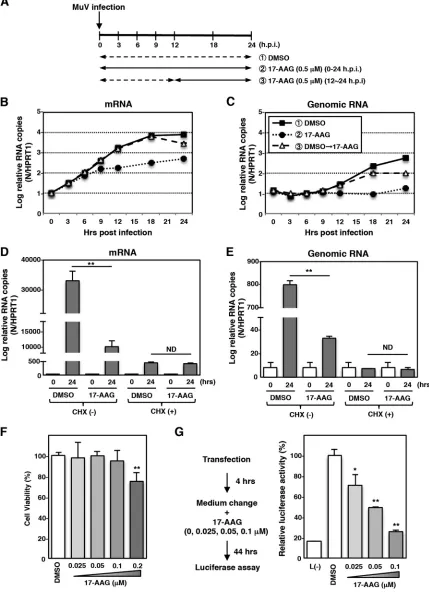
Hsp90 activity is required for MuV RNA synthesis.
The importance of Hsp90
activity in viral RNA synthesis was analyzed. Vero cells were infected with MuV and cultured
in the presence (0.5
M) of 17-AAG or in dimethyl sulfoxide (DMSO) without 17-AAG (Fig.
2A to C). At 0, 3, 6, 9, 12, 18, and 24 hpi, total RNA was extracted, and the levels of N mRNA
and genomic RNA were determined by quantitative reverse transcription-PCR (qRT-PCR).
When Vero cells were cultured in the absence of 17-AAG (Fig. 2A to C, DMSO), the N mRNA
level increased rapidly shortly after the virus infection (Fig. 2B), while the genomic RNA level
remained unchanged until 9 hpi (Fig. 2C). These data suggested that until 9 hpi RdRp was
devoted to transcription but not genome replication. As shown in Fig. 2B, the reduction of
the N mRNA level in 17-AAG-treated cells was evident at 9 hpi, suggesting that Hsp90
activity was important for viral RNA transcription. Although the genomic RNA level
re-mained unchanged until 9 hpi, it increased significantly at 12 hpi (Fig. 2C). These data
indicated that genome replication started before 12 hpi. Vero cells were cultured in the
absence of 17-AAG for 12 h to avoid any effect on transcription prior to genome replication.
Then, during the following 12 h (between 12 and 24 hpi), cells were cultured in the
presence of 0.5
M 17-AAG (Fig. 2A to C, DMSO
¡
17-AAG). Under this experimental
condition, the transcription level was not affected until 18 hpi (Fig. 2B). Nevertheless, the
genomic RNA level was already reduced at 18 hpi (Fig. 2C). These data suggested that
Hsp90 activity was important for not only viral mRNA transcription but also genome
replication. The effect of 17-AAG on MuV RNA synthesis was also analyzed in the presence
of a protein synthesis inhibitor, cycloheximide (CHX). Under the condition in which cells
were treated with CHX, neither the viral mRNA level nor the genomic RNA level was
affected by 17-AAG treatment (Fig. 2D and E), indicating that Hsp90 activity was not
required for MuV primary transcription. To further confirm the requirement of Hsp90 for
viral RNA synthesis, an MuV minigenome assay was performed. The viability of BHK/T7-9
cells used for the minigenome assay was not affected by 17-AAG at concentrations of up
to 0.1
M (Fig. 2F). In the range of 0 to 0.1
M, luciferase expression was reduced in a
dose-dependent manner by 17-AAG (Fig. 2G). Collectively, these results suggested that
Hsp90 activity was important for MuV RNA synthesis conducted by
de novo
-synthesized
polymerases.
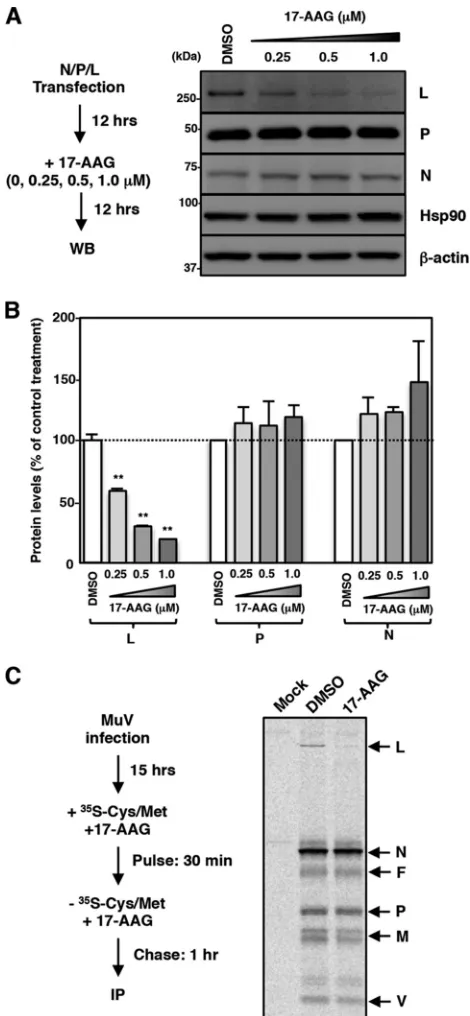
MuV L protein is an Hsp90 client protein.
Many Hsp90 client proteins are
destabilized when Hsp90 is dysfunctional (37). Next, therefore, we examined the effects
of 17-AAG on the expression levels of the N, P, and L proteins. The N, P, and L proteins
were expressed using expression plasmids. 17-AAG decreased the amount of L protein
in a dose-dependent manner, while the amounts of the N and P proteins were
unchanged (Fig. 3A and B). To further evaluate the change in the amount of the L
protein as a result of 17-AAG treatment, a pulse-chase experiment was performed. After
incubation with MuV for 15 h, Vero cells were radiolabeled for 30 min, and proteins in
Hsp90 Is Required for MuV Polymerase Formation Journal of Virology
March 2017 Volume 91 Issue 6 e02220-16 jvi.asm.org 3
on November 7, 2019 by guest
http://jvi.asm.org/
FIG 1Hsp90 activity is required for MuV propagation. (A) Schematic of the rOdate and rOdate/AcGFP genes. SH, small hydrophobic gene. (B) Vero cells infected with rOdate or rOdate/AcGFP were observed under phase-contrast and a fluorescence microscopes at 48 hpi. (C) Vero cells were infected with rOdate or rOdate/AcGFP at an MOI of 0.01. The supernatants were collected at 24, 48, 72, and 96 hpi, and the infectious titers were determined by plaque assay. (D) Vero cells were treated with the indicated concentrations of 17-AAG for 96 h, and then cell viability was determined and calculated as a percentage of the viability of cells treated with DMSO. (E and F) Vero cells were infected with rOdate/AcGFP at an MOI of 0.01 and treated with the indicated concentrations of 17-AAG. At 96 hpi, the cells were observed under a fluorescence microscope (E), and the infectious titers in the supernatants were determined (F). (G) A549 cells were treated with 0.1 and 0.2M 17-AAG for 96 h, and then cell viability was determined and calculated as a percentage of the viability of cells treated with DMSO. (H and I) A549 cells were infected with rOdate/AcGFP at an MOI of 0.01 and treated with 0.1M 17-AAG. At 96 hpi, the cells were observed under a fluorescence microscope (H), and the infectious titers in the supernatants were determined (I). Error bars indicate the standard deviations of triplicates. The significance of differences between the means was determined by Student’sttest.*,P⬍0.05;**, P⬍0.01. ND, not detected; DAPI, 4=,6=-diamidino-2-phenylindole.
on November 7, 2019 by guest
http://jvi.asm.org/
[image:4.585.39.476.69.618.2]FIG 2Hsp90 activity is required for MuV RNA synthesis after the secondary transcription phase. (A) Schematic of the experimental procedures showing the times of RNA collection and 17-AAG treatment after virus infection. (B and C) Vero cells were infected with MuV at an MOI of 5.0. At the indicated times postinfection, total cellular RNA was extracted and subjected to an RT reaction using oligo(dT) and strand-specific primers. The levels of N mRNA and genomic RNA were determined by qPCR. The values were normalized to the level of the control gene HPRT1 and are shown as the log fold change. Three independent experiments were performed, and the most representative data are shown. (D and E) Vero cells were infected with MuV at an MOI of 5.0 and cultured with or without 50g/ml of CHX and 0.5M 17-AAG or DMSO. At 0 and 24 hpi, total cellular RNA was extracted and subjected to an RT reaction using oligo(dT) and
(Continued on next page)
Hsp90 Is Required for MuV Polymerase Formation Journal of Virology
March 2017 Volume 91 Issue 6 e02220-16 jvi.asm.org 5
on November 7, 2019 by guest
http://jvi.asm.org/
[image:5.585.41.470.77.670.2]the cells were chased for 1 h in the presence or absence of 17-AAG. Viral proteins were
then immunoprecipitated with a polyclonal antibody raised against whole MuV virions.
All viral proteins except for the hemagglutinin-neuraminidase (HN) protein were
pre-cipitated and analyzed by SDS-PAGE and autoradiography. The levels of the N, P, V, M,
and F proteins were unchanged by 17-AAG, while the level of the L protein was
reduced, suggesting that the L protein was specifically destabilized by 17-AAG (Fig. 3C).
These data demonstrated that the MuV L protein is a client protein of Hsp90.
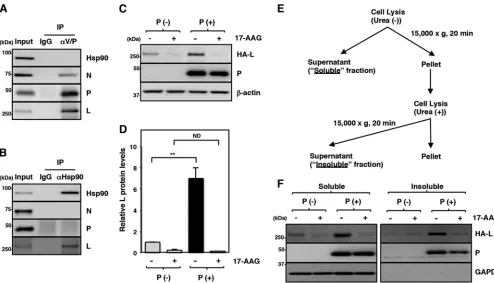
The maturation of the L protein is regulated by Hsp90 and the P protein.
The
interaction between Hsp90 and the N, P, and L proteins in MuV-infected cells was
analyzed. When the P protein was immunoprecipitated by an antibody targeting the
amino terminus of the P protein, the N and L proteins were coimmunoprecipitated with
the P protein, suggesting that the precipitated protein complexes contain RNP
com-plexes (Fig. 4A). However, since this antibody also binds to the V protein, which has the
same amino terminus sequence as the P protein, involvement of the V protein cannot
be excluded. Although all the N, P, and L proteins, probably in the form of RNP
complexes, were precipitated in this assay, Hsp90 was not coimmunoprecipitated with
these proteins (Fig. 4A). Then, Hsp90 in MuV-infected cells was immunoprecipitated
with an anti-Hsp90 antibody, and the levels of N, P, and L proteins coprecipitated with
Hsp90 were analyzed by immunoblotting. In this assay, the L protein was specifically
detected in the immunoprecipitated complexes (Fig. 4B). These data suggested that a
free form of the L protein, but not the L protein in RNP complexes, interacts with Hsp90.
Previous studies of paramyxoviruses have shown that the P protein is required for
proper folding of the L protein and prevention of L-protein aggregation (10–12). In the
present study, therefore, we used expression plasmids to express the MuV L protein
alone or together with the MuV P protein. A hemagglutinin (HA)-tagged L protein was
used in further experiments for higher detection sensitivity. Cells transfected with the
L-protein expression plasmid with or without the P-protein expression plasmid were
lysed and centrifuged, and the amount of the L protein in the soluble fraction was
analyzed. The data showed that the amount of the L protein in the soluble fraction was
increased when the L and P proteins were expressed together (Fig. 4C and D),
suggesting that the MuV P protein also functions as a chaperone for the MuV L protein,
as shown for other paramyxoviruses (10–12). Therefore, both Hsp90 and the P protein
contributed to the stability of the L protein.
Our data demonstrated that the level of L protein detected in the soluble fraction
in our assays was reduced in the absence of the P protein or when the Hsp90 activity
was inhibited. These results may be due to degradation of the L protein or aggregation
in the insoluble fraction. To clarify this, the cells were divided into soluble and insoluble
fractions (Fig. 4E), and the amounts of the L protein in both fractions were analyzed. In
the absence of the P protein, the amounts of the L protein in both the soluble and
insoluble fractions were reduced compared with those in the presence of the P protein
(Fig. 4F). In addition, the amounts of the L protein in both the soluble and insoluble
fractions also declined with 17-AAG treatment. These data suggested that the L protein
was degraded but not aggregated when Hsp90 was dysfunctional or when the P
protein was absent.
Hsp90 is dispensable for maintenance and function of the P/L polymerase
complex.
Our data demonstrated that Hsp90 did not interact with the L protein once
the L protein formed the P/L polymerase complex. The roles of Hsp90 for the stability
and functions of the P/L polymerase complex were further studied. Using expression
FIG 2Legend (Continued)
strand-specific primers. The levels of N mRNA and genomic RNA were determined by qPCR. The values were normalized to the value for the control gene HPRT1. (F) BHK/T7-9 cells were treated with the indicated concentrations of 17-AAG for 48 h, and then cell viability was determined and calculated as a percentage of the viability of cells treated with DMSO. (G) BHK/T7-9 cells were transfected with the minigenome plasmid (pFL-MuV-MG) and three support plasmids (pCR-N, -P, and -L). At 4 h posttransfection, the medium was replaced with fresh medium containing 17-AAG, as indicated in the figure. pCR-L was omitted from the transfection mixture to establish a negative control. The luciferase activity in DMSO-treated cells was set to 100%. Error bars indicate the standard deviations of triplicates. The significance of differences between the means was determined by Student’sttest.*,P⬍0.05;**,P⬍0.01. ND, no significant difference.
on November 7, 2019 by guest
http://jvi.asm.org/
plasmids, the L protein was expressed in 293T cells with or without the P protein. At
23.5 h posttransfection,
de novo
synthesis of the L and P proteins was blocked by CHX,
and cells were cultured for an additional 30 min to ensure the maturation of the L
protein and the formation of the P/L polymerase complex (Fig. 5A). Then, the cells were
FIG 3MuV L protein is a client of Hsp90. (A and B) 293T cells were cotransfected with three plasmids, pCAGGS-N, -P, and -L, and the medium was replaced with fresh medium containing the indicated concen-trations of 17-AAG at 12 h posttransfection. At 24 h posttransfection, the cell lysates were subjected to immunoblotting with the indicated antibodies (A). The relative band intensities of each viral protein were normalized by the-actin level, and the relative expression levels of each viral protein based on the levels of DMSO-treated cells are shown in panel B. (C) Vero cells were infected with MuV at an MOI of 5.0. At 15 hpi, the cells were radiolabeled for 30 min with [35S]methionine-cysteine and incubated in nonradioactive
medium for 1 h before lysis and analysis by immunoprecipitation with an anti-MuV antibody. 17-AAG was incubated at 0.5M during the pulse and chase periods. WB, Western blotting; IP, immunoprecipitation.
Hsp90 Is Required for MuV Polymerase Formation Journal of Virology
March 2017 Volume 91 Issue 6 e02220-16 jvi.asm.org 7
on November 7, 2019 by guest
http://jvi.asm.org/
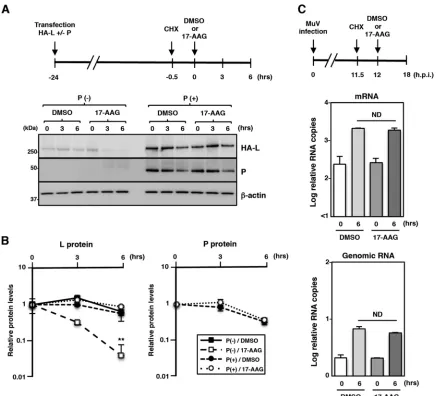
[image:7.585.87.322.70.578.2]cultured in the presence or absence of 17-AAG to assess the stability of the L protein
in the P/L polymerase complex. In the presence of 17-AAG, the L protein was degraded
rapidly when expressed alone (Fig. 5A and B). In contrast, the L protein complexed with
the P protein remained mostly stable under the 17-AAG treatment (Fig. 5A and B).
Our data suggest that once the L protein forms the P/L polymerase complex, Hsp90
activity is no longer required for L protein stability. Next, the role of Hsp90 in the P/L
polymerase functions was evaluated. Vero cells were infected with MuV, and
de novo
protein synthesis was blocked at 11.5 hpi. The cells were further cultured for 30 min and
maintained in the presence or absence of 17-AAG for 6 h (between 12 and 18 hpi) (Fig.
5C). Then, the levels of viral N mRNA and genomic RNA in the cells at 12 and 18 hpi
were determined to assess viral RNA synthesis by the preexisting P/L polymerase
complexes during the 6-h incubation period. The levels of viral RNAs (both N mRNA and
genomic RNA) were similar in 17-AAG-treated and untreated cells (Fig. 5C). Therefore,
we concluded that the viral polymerase activity was not affected by 17-AAG. Together,
these findings demonstrated that Hsp90 was no longer required for the stability and
polymerase activity of the L protein once the L protein formed the P/L polymerase
complex.
Hsp90 dysfunction induces proteasomal degradation of the L protein
medi-ated by CHIP.
17-AAG blocks Hsp90 chaperone activity by interfering with the ATP
hydrolysis cycle of Hsp90 and induces proteasomal degradation of misfolded Hsp90
clients. Hsp70 and CHIP are components of an Hsp90 multichaperone complex and are
responsible for ubiquitination and degradation of certain misfolded Hsp90 clients (38).
We therefore examined the involvement of Hsp70 and CHIP in L-protein degradation by
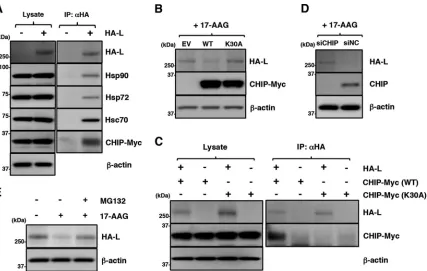
17-AAG. First, interaction of the L protein with Hsp70 and CHIP was analyzed. Hsp72 (a
FIG 4The maturation of the L protein is sequentially regulated by Hsp90 and the P protein. (A and B) At 24 hpi, Vero cells infected with MuV at an MOI of 5.0 were subjected to immunoprecipitation with anti-MuV V/P or anti-Hsp90 antibody and immunoblotting with the indicated antibodies. (C and D) 293T cells were transfected with pCAGGS-HA-MuV-L alone or cotransfected with pCAGGS-HA-MuV-L and pCAGGS-P, and the medium was replaced with fresh medium containing 0.5M 17-AAG at 12 h posttransfection. At 24 h posttransfection, the cell lysates were prepared using a urea-free cell lysis buffer and subjected to immunoblotting with the indicated antibodies. The relative band intensities of L protein were normalized to the-actin level, and the relative expression levels of L protein based on the levels of DMSO-treated cells without P are shown (D). (E and F) At 24 h posttransfection, soluble and insoluble fractions of the cells were prepared as indicated in panel E and subjected to immunoblotting with the antibodies indicated in panel F. Error bars indicate the standard deviations of triplicates. The significance of differences between the means was determined by Student’sttest.**,P⬍0.01. ND, no significant difference.
on November 7, 2019 by guest
http://jvi.asm.org/
[image:8.585.42.536.69.352.2]stress-inducible Hsp70 isoform), Hsc70 (a constitutively expressed Hsp70 isoform), and
CHIP were coimmunoprecipitated with the L protein (Fig. 6A). The findings indicated
that the L protein interacted with these chaperone and cochaperone proteins.
How-ever, the results did not differentiate between independent interactions of the L protein
with each chaperone or cochaperone protein and a multimeric interaction of the L
protein with these chaperone and cochaperone proteins in the same complex.
The involvement of CHIP in L-protein degradation was examined further. CHIP is an
E3 ubiquitin ligase and interacts with both Hsp90 and Hsp70 via its amino-terminal
tetratricopeptide repeat (TPR) domain (38). Both the wild-type (WT) CHIP and a mutant
CHIP carrying a K30A substitution in the TPR domain (K30A CHIP) (39) were coexpressed
with the L protein. The expression of the WT CHIP, but not the K30A CHIP, further
reduced the expression level of the L protein, suggesting that the WT CHIP promoted
L-protein degradation (Fig. 6B). Immunoprecipitation assays detected the WT CHIP but
FIG 5Hsp90 is dispensable for stability of the L protein and the polymerase activity after an interaction with the P protein. (A and B) 293T cells were transfected with pCAGGS-HA-MuV-L alone or cotransfected with pCAGGS-HA-MuV-L and pCAGGS-P. At 23.5 h posttransfection, the cells were treated with 50g/ml CHX for 30 min. Then, 0.5M 17-AAG was added to the cells, and the cell lysates were collected at the indicated times and subjected to immunoblotting. The relative band intensities of L and P proteins were normalized to the-actin level, and the relative expression levels of each viral protein based on the levels of cells collected at 0 h are shown in panel B. (C) Vero cells were infected with MuV at an MOI of 5.0. At 11.5 hpi, the cells were treated with 50g/ml of CHX for 30 min. Then, 0.5M 17-AAG was added to the cells, and total cellular RNA was extracted at the indicated times and subjected to an RT reaction using oligo(dT) and strand-specific primers. The levels of N mRNA and genomic RNA were determined by qPCR, and the values were normalized to the level of the control gene HPRT1 and shown as log fold changes. Error bars indicate the standard deviations of triplicates. The significance of differences between the means was determined by Student’sttest.**,P⬍0.01. ND, no significant difference.
Hsp90 Is Required for MuV Polymerase Formation Journal of Virology
March 2017 Volume 91 Issue 6 e02220-16 jvi.asm.org 9
on November 7, 2019 by guest
http://jvi.asm.org/
[image:9.585.40.476.69.465.2]not the K30A CHIP in the complexes precipitated by a specific antibody targeting the
HA tag of the L protein (Fig. 6C). In addition, CHIP knockdown increased the level of L
protein (Fig. 6D). These data revealed that CHIP functioned as part of the chaperone
complex and was involved in the degradation of the L protein. Finally, 17-AAG-induced
L-protein degradation was canceled by treatment with a proteasome inhibitor, MG-132
(Fig. 6E).
Single treatment of an Hsp70 inhibitor does not affect MuV propagation or
L-protein expression.
The roles of Hsp70 in MuV propagation and L-protein expression
were analyzed by using an Hsp70 inhibitor, VER155008, which targets the
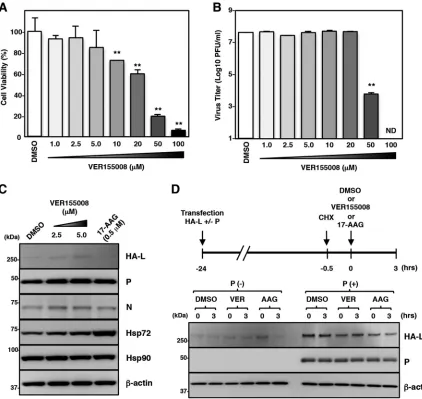
ATPase-binding domain of Hsp70. Vero cells were infected with rOdate/AcGFP and incubated
for 96 h with various concentrations of VER155008. Virus production was not affected
by VER155008 at concentrations of up to 20
M but was suppressed at concentrations
of 50 and 100
M. However, VER155008 exhibited strong cytotoxicity at these
concen-trations (Fig. 7A and B). Our data also showed that the expression level of the L protein
as well as the levels of the N and P proteins were unchanged by VER155008 (Fig. 7C).
Finally, VER155008 did not affect the stability of either the free or complex forms of the
L protein (Fig. 7D).
An Hsp70 inhibitor potentiates degradation of the MuV L protein under Hsp90
inhibitor treatment.
Previous studies have shown that the inhibition of Hsp70
activity potentiates proteasome-dependent degradation of Hsp90 client proteins
(40, 41). Therefore, the effects of the combined use of VER155008 and 17-AAG on
L-protein expression and MuV propagation were analyzed. VER155008 accelerated
the 17-AAG-induced degradation of the L protein (Fig. 8A and B). The combined use
FIG 6Disruption of Hsp90 function induces degradation of the MuV L protein through a CHIP-mediated proteasomal pathway. (A) 293T cells were transfected with pcDNA-CHIP-Myc/His (WT) and/or pCAGGS-HA-MuV-L. At 24 h posttransfection, the cell lysates were subjected to immunoprecipitation with anti-HA antibody and to immunoblotting with the indicated antibodies. (B) 293T cells were cotransfected with pCAGGS-HA-MuV-L and pcDNA-CHIP-Myc/His (WT or K30A). At 24 h posttransfection, the cells were treated with 0.5M 17-AAG for 4 h, and then the cell lysates were subjected to immunoblotting with the indicated antibodies. EV, empty vector. (C) HA-MuV-L was coexpressed with CHIP-Myc (WT or K30A) in 293T cells, immunoprecipitated with anti-HA antibody, and immunoblotted with the indicated antibodies. (D) At 48 h posttransfection with either siCHIP or siNC, 293T cells were transfected with pCAGGS-HA-MuV-L. After 24 h, the cells were treated with 0.5M 17-AAG for 4 h, and then the cell lysates were subjected to immunoblotting with the indicated antibodies. (E) 293T cells were cotransfected with pCAGGS-HA-MuV-L and pcDNA-CHIP-Myc/His (WT). At 24 h posttransfection, the cells were treated with 0.5M 17-AAG and/or 10M MG-132 for 4 h, and then the cell lysates were subjected to immunoblotting with anti-HA and anti--actin antibodies.
on November 7, 2019 by guest
http://jvi.asm.org/
[image:10.585.47.474.69.340.2]of VER155008 and 17-AAG did not affect the levels of other viral proteins (N, P, V,
M, and F proteins) (Fig. 8C). The stability of the complex form of the L protein was
also unchanged (Fig. 8D).
The effects of the combined use of VER155008 and 17-AAG on MuV infection were
also analyzed. The viability of Vero cells was not significantly affected by VER155008 at
concentrations of up to 5.0
M in combination with 0.2
M 17-AAG (Fig. 8E). However,
VER155008 additively inhibited virus production when it was used in combination with
17-AAG (Fig. 8F). To analyze the effects of this combination on viral RNA synthesis, Vero
cells were infected with MuV and cultured with 5.0
M VER155008 singly or in
combination with 0.2 or 0.5
M 17-AAG. The levels of N mRNA and genomic RNA were
not affected by VER155008 when VER155008 was used singly (Fig. 8G, DMSO). However,
VER155008 additively inhibited viral RNA synthesis when used in combination with
17-AAG (Fig. 8G). Similar results were obtained when A549 cells were used (Fig. 8H and
FIG 7An Hsp70 inhibitor does not affect MuV propagation or the L protein expression. (A) Vero cells were treated with the indicated concentrations of VER155008 for 96 h, and then cell viability was determined and calculated as a percentage of the viability of cells treated with DMSO. (B) Vero cells were infected with rOdate/AcGFP at an MOI of 0.01 and treated with the indicated concentrations of VER155008. At 96 hpi, the infectious titers in the supernatants were determined. (C) 293T cells were cotransfected with three plasmids, pCAGGS-N, -P, and -L, and the medium was replaced with fresh medium containing 2.5 or 5.0M VER155008 at 12 h posttransfection. At 24 h posttransfection, the cell lysates were subjected to immunoblotting with the indicated antibodies. (D) 293T cells were transfected with pCAGGS-HA-MuV-L alone or cotransfected with pCAGGS-HA-MuV-L and pCAGGS-P. At 23.5 h posttransfection, the cells were treated with 50g/ml CHX for 30 min. Then, 5.0M VER155008 (VER) or 0.5M 17-AAG (AAG) was added to the cells for 3 h, and the cell lysates were collected and subjected to immunoblotting. Error bars indicate the standard deviations of triplicates. The significance of differences between the means was determined by Student’sttest.**,P⬍0.01. ND, not detected.
Hsp90 Is Required for MuV Polymerase Formation Journal of Virology
March 2017 Volume 91 Issue 6 e02220-16 jvi.asm.org 11
on November 7, 2019 by guest
http://jvi.asm.org/
[image:11.585.43.465.71.470.2]FIG 8The Hsp70 inhibitor potentiates degradation of the MuV L protein under Hsp90 inhibitor treatment. (A and B) 293T cells were cotransfected with pCAGGS-HA-MuV-L and pCAGGS-P, and the medium was replaced with fresh medium containing 0.5 M 17-AAG and/or 5.0 M VER155008 at 12 h posttransfection. At 24 h posttransfection, the cell lysates were subjected to immunoblotting (A) with the indicated antibodies. The relative band intensities of HA-L protein were normalized to the-actin level, and the relative expression levels of L protein based on the levels of DMSO-treated cells are shown in panel B. D, DMSO; V, VER155008; A, 17-AAG; V⫹A, VER155008 and 17-AAG. (C) Vero cells were infected with MuV at an MOI of 5.0. At 15 hpi, the cells were radiolabeled for 30 min with [35S]methionine-cysteine and incubated in nonradioactive medium for 1 h before lysis and analysis by immunoprecipitation with
an anti-MuV antibody. 17-AAG (0.5M) and/or VER155008 (5.0M) was incubated during the pulse and chase periods. (D) 293T cells were transfected with pCAGGS-HA-MuV-L alone or cotransfected with pCAGGS-HA-MuV-L and pCAGGS-P. At 23.5 h posttransfection, the cells were treated with 50g/ml of CHX for 30 min. Then, 17-AAG (0.5M) and/or VER155008 (5.0M) was added to the cells for 3 h, and the cell lysates were collected and subjected to immunoblotting. AAG,17-AAG; AAG/VER, 17-AAG and VER155008. (E) Vero cells were treated with the indicated concentrations of VER155008 with/without 0.2M 17-AAG for 96 h, and then cell viability was determined and calculated as a percentage of the viability of cells treated with DMSO alone. (F) Vero cells were infected with rOdate/AcGFP at an MOI of 0.01 and treated with the indicated concentrations of VER155008 with/without 0.2M 17-AAG. At 96 hpi, the infectious titers in the supernatants were determined. (G) Vero cells were infected with MuV at an MOI of 5.0 and treated with 5.0M VER155008 and the indicated concentrations of 17-AAG. At 24 hpi, total cellular RNA was extracted and subjected to an RT reaction using oligo(dT) and strand-specific primers. The levels of N mRNA and genomic RNA were determined by qPCR, and the values were normalized to the value for the control gene HPRT1 and are shown as log fold changes. (H) A549 cells were treated with 5.0M VER155008 with/without 0.1M 17-AAG for 96 h, and then cell viability was determined and calculated as a percentage of the viability of cells treated with DMSO alone. (I) A549 cells were infected with rOdate/AcGFP at an MOI of 0.01 and treated with 5.0M VER155008 with/without 0.1M 17-AAG. At 96 hpi, the infectious titers in the supernatants were determined. Error bars indicate the standard deviations of triplicates. The significance of differences between the means was determined by Student’sttest.*,P⬍0.05;**,P⬍0.01. ND, no significant difference.
on November 7, 2019 by guest
http://jvi.asm.org/
[image:12.585.41.538.70.558.2]I). Importantly, 17-AAG at the high concentrations (0.2
M or higher) required when it
was used singly to block MuV propagation showed strong cytotoxicity to A549 cells
(Fig. 1G), but when 17-AAG was applied in combination with VER155008, 17-AAG could
be used at a much lower concentration with minimal cytotoxicity and still block MuV
propagation.
Hsp70 and CHIP are also associated with measles virus (MeV) L protein and
involved in MeV propagation.
We next analyzed whether the involvement of Hsp70
and CHIP in the maturation and degradation of the L protein was common among
other paramyxoviruses. Consistent with a previous study (12), we found that the MeV
L protein was also degraded by 17-AAG (Fig. 9A). The immunoprecipitation assay
showed that Hsp90, Hsp72, and CHIP were associated with the MeV L protein (Fig. 9B).
The effects of 17-AAG and VER155008 on MeV were similar to or even greater than
those on MuV. Vero/hSLAM cells were infected with MeV at an MOI of 0.01 and cultured
with various concentrations of VER155008 alone or in combination with 0.2
M
17-AAG. Virus titers in the culture supernatants were determined at 72 hpi (Fig. 9C).
Even when used singly, 17-AAG reduced the virus titer by
⬃
100-fold (Fig. 9C). On the
other hand, VER155008 showed no effect on virus titers when used singly. However,
when used in combination with 17-AAG, VER155008 effectively inhibited MeV
produc-FIG 9Hsp70 and CHIP are also associated with measles virus (MeV) L protein and involved in MeV infection. (A) 293T cells were transfected with pCAGGS-HA-MeV-L, and the medium was replaced with fresh medium containing 0.5M 17-AAG at 12 h posttransfection. At 24 h posttransfection, the cell lysates were subjected to immunoblotting with anti-HA and anti--actin antibodies. (B) 293T cells were transfected with pcDNA-CHIP-Myc/His (WT) and/or pCAGGS-HA-MeV-L. At 24 h posttransfection, the cell lysates were subjected to immunoprecipitation with anti-HA antibody and to immunoblotting with the indicated antibodies. (C) Vero/hSLAM cells were infected with IC323-EGFP at an MOI of 0.01 and treated with the indicated concentrations of VER155008 with/without 0.2M 17-AAG. At 72 hpi, the infectious titers in the supernatants were determined. Error bars indicate the standard deviations of triplicates. ND, not detected.
Hsp90 Is Required for MuV Polymerase Formation Journal of Virology
March 2017 Volume 91 Issue 6 e02220-16 jvi.asm.org 13
on November 7, 2019 by guest
http://jvi.asm.org/
[image:13.585.86.326.67.400.2]tion, and the virus titers were reduced to an undetectable level (Fig. 9C). These data
demonstrated that the regulation of L-protein biogenesis by Hsp90/Hsp70/CHIP was
conserved in certain paramyxoviruses.
DISCUSSION
Hsp90 has been shown to play a role in the propagation of many viruses, including
both DNA and RNA viruses (25). The data in the present study showed that Hsp90
dysfunction suppressed MuV RNA synthesis and viral production in cultured cells.
Importantly, the viral polymerase L protein was destabilized in cells treated with
17-AAG. 17-AAG disrupts the Hsp90 chaperone activity by binding to the ATP-binding
pocket of Hsp90 and subsequently induces proteasomal degradation of misfolded
Hsp90 client proteins. Therefore, our data indicated that the MuV L protein was a client
protein of Hsp90. Our data for MuV were in line with previous reports on other
monon-egaviruses (12, 33–36). Thus, the requirement of Hsp90 chaperone activity for polymerase
stability seems to be conserved among mononegavirus members (12, 33–36).
Some Hsp90 client proteins, referred to as “weak clients,” associate transiently with
Hsp90 during maturation. Once such client proteins have achieved their mature
folding, their stability and functions no longer rely on Hsp90. In contrast, other “strong
clients” repeatedly access the Hsp90-susceptible conformation after their initial folding
(42). Recently, Bloyet et al. demonstrated that Hsp90 is not required for maintenance of
the P/L polymerase complexes of MeV and NiV once they have been assembled (12).
Much as for the L proteins of MeV and NiV, Hsp90 activity was not required for the
stability and polymerase functions of the MuV L protein when the L protein interacted
with the P protein. Our observation that the primary transcription performed by
preformed polymerases contained in virions was not dependent on Hsp90 activity was
also consistent with the notion that the MuV L protein is a weak client of Hsp90.
The paramyxoviral P protein contributes to stabilization of the L protein (10–12).
Without the P protein, the L proteins of MeV and HPIV type 3 undergo aggregation into
the insoluble forms (11, 12). On the other hand, the amounts of the MuV L protein in
both the soluble and insoluble fractions were decreased in the absence of the MuV P
protein. These data suggested that the MuV P protein functioned as a chaperone for
the L protein to prevent degradation but not aggregation. There may be some
functional differences among the paramyxoviral P proteins in the process of the
polymerase formation.
In eukaryotic cells, more than 20 cochaperones, including Hsp70, Hsp40, HOP, CHIP,
Cdc37, and p23, have been identified for Hsp90 (18). Although the biological roles of
these cochaperones remain to be fully elucidated, some of them are known to guide
the recognition of client proteins and modulate the biological activities of Hsp90 (18).
CHIP is an E3 ubiquitin ligase that mediates the ubiquitination and degradation of
various chaperone-bound proteins through its interaction with Hsp70 or Hsp90 (43).
Our data indicated that CHIP was a part of the chaperone complex of the MuV L protein
and promoted L-protein degradation via a proteasome pathway when Hsp90 was
dysfunctional. The requirement of the chaperone-binding TPR domain in CHIP for the
complex formation with the L protein also supports the function of CHIP as an
Hsp90-cochaperone in the biogenesis of the L protein.
In the present study, Hsp70 was also identified as a binding partner of the MuV
and MeV L proteins. However, an Hsp70 inhibitor, VER155008, had little effect on
L-protein expression and MuV and MeV propagation when used alone. In the
case of RSV infection, VER155008 reduces virus polymerase activity (35), indicating
that the paramyxovirus family members show different levels of sensitivity to
VER155008. Hsp70 is a ubiquitously expressed multifunctional chaperone. In addition,
when Hsp90 is dysfunctional, Hsp70 is induced and compensates for the loss of Hsp90
function by acting as a cochaperone for Hsp90 (40, 41). Our data showed that the level
of Hsp70 expression was increased by 17-AAG (Fig. 7C and 8A). Moreover, the effects
of VER155008 became evident only when Hsp90 activity was inhibited. These results
suggest that Hsp70 functions as a cochaperone of Hsp90 for L protein maturation.
on November 7, 2019 by guest
http://jvi.asm.org/
Collectively, these findings indicate that biogenesis of the paramyxovirus L proteins
was regulated by the Hsp90/Hsp70/CHIP chaperone system.
Although certain viral polymerases do not require Hsp90 activity (26), the stability
and activity of many viral polymerases, including the RdRp of RNA viruses (31, 33), DNA
polymerase of DNA viruses (27), and reverse transcriptase of hepadnaviruses (28, 29),
depend on Hsp90. Despite the evolutionary, structural, and functional divergence of
these viruses, the general dependence of viral polymerases on Hsp90 suggests that the
chaperones may recognize a conserved feature of the viral polymerases, thereby
assisting with a common step in their maturation. To date, the molecular basis of client
recognition by Hsp90 remains to be fully understood. However, increasing evidence
suggests that recognition by Hsp90 is related to the conformation or stability of the
client protein rather than the primary amino acid sequence of these proteins (19).
Cochaperones and other accessory factors also play an important part in client
recog-nition. In fact, Cdc37, which is a kinase-specific cochaperone of Hsp90, binds to an
unstable kinase domain of client kinases and recruits them to Hsp90 (44). In the present
study, we identified Hsp70 and CHIP as cochaperones of Hsp90 for MuV L maturation
and degradation. Further studies will be needed to identify other cochaperones
in-volved in recognition and folding of the MuV L protein and to reveal the detailed
molecular mechanisms by which the varieties of viral polymerases depend on the
Hsp90 chaperone machinery.
Hsp90 is an interesting antiviral target because the activity of Hsp90 is important for
a wide variety of virus infections (25). Hsp90 inhibitors show strong antiviral activities
and are probably less likely to induce drug resistance mutations on viruses (26, 34).
Therefore, they may be particularly useful for the treatment of RNA viruses for which
the emergence of drug resistance is common. Previous studies have suggested that the
suppression of Hsp70 potentiates the proteasomal degradation of Hsp90 client proteins
induced by Hsp90 inhibition (40, 41). In the present study, enhanced degradation of the
MuV L protein was also observed in cells treated with both Hsp90 and Hsp70 inhibitors.
Moreover, the combined use of Hsp90 and Hsp70 inhibitors was successful in further
reducing MuV and MeV production without inducing cytotoxicity to the cells.
There-fore, the combined use of Hsp90 and Hsp70 inhibitors may be a useful approach
against virus infections in humans. Moreover, because Hsp90 functions in concert with
many kinds of cochaperones (18), further investigation into the additional Hsp90
cochaperones involved in maintenance of the L protein will also expand the
possibil-ities for development of antiviral therapies.
Molecular chaperones, such as Hsp70 and Hsp90, have been identified as binding
partners of viral proteins in the RNP complex and/or as essential factors for viral RNA
replication in many paramyxovirus infections. It has been well established that Hsp90
activity is required for functional formation of the polymerase in paramyxovirus
infec-tions (12, 33, 35). In addition, Hsp70 has been reported to regulate viral polymerase
activity through interaction with the N proteins of MeV (45), respiratory syncytial virus
(RSV) (35), and canine distemper virus (46). Hsp70 also interacts with the P proteins of
MuV (47) and RSV (48). In the present study, we demonstrated that Hsp90 is required
for the correct folding of the MuV L protein and that Hsp70 assists in the biogenesis of
the L protein as a cochaperone of Hsp90. These findings should provide insights into
the roles of chaperone proteins in paramyxovirus RNA replication.
MATERIALS AND METHODS
Cells and viruses. Vero (African green monkey kidney), Vero/hSLAM (49), A549 (human lung epithelial), and 293T (human kidney) cells were maintained in Dulbecco’s modified Eagle’s minimal essential medium (DMEM) (Nacalai Tesque, Kyoto, Japan) supplemented with 100 U/ml penicillin, 100 mg/ml streptomycin, and 10% fetal bovine serum (FBS). Baby hamster kidney (BHK) cells constitutively expressing T7 RNA polymerase (BHK/T7-9 cells) were kindly provided by Naoto Ito (50) and maintained in Glasgow minimal essential medium (GMEM) (Nacalai Tesque) with 5% FBS and 5% tryptose phosphate broth (TPB). Cell viability was measured by using a CellTiter-Glo Luminescent Cell Viability Assay kit (Promega, Madison, WI).
The MuV strain Odate, which was isolated from a patient who developed aseptic meningitis (51), and a recombinant MeV expressing enhanced GFP (IC323-EGFP) (52) were used in this study.
Hsp90 Is Required for MuV Polymerase Formation Journal of Virology
March 2017 Volume 91 Issue 6 e02220-16 jvi.asm.org 15
on November 7, 2019 by guest
http://jvi.asm.org/
Plasmids. The plasmids pCAGGS-N, -P, and -L and pcDNA-FLAG-Hsp72 (for expression of an N-terminal FLAG-tagged Hsp72) were generated as previously described (47). The cDNAs of the MuV Odate N, P, and L proteins were amplified from pCAGGS-N, -P, and -L by PCR and cloned into pCR2.1 (Invitrogen/Thermo Fisher Scientific Inc., Waltham, MA), resulting in pCR-N, -P, and -L, respectively. The cDNAs of the L protein of MuV Odate and MeV 9301B were cloned into pCAGGS-HA, which encodes an N-terminal HA tag, resulting in pCAGGS-HA-MuV-L and -MeV-L, respectively. The cDNA of human CHIP was amplified from 293T cells by RT-PCR and cloned into pcDNA3.1/myc-His (Invitrogen), which encodes C-terminal Myc and His tags, resulting in pcDNA-CHIP-Myc/His. The nucleotide residues of the adenines at positions 88 and 89 of the CHIP gene in pcDNA-CHIP-Myc/His were replaced with guanine and cytosine by PCR-based mutagenesis to change Lys30to Ala, yielding pcDNA-CHIP(K30A)-Myc/His.
Based on the MeV minigenome expression plasmid p18MGFLuc01 (53), a plasmid containing a minigenome with a luciferase reporter gene for MuV (pFL-MuV-MG) was constructed using MuV Odate leader and trailer sequences.
The plasmid pMuV-Odate, which carries the antigenome of MuV Odate, was previously reported (54). pMuV-Odate/AcGFP was constructed by inserting an AcGFP gene as an additional transcriptional unit between the V/P and M genes of pMuV-Odate. All the plasmids were confirmed by sequencing with an ABI Prism 3130xl genetic analyzer (Applied Biosystems/Thermo Fisher Scientific, Inc.).
Virus rescue.The plasmid pMuV-Odate/AcGFP (5g) was cotransfected with the helper plasmids pC-N (300 ng), pCR-P (50 ng), and pCR-L (200 ng) in BHK/T7-9 cells by using TransIT LT1 (Mirus, Madison, WI). At 48 h posttransfection, the transfected BHK/T7-9 cells were mixed with Vero cells at a 1:1 dilution. At the height of the cytopathic effect, usually 4 to 7 days posttransfection, the supernatants were collected, and then the entire genome sequences of the rescued viruses were determined.
Reagents and antibodies.17-AAG, VER155008, MG-132, and cycloheximide (CHX) were purchased from Focus Biomolecules (Plymouth Meeting, PA), AdooQ Bioscience (Irvine, CA), Cell Signaling Tech-nology (Danvers, MA), and Sigma (St. Louis, MO), respectively. Anti-MuV N, V/P (T61), and L (L17) rabbit polyclonal antibodies (PAbs) were prepared as described previously (47, 55). Anti-Hsp72 (C92F3A-5), anti-Hsc70 (1F2-H5), and anti-Hsp90 (Hyb-K41009) mouse monoclonal antibodies (MAbs) were purchased from StressMarq Bioscience, Inc. (Victoria, Canada). Anti-FLAG (M2) and anti--actin (AC-15) mouse MAbs were purchased from Sigma. Anti-HA (HA11), anti-Myc (My3), and anti-glyceraldehyde-3-phosphate dehydrogenase (GAPDH; 3E12) mouse MAbs and anti-CHIP rabbit PAb (C3B6) were purchased from BioLegend, Inc. (San Diego, CA), MBL (Nagoya, Japan), Bioss, Inc. (Woburn, MA), and Cell Signaling Technology, respectively.
Plaque assay.Infectious titers of recombinant MuVs (rMuVs) were determined in triplicate by plaque assay using Vero cells in 12-well plates. After 1 h of virus adsorption, the cells were cultured in DMEM with 5% FBS and 1% agarose. At 5 days postinfection, the cells were stained with neutral red solution (Sigma), and the plaque counts were determined.
Cell extracts, immunoblotting, and immunoprecipitation. For the preparation of soluble cell extracts, cells were washed twice with cold phosphate-buffered saline (PBS) and then lysed in urea-free cell lysis buffer (20 mM Tris-HCl, pH 7.5, 135 mM NaCl, 1% Triton X-100, and protease inhibitor cocktail [Complete Mini; Roche, Mannheim, Germany]). For the preparation of insoluble cell extracts, 8 M urea-containing cell lysis buffer (20 mM Tris-HCl, pH 7.5, 135 mM NaCl, 1% Triton X-100, 8 M urea, and protease inhibitor cocktail) was used. For immunoblotting, the cell lysate was boiled in sodium dodecyl sulfate (SDS) sample buffer and subjected to SDS-polyacrylamide gel electrophoresis (SDS-PAGE). The proteins were transferred to polyvinylidene difluoride membranes (Millipore, Bedford, MA) and incu-bated with the appropriate antibodies. Each protein was visualized with SuperSignal West Femto Maximum Sensitivity Substrate (Thermo Scientific/Thermo Fisher Scientific, Inc.) and detected by use of an LAS-3000 image analyzer system (Fuji Film, Tokyo, Japan). For immunoprecipitation, the cell lysate was precleaned with protein G-Sepharose (GE Healthcare, Buckinghamshire, UK). Antibody-protein complexes were purified with protein G beads and washed with cell lysis buffer three times. After the complexes were boiled in SDS sample buffer, the proteins were separated by SDS-PAGE and processed for immunoblotting. Each cell extract preparation and immunoprecipitation assay were performed at least thee times, and the most representative data are shown.
Quantitative RT-PCR.Total RNA was prepared by use of an RNeasy minikit (Qiagen, Hilden, Germany), and first-strand cDNA was synthesized using PrimeScript RTase (TaKaRa Bio, Inc., Shiga, Japan). The RT reaction was performed using the primer 5=-ACCAAGGGGAGAAAGTAAAATCAATTTTTT-3= (at nucleotide positions 1 to 21 of the MuV genome) for MuV genomic RNA and an oligo(dT) primer for viral and cellular mRNAs. The amount of each cDNA was measured using a Universal ProbeLibrary and a LightCycler 480 system (Roche) according to the manufacturer’s instructions. The value of each RNA was normalized to that of hypoxanthine phosphoribosyltransferase 1 (HPRT1) mRNA. Quantitative PCR (qPCR) was performed using a Universal ProbeLibrary probe 62 (Roche) and the following primers: 5=-TTCCTC CAGTTCAACAGCAA-3=and 5=-ACCGTCGTCATCTGATTCCT-3=for primers targeted to a fragment consist-ing of nucleotides 1398 to 1474 in the MuV genome and 5=-GGGAGGCCATCACATTGTAG-3=and 5=-CACTA TTTCTATTCAGTGCTTTGA-3=for primers targeted to a fragment consisting of nucleotides 256 to 353 in the HPRT1 gene.
Minigenome assay.BHK/T7-9 cells in 24-well plates were transfected with 50 ng of pCR-N, 100 ng of pCR-P, 500 ng of pCR-L, 200 ng of pFL-MuV-MG, and 50 ng of pRL-TK (Promega) by using TransIT LT1 (Mirus). After 48 h, enzymatic activities of the firefly andRenillaluciferases (FL and RL, respectively) were measured by a Dual-Luciferase Reporter Assay System (Promega) and TR717 Microplate luminometer (Applied Biosystems). Relative luciferase activity was defined as the ratio of firefly luciferase toRenilla luciferase activity.
on November 7, 2019 by guest
http://jvi.asm.org/
Pulse-chase labeling.At 14 h postinfection (hpi), MuV-infected Vero cells were cultured in methionine-cysteine-deficient MEM (Gibco/Thermo Fisher Scientific, Inc.) for 1 h. The cells were then pulse-labeled with [35S]methionine-cysteine using an EasyTag EXPRESS35S protein labeling mix (PerkinElmer, Waltham, MA)
for 30 min and chased in DMEM with/without 17-AAG for 1 h. The cell lysates were prepared in cell lysis buffer and subjected to an immunoprecipitation assay with a rabbit anti-MuV PAb raised against the MuV particle. The immunoprecipitated proteins were analyzed by SDS-PAGE and autoradiography using a Typhoon FLA 7000 (GE Healthcare).
Gene silencing.The small interfering RNA (siRNA) siCHIP (5=-CGCUGGUGGCCGUGUAUUA-3=) was used for knockdown of endogenous CHIP (56). A nontargeting siRNA pool (siNC) (Dharmacon/GE Healthcare) was used as a negative control. Each siRNA was transfected using Lipofectamine RNAiMax (Invitrogen/Thermo Fisher Scientific, Inc.) according to the manufacturer’s protocol.
ACKNOWLEDGMENTS
We thank Naoto Ito of Gifu University for providing BHK/T7-9 cells. We also thank all
the members of the Department of Virology III, National Institute of Infectious Diseases,
for their technical advice and critical input.
This work was supported by grants from the Japan Society for the Promotion of
Science [Grant-in-Aid for Young Scientists (B), 16K19146] and the Japan Agency for
Medical Research and Development to Hiroshi Katoh and from the Takeda Science
Foundation to Makoto Takeda.
We declare that we have no conflicts of interest.
REFERENCES
1. Hviid A, Rubin S, Muhlemann K. 2008. Mumps. Lancet 371:932–944. https://doi.org/10.1016/S0140-6736(08)60419-5.
2. Lamb RA, Parks GD. 2006.Paramyxoviridae: the viruses and their repli-cation, p 1449 –1496.InKnipe DM, Howley PM, Griffin DE, Lamb RA, Martin MA, Roizman B, Straus SE (ed), Fields virology, 5th ed. Lippincott Williams & Wilkins, Philadelphia, PA.
3. Whelan SP, Barr JN, Wertz GW. 2004. Transcription and replication of nonsegmented negative-strand RNA viruses. Curr Top Microbiol Immu-nol 283:61–119.
4. Emerson SU, Wagner RR. 1973. L protein requirement for in vitro RNA synthesis by vesicular stomatitis virus. J Virol 12:1325–1335.
5. Hercyk N, Horikami SM, Moyer SA. 1988. The vesicular stomatitis virus L protein possesses the mRNA methyltransferase activities. Virology 163: 222–225.https://doi.org/10.1016/0042-6822(88)90253-X.
6. Hunt DM, Smith EF, Buckley DW. 1984. Aberrant polyadenylation by a vesicular stomatitis virus mutant is due to an altered L protein. J Virol 52:515–521.
7. Emerson SU, Schubert M. 1987. Location of the binding domains for the RNA polymerase L and the ribonucleocapsid template within different halves of the NS phosphoprotein of vesicular stomatitis virus. Proc Natl Acad Sci U S A 84:5655–5659.https://doi.org/10.1073/pnas.84.16.5655. 8. Kingston RL, Baase WA, Gay LS. 2004. Characterization of nucleocapsid
binding by the measles virus and mumps virus phosphoproteins. J Virol 78:8630 – 8640.https://doi.org/10.1128/JVI.78.16.8630-8640.2004. 9. Cox R, Pickar A, Qiu S, Tsao J, Rodenburg C, Dokland T, Elson A, He B, Luo
M. 2014. Structural studies on the authentic mumps virus nucleocapsid showing uncoiling by the phosphoprotein. Proc Natl Acad Sci U S A 111:15208 –15213.https://doi.org/10.1073/pnas.1413268111.
10. Smallwood S, Ryan KW, Moyer SA. 1994. Deletion analysis defines a carboxyl-proximal region of Sendai virus P protein that binds to the polymerase L protein. Virology 202:154 –163.https://doi.org/10.1006/ viro.1994.1331.
11. Chattopadhyay S, Banerjee AK. 2009. Phosphoprotein, P of human para-influenza virus type 3 prevents self-association of RNA-dependent RNA polymerase, L. Virology 383:226 –236. https://doi.org/10.1016/j.virol .2008.10.019.
12. Bloyet LM, Welsch J, Enchery F, Mathieu C, de Breyne S, Horvat B, Grigorov B, Gerlier D. 2016. HSP90 chaperoning in addition to phospho-protein required for folding but not for supporting enzymatic activities of measles and Nipah virus L polymerases. J Virol 90:6642– 6656.https:// doi.org/10.1128/JVI.00602-16.
13. Masters PS, Banerjee AK. 1988. Complex formation with vesicular sto-matitis virus phosphoprotein NS prevents binding of nucleocapsid pro-tein N to nonspecific RNA. J Virol 62:2658 –2664.
14. Yabukarski F, Lawrence P, Tarbouriech N, Bourhis JM, Delaforge E, Jensen MR, Ruigrok RW, Blackledge M, Volchkov V, Jamin M. 2014.
Structure of Nipah virus unassembled nucleoprotein in complex with its viral chaperone. Nat Struct Mol Biol 21:754 –759.https://doi.org/10.1038/ nsmb.2868.
15. Moyer SA, Baker SC, Lessard JL. 1986. Tubulin: a factor necessary for the synthesis of both Sendai virus and vesicular stomatitis virus RNAs. Proc Natl Acad Sci U S A 83:5405–5409.https://doi.org/10.1073/pnas.83.15 .5405.
16. Bose S, Banerjee AK. 2004. Beta-catenin associates with human parain-fluenza virus type 3 ribonucleoprotein complex and activates transcrip-tion of viral genome RNA in vitro. Gene Expr 11:241–249.
17. Ito M, Iwasaki M, Takeda M, Nakamura T, Yanagi Y, Ohno S. 2013. Measles virus nonstructural C protein modulates viral RNA polymerase activity by interacting with host protein SHCBP1. J Virol 87:9633–9642. https:// doi.org/10.1128/JVI.00714-13.
18. Taipale M, Jarosz DF, Lindquist S. 2010. HSP90 at the hub of protein homeostasis: emerging mechanistic insights. Nat Rev Mol Cell Biol 11: 515–528.https://doi.org/10.1038/nrm2918.
19. Taipale M, Krykbaeva I, Koeva M, Kayatekin C, Westover KD, Karras GI, Lindquist S. 2012. Quantitative analysis of HSP90-client interactions reveals principles of substrate recognition. Cell 150:987–1001.https:// doi.org/10.1016/j.cell.2012.06.047.
20. Kirschke E, Goswami D, Southworth D, Griffin PR, Agard DA. 2014. Glucocorticoid receptor function regulated by coordinated action of the Hsp90 and Hsp70 chaperone cycles. Cell 157:1685–1697.https://doi.org/ 10.1016/j.cell.2014.04.038.
21. Morishima Y, Murphy PJ, Li DP, Sanchez ER, Pratt WB. 2000. Stepwise assembly of a glucocorticoid receptor · hsp90 heterocomplex resolves two sequential ATP-dependent events involving first hsp70 and then hsp90 in opening of the steroid binding pocket. J Biol Chem 275: 18054 –18060.https://doi.org/10.1074/jbc.M000434200.
22. Chen S, Prapapanich V, Rimerman RA, Honore B, Smith DF. 1996. Inter-actions of p60, a mediator of progesterone receptor assembly, with heat shock proteins hsp90 and hsp70. Mol Endocrinol 10:682– 693.https:// doi.org/10.1210/mend.10.6.8776728.
23. Dittmar KD, Demady DR, Stancato LF, Krishna P, Pratt WB. 1997. Folding of the glucocorticoid receptor by the heat shock protein (hsp) 90-based chaperone machinery. The role of p23 is to stabilize receptor·hsp90 hetero-complexes formed by hsp90 · p60 · hsp70. J Biol Chem 272:21213–21220. 24. Kundrat L, Regan L. 2010. Balance between folding and degradation for Hsp90-dependent client proteins: a key role for CHIP. Biochemistry 49:7428 –7438.https://doi.org/10.1021/bi100386w.
25. Geller R, Taguwa S, Frydman J. 2012. Broad action of Hsp90 as a host chaperone required for viral replication. Biochim Biophys Acta 1823: 698 –706.https://doi.org/10.1016/j.bbamcr.2011.11.007.
26. Geller R, Vignuzzi M, Andino R, Frydman J. 2007. Evolutionary constraints on chaperone-mediated folding provide an antiviral approach refractory
Hsp90 Is Required for MuV Polymerase Formation Journal of Virology
March 2017 Volume 91 Issue 6 e02220-16 jvi.asm.org 17
on November 7, 2019 by guest
http://jvi.asm.org/
to development of drug resistance. Genes Dev 21:195–205. https:// doi.org/10.1101/gad.1505307.
27. Burch AD, Weller SK. 2005. Herpes simplex virus type 1 DNA polymerase requires the mammalian chaperone hsp90 for proper localization to the nucleus. J Virol 79:10740 –10749.https://doi.org/10.1128/JVI.79.16.10740 -10749.2005.
28. Hu J, Toft DO, Seeger C. 1997. Hepadnavirus assembly and reverse transcription require a multi-component chaperone complex which is incorporated into nucleocapsids. EMBO J 16:59 – 68.https://doi.org/ 10.1093/emboj/16.1.59.
29. Hu J, Anselmo D. 2000. In vitro reconstitution of a functional duck hepatitis B virus reverse transcriptase: posttranslational activation by Hsp90. J Virol 74:11447–11455.https://doi.org/10.1128/JVI.74.24.11447 -11455.2000.
30. Waxman L, Whitney M, Pollok BA, Kuo LC, Darke PL. 2001. Host cell factor requirement for hepatitis C virus enzyme maturation. Proc Natl Acad Sci U S A 98:13931–13935.https://doi.org/10.1073/pnas.241510898. 31. Naito T, Momose F, Kawaguchi A, Nagata K. 2007. Involvement of Hsp90
in assembly and nuclear import of influenza virus RNA polymerase subunits. J Virol 81:1339 –1349.https://doi.org/10.1128/JVI.01917-06. 32. Chase G, Deng T, Fodor E, Leung BW, Mayer D, Schwemmle M, Brownlee
G. 2008. Hsp90 inhibitors reduce influenza virus replication in cell cul-ture. Virology 377:431– 439.https://doi.org/10.1016/j.virol.2008.04.040. 33. Connor JH, McKenzie MO, Parks GD, Lyles DS. 2007. Antiviral activity and
RNA polymerase degradation following Hsp90 inhibition in a range of negative strand viruses. Virology 362:109 –119.https://doi.org/10.1016/ j.virol.2006.12.026.
34. Geller R, Andino R, Frydman J. 2013. Hsp90 inhibitors exhibit resistance-free antiviral activity against respiratory syncytial virus. PLoS One 8:e56762.https://doi.org/10.1371/journal.pone.0056762.
35. Munday DC, Wu W, Smith N, Fix J, Noton SL, Galloux M, Touzelet O, Armstrong SD, Dawson JM, Aljabr W, Easton AJ, Rameix-Welti MA, de Oliveira AP, Simabuco FM, Ventura AM, Hughes DJ, Barr JN, Fearns R, Digard P, Eleouet JF, Hiscox JA. 2015. Interactome analysis of the human respiratory syncytial virus RNA polymerase complex identifies protein chaperones as important cofactors that promote L-protein stability and RNA synthesis. J Virol 89:917–930.https://doi.org/10.1128/JVI.01783-14. 36. Smith DR, McCarthy S, Chrovian A, Olinger G, Stossel A, Geisbert TW, Hensley LE, Connor JH. 2010. Inhibition of heat-shock protein 90 reduces Ebola virus replication. Antiviral Res 87:187–194. https://doi.org/ 10.1016/j.antiviral.2010.04.015.
37. Isaacs JS, Xu W, Neckers L. 2003. Heat shock protein 90 as a molecular target for cancer therapeutics. Cancer Cell 3:213–217.https://doi.org/ 10.1016/S1535-6108(03)00029-1.
38. Ballinger CA, Connell P, Wu Y, Hu Z, Thompson LJ, Yin LY, Patterson C. 1999. Identification of CHIP, a novel tetratricopeptide repeat-containing protein that interacts with heat shock proteins and negatively regulates chaperone functions. Mol Cell Biol 19:4535– 4545. https://doi.org/ 10.1128/MCB.19.6.4535.
39. Xu W, Marcu M, Yuan X, Mimnaugh E, Patterson C, Neckers L. 2002. Chaperone-dependent E3 ubiquitin ligase CHIP mediates a degradative pathway for c-ErbB2/Neu. Proc Natl Acad Sci U S A 99:12847–12852. https://doi.org/10.1073/pnas.202365899.
40. Powers MV, Clarke PA, Workman P. 2008. Dual targeting of HSC70 and HSP72 inhibits HSP90 function and induces tumor-specific apoptosis. Cancer Cell 14:250 –262.https://doi.org/10.1016/j.ccr.2008.08.002. 41. Massey AJ, Williamson DS, Browne H, Murray JB, Dokurno P, Shaw T,
Macias AT, Daniels Z, Geoffroy S, Dopson M, Lavan P, Matassova N, Francis GL, Graham CJ, Parsons R, Wang Y, Padfield A, Comer M, Drysdale MJ, Wood M. 2010. A novel, small molecule inhibitor of Hsc70/Hsp70 potentiates Hsp90 inhibitor induced apoptosis in HCT116 colon
carci-noma cells. Cancer Chemother Pharmacol 66:535–545.https://doi.org/ 10.1007/s00280-009-1194-3.
42. Citri A, Alroy I, Lavi S, Rubin C, Xu W, Grammatikakis N, Patterson C, Neckers L, Fry DW, Yarden Y. 2002. Drug-induced ubiquitylation and degradation of ErbB receptor tyrosine kinases: implications for cancer therapy. EMBO J 21:2407–2417.https://doi.org/10.1093/emboj/21.10 .2407.
43. Cyr DM, Hohfeld J, Patterson C. 2002. Protein quality control: U-box-containing E3 ubiquitin ligases join the fold. Trends Biochem Sci 27: 368 –375.https://doi.org/10.1016/S0968-0004(02)02125-4.
44. Vaughan CK, Gohlke U, Sobott F, Good VM, Ali MM, Prodromou C, Robinson CV, Saibil HR, Pearl LH. 2006. Structure of an Hsp90-Cdc37-Cdk4 complex. Mol Cell 23:697–707. https://doi.org/10.1016/ j.molcel.2006.07.016.
45. Carsillo T, Zhang X, Vasconcelos D, Niewiesk S, Oglesbee M. 2006. A single codon in the nucleocapsid protein C terminus contributes to in vitro and in vivo fitness of Edmonston measles virus. J Virol 80: 2904 –2912.https://doi.org/10.1128/JVI.80.6.2904-2912.2006.
46. Oglesbee MJ, Liu Z, Kenney H, Brooks CL. 1996. The highly inducible member of the 70 kDa family of heat shock proteins increases canine distemper virus polymerase activity. J Gen Virol 77:2125–2135.https:// doi.org/10.1099/0022-1317-77-9-2125.
47. Katoh H, Kubota T, Kita S, Nakatsu Y, Aoki N, Mori Y, Maenaka K, Takeda M, Kidokoro M. 2015. Heat shock protein 70 regulates degradation of the mumps virus phosphoprotein via the ubiquitin-proteasome pathway. J Virol 89:3188 –3199.https://doi.org/10.1128/JVI.03343-14.
48. Oliveira AP, Simabuco FM, Tamura RE, Guerrero MC, Ribeiro PG, Liber-mann TA, Zerbini LF, Ventura AM. 2013. Human respiratory syncytial virus N, P and M protein interactions in HEK-293T cells. Virus Res 177:108 –112.https://doi.org/10.1016/j.virusres.2013.07.010.
49. Ono N, Tatsuo H, Hidaka Y, Aoki T, Minagawa H, Yanagi Y. 2001. Measles viruses on throat swabs from measles patients use signaling lymphocytic activation molecule (CDw150) but not CD46 as a cellular receptor. J Virol 75:4399 – 4401.https://doi.org/10.1128/JVI.75.9.4399-4401.2001. 50. Ito N, Takayama-Ito M, Yamada K, Hosokawa J, Sugiyama M, Minamoto
N. 2003. Improved recovery of rabies virus from cloned cDNA using a vaccinia virus-free reverse genetics system. Microbiol Immunol 47: 613– 617.https://doi.org/10.1111/j.1348-0421.2003.tb03424.x. 51. Saito H, Takahashi Y, Harata S, Tanaka K, Sano T, Suto T, Yamada A,
Yamazaki S, Morita M. 1996. Isolation and characterization of mumps virus strains in a mumps outbreak with a high incidence of aseptic meningitis. Microbiol Immunol 40:271–275.https://doi.org/10.1111/ j.1348-0421.1996.tb03346.x.
52. Hashimoto K, Ono N, Tatsuo H, Minagawa H, Takeda M, Takeuchi K, Yanagi Y. 2002. SLAM (CD150)-independent measles virus entry as re-vealed by recombinant virus expressing green fluorescent protein. J Virol 76:6743– 6749.https://doi.org/10.1128/JVI.76.13.6743-6749.2002. 53. Komase K, Nakayama T, Iijima M, Miki K, Kawanishi R, Uejima H. 2006.
The phosphoprotein of attenuated measles AIK-C vaccine strain contrib-utes to its temperature-sensitive phenotype. Vaccine 24:826 – 834. https://doi.org/10.1016/j.vaccine.2005.06.036.
54. Katoh H, Kubota T, Ihara T, Maeda K, Takeda M, Kidokoro M. 2016. Cross-neutralization between human and African bat mumps viruses. Emerg Infect Dis 22:703–706.https://doi.org/10.3201/eid2204.151116. 55. Takeuchi K, Tanabayashi K, Hishiyama M, Yamada YK, Yamada A, Sugiura
A. 1990. Detection and characterization of mumps virus V protein. Virology 178:247–253.https://doi.org/10.1016/0042-6822(90)90400-L. 56. Lee JH, Khadka P, Baek SH, Chung IK. 2010. CHIP promotes human
telomerase reverse transcriptase degradation and negatively regulates telomerase activity. J Biol Chem 285:42033– 42045. https://doi.org/ 10.1074/jbc.M110.149831.
on November 7, 2019 by guest
http://jvi.asm.org/