Restriction of Human Cytomegalovirus
Replication by ISG15, a Host Effector
Regulated by cGAS-STING
Double-Stranded-DNA Sensing
Christopher Bianco,aIan Mohra,b
Department of Microbiologyaand Laura and Isaac Perlmutter Cancer Institute,bNYU School of Medicine, New
York, New York, USA
ABSTRACT Accumulation of the interferon-stimulated gene 15 (ISG15) protein product, which is reversibly conjugated to numerous polypeptide targets, impacts the proteome and physiology of uninfected and infected cells. While many viruses, including human cytomegalovirus (HCMV), blunt host antiviral defenses by limiting ISG expression, the overall abundance of ISG15 monomer and protein conjugates rises in HCMV-infected cells. However, the molecular signals underlying ISG15 accu-mulation and whether the ISG15 polypeptide itself influences HCMV infection biol-ogy remain unknown. Here, we establish that the ISG15 gene product itself directly regulates HCMV replication and that its accumulation restricts productive virus growth. Although ISG15 monomer and protein conjugate accumulation was induced in cells infected with UV-inactivated HCMV, it was subsequently reduced, but not eliminated, by an immediate-early (IE) or early (E) virus-encoded function(s). Instead, HCMV-induced ISG15 monomer and protein conjugate accumulation was dependent upon the double-stranded DNA (dsDNA) sensor cyclic GMP-AMP synthase (cGAS), the innate immune adaptor STING, and interferon signaling. Significantly, dsDNA itself was sufficient to induce cGAS-, STING-, and interferon signaling-dependent ISG15 monomer and conjugate protein accumulation in uninfected cells. Accumula-tion of ISGylated proteins in uninfected cells treated with dsDNA was prevented by expressing the HCMV multifunctional IE1 transactivator. This demonstrates that ex-pression of a single host interferon-stimulated gene, ISG15, restricts HCMV replica-tion, and that IE1 is sufficient to blunt ISGylation in response to dsDNA sensing in uninfected cells. Moreover, it establishes that ISGylation modifies the proteomes of virus-infected and uninfected normal cells in response to cell-intrinsic dsDNA sens-ing dependent upon cGAS-STING.
IMPORTANCE By antagonizing type I interferon production and action, many vi-ruses, including human cytomegalovirus (HCMV), evade host defenses. However, lev-els of the interferon-induced ISG15 protein, which is covalently conjugated to host and viral proteins, increase in HCMV-infected cells. How ISG15 accumulation is regu-lated and whether the ISG15 polypeptide influences HCMV replication remain un-known. This study establishes that ISG15 itself restricts HCMV replication and that HCMV-induced ISG15 accumulation is triggered by host defenses that detect cytoplas-mic double-stranded DNA (dsDNA). Remarkably, dsDNA triggered ISG15 accumulation even in uninfected cells, and this was reduced by HCMV IE1 expression. This shows that ISG15 itself controls the replication of HCMV, which causes life-threatening disease among the immunocompromised and is a significant source of congenital morbidity and mortality among newborns. Moreover, it demonstrates that ISG15 modifies the un-infected cell proteome in response to dsDNA, potentially impacting responses to DNA vaccines, gene therapy, and autoimmune disease pathogenesis.
Received29 December 2016Accepted8
February 2017
Accepted manuscript posted online15
February 2017
CitationBianco C, Mohr I. 2017. Restriction of
human cytomegalovirus replication by ISG15, a host effector regulated by cGAS-STING double-stranded-DNA sensing. J Virol 91:e02483-16.
https://doi.org/10.1128/JVI.02483-16.
EditorRozanne M. Sandri-Goldin, University of
California, Irvine
Copyright© 2017 American Society for
Microbiology.All Rights Reserved. Address correspondence to Ian Mohr, [email protected].
crossm
on November 7, 2019 by guest
http://jvi.asm.org/
KEYWORDS DNA sensing, HCMV, ISG15, STING, cGAS
U
pon detecting double-stranded DNA (dsDNA) within the cytoplasm, a panoply of powerful cell-intrinsic host responses are triggered that collectively defend against virus infection. Recognition of dsDNA in the host cell cytoplasm by cyclic GMP-AMP (cGAMP) synthase (cGAS), one of several dsDNA-sensing receptors, results in cGAMP production in response to dsDNA binding (1). Subsequent stimulation of the innate immune adaptor protein STING by the second messenger cGAMP activates IRF3 and NF-B, which in turn results in type I interferon (IFN) production (1). By signaling through specific cell surface receptors on neighboring cells, IFN ultimately induces expression of several hundred interferon-stimulated genes (ISGs.) (2–4) One of these, ISG15, encodes a ubiquitin (Ub)-like protein that can be conjugated to host and viral protein targets in a manner similar to ubiquitin (5–7). Although ISG15 conjugation or ISGylation requires the activity of interferon-stimulated E1, E2, and E3 ligases (8, 9), the major ISG15 E3 ligase, Herc5, associates with polyribosomes, and ISGylation occurs cotranslationally on nascent polypeptides with little specificity (10). Indeed, pleiotropic effects of ISGylation on target protein function have been reported to have potential opposing pro- and antiviral roles (11–14). Likewise, roles for unconjugated ISG15 during viral infection have been suggested, including controlling inflammatory responses and inhibiting virus-like-particle release (15, 16). Besides being induced by IFN, ISG15 accumulated in IFN receptor-deficient cells, suggesting alternative, noncanonical path-ways also regulate ISG15 levels (17, 18). While poised to broadly influence host and viral protein functions during infection, mechanisms controlling ISG15 accumulation and action are incompletely understood.Replication of representative alpha- and gammaherpesvirus subfamily members is restricted by ISG15 (19, 20). To antagonize host defenses, these viruses impair ongoing host protein synthesis (21), likely limiting roles for ISG15 on newly synthesized host proteins. In contrast, host protein synthesis proceeds in cells infected with the beta-herpesvirus human cytomegalovirus (HCMV) (22). While infection is predominately asymptomatic in healthy individuals (23, 24), HCMV causes life-threatening disease among the immunocompromised, including solid-organ or stem cell transplant recip-ients (25, 26), and is a significant source of congenital morbidity and mortality among newborn infants in the developed world (27, 28). Importantly, changes in ongoing host mRNA translation play key roles in regulating HCMV productive replication (22, 29), potentially exposing newly synthesized host proteins to ISG15. Although HCMV limits the IFN response (30), it does not eliminate ISG15 production or conjugation (31–33) and reportedly encodes an ISG15 binding protein (33). Furthermore, signaling path-ways (IFN mediated or noncanonical) that induce ISG15 production and conjugation during HCMV infection have not been elucidated. Although ISG15 conjugation en-zymes have at best only a limited impact on productive HCMV growth in fibroblasts (33), any contributions of ISG15 itself to host antiviral defenses against HCMV remain undefined.
Here, we show that ISG15 directly regulates HCMV replication and that its accumu-lation restricts productive virus growth. Significantly, depleting either cGAS or STING abrogated accumulation of ISG15 monomer and protein conjugates in response to HCMV, establishing that HCMV-induced ISG15 accumulation is dependent upon cyto-plasmic dsDNA-sensing pathways. Furthermore, dsDNA itself was sufficient to induce cGAS- and STING-dependent ISG15 accumulation and conjugation in uninfected cells, and the HCMV IE1 transactivator blocked ISGylation in response to dsDNA in uninfected cells. This demonstrates that HCMV replication is restricted by the product of a single ISG, ISG15, and that IE1 is sufficient to blunt ISGylation in response to dsDNA sensing in uninfected cells. Moreover, it establishes that cell-intrinsic dsDNA sensing by cGAS-STING shapes the proteome through ISGylation in virus-infected and unin-fected normal cells.
on November 7, 2019 by guest
http://jvi.asm.org/
RESULTS
ISG15 monomer and conjugate protein accumulation is triggered by HCMV and subsequently limited by viral gene expression. Although ISG15 and ISG15 protein conjugates reportedly accumulated during HCMV infection (31–33), how the ISG15-encoded polypeptide itself might impact productive HCMV replication and contribute to host anti-HCMV defenses remains unknown. To better define ISG15 behavior in primary human fibroblasts (normal human dermal fibroblasts [NHDFs]) infected with HCMV, ISG15 abundance and conjugation were evaluated over a 96-h time course by immunoblotting (Fig. 1A). Unconjugated ISG15 monomer was detected beginning at 6 h postinfection (hpi). HCMV immediate-early proteins IE1/2 were also readily detected at this time. Accumulation of high-molecular-weight ISG15-conjugated proteins was observed beginning at 12 hpi. Compared to mock-infected cells, elevated ISG15 monomer and protein conjugates persisted throughout a 96-h time course, reaching peak levels at 48 hpi. By 72 hpi, overall levels of ISG15 monomer and conjugated proteins were reduced compared to levels observed at 48 hpi and were reduced even further by 96 hpi. The decline in ISG15 monomer/conjugate abundance
FIG 1Regulation of ISG15 accumulation and conjugation in HMCV-infected cells. (A) Total protein from NHDFs mock infected or infected with HCMV (MOI⫽ 3 PFU/cell) was collected at the indicated times, fractionated by SDS-PAGE, and analyzed by immunoblotting using antibodies specific for total ISG15, HCMV IE1/2, HCMV pp28, or-actin. Migration of molecular mass standards is shown on the right. (B) As in panel A, except total protein was isolated at 72 hpi from cultures left untreated or treated with PAA. HMCV pp65 is a late protein control whose accumulation is sensitive to PAA. (C) As in panel A, except NHDFs were infected with HCMV or UV-inactivated HCMV. (D) As in panel C, except total RNA was collected at 24 hpi and reverse transcription (RT)-qPCR was performed to measure ISG15 mRNA abundance. The error bars indicate standard errors of the mean (SEM).*,Pⱕ0.05 by Student’sttest. (E) NHDFs were mock infected or infected with HCMV at the indicated MOI. Total protein was collected 24 hpi and analyzed by immunoblotting as for panel A.
on November 7, 2019 by guest
http://jvi.asm.org/
[image:3.585.45.539.73.455.2]was correlated with increased abundance of a representative viral late protein, pp28, by 72 h (Fig. 1A.) This suggested that ISG15 accumulation was induced very early in the viral productive growth cycle yet began to be suppressed with the onset of late viral gene expression or an event associated with late viral gene expression, such as viral DNA synthesis.
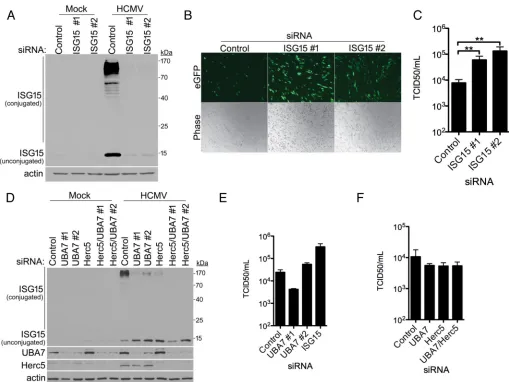
To investigate if viral DNA synthesis might suppress ISG15 monomer and protein conjugate accumulation, HCMV-infected cultures were treated with phosphonoacetic acid (PAA) to block viral DNA replication and late gene expression. While PAA effec-tively reduced steady-state levels of the HCMV late protein pp65, ISG15 monomer and protein conjugate abundances remained similar (Fig. 1B). Thus, inhibiting late viral gene expression with PAA did not significantly increase ISG15 accumulation, implying that late viral protein production or viral DNA synthesis was not required to suppress virus-induced ISG15 monomer and conjugate accumulation. In contrast, infection of NHDFs with UV-inactivated HCMV stimulated ISG15 monomer accumulation and re-sulted in a dramatic rise in ISG15-conjugated protein abundance (Fig. 1C). In addition, at 24 and 48 hpi, ISG15 monomer accumulated to similar levels in cultures infected with UV-inactivated or unirradiated virus. However, the overall abundance of ISG15-conjugated proteins was greater at 24 and 48 h in cultures infected with UV-inactivated HCMV than in those infected with unirradiated virus (Fig. 1C). Unlike cultures infected with unirradiated virus, ISG15 monomer and conjugated-protein levels in NHDFs infected with UV-inactivated HCMV did not detectably decrease by 72 hpi (Fig. 1C). In addition, ISG15 mRNA increased to 16-fold-greater levels in NHDFs infected with UV-inactivated HCMV than in cultures infected with active, unirradiated HCMV (Fig. 1D). Finally, even though HCMV reduced ISG15 conjugate protein accumulation, similar levels of ISGylated proteins were readily detected at 24 hpi at multiplicities of infection (MOI) ranging from 3 to 12, reducing the likelihood that uninfected cells were the source of residual ISGylation and demonstrating that HCMV is incapable of completely blocking ISGylation under these conditions (Fig. 1E). Taken together, this suggests that HCMV infection stimulates ISG15 protein accumulation by increasing ISG15 mRNA abundance, likely through a general mechanism of ISG induction. While overall levels of ISG15-conjugated proteins rise over 48 h, one or more viral IE or E gene products likely limit, but do not eliminate, ISG15 monomer and protein conjugate accumulation. Preventing ISG15 accumulation enhances HCMV replication. To investigate whether ISG15 itself impacted HCMV replication, ISG15 mRNA was depleted using RNA interference. Following transfection of NHDFs with either nonsilencing (NS) small interfering RNA (siRNA) or two different siRNAs targeting ISG15, cultures were either mock infected or infected with HCMV, and levels of ISG15 were evaluated by immu-noblotting. Figure 2A shows that both ISG15 siRNAs effectively reduced HCMV-induced ISG monomer and protein conjugate accumulation. To evaluate HCMV replication and spread, NHDFs transfected with NS control or ISG15-targeting siRNA were infected with an HCMV reporter expressing enhanced green fluorescent protein (eGFP) at a low MOI, and virus productive growth and dissemination were monitored by visualizing eGFP fluorescence in live cells. Compared to cultures treated with control NS siRNA, a substantial increase in eGFP-positive cells was observed following ISG15 depletion with either of two ISG15-specific siRNAs (Fig. 2B), consistent with ISG15 acting to limit virus reproduction and spread. Quantifying infectious-virus production after 6 days revealed that ISG15 depletion enhanced HCMV replication up to 20-fold (Fig. 2C). This demon-strates that the ISG15-encoded polypeptide itself plays a significant role in restricting productive HCMV replication.
To investigate the impact of ISG15 conjugation on HCMV replication, the ISG15-conjugating enzymes UBA7 (E1) and Herc5 (E3) were depleting using RNA interference. NHDFs treated with NS control, UBA7, or Herc5 siRNA were infected with HCMV, and ISG15 monomer and conjugate abundances were measured by immunoblotting. Com-pared to NS control siRNA-treated cultures, ISG15 high-molecular-weight protein con-jugates accumulated to lower levels in cells treated with UBA7 or Herc5 siRNAs (Fig. 2D).
on November 7, 2019 by guest
http://jvi.asm.org/
The abundance of ISG15 protein conjugates was further reduced by codepleting both Herc5 and UBA7 (Fig. 2D). In all cases where ISG15 conjugation was impaired by siRNA depletion of UBA7 and/or Herc5, ISG15 monomer abundance increased (Fig. 2D). However, whereas ISG15 depletion stimulated infectious-virus production by up to 20-fold, depletion of UBA7 at best minimally impacted productive viral replication, even though ISG15 protein conjugate levels were effectively reduced (Fig. 2E). Similarly, although UBA7, Herc5, or UBA7/Herc5 codepletion reduced ISG15 protein conjugate accumulation, significant differences in HCMV productive growth were not detected (Fig. 2F).
Sensing dsDNA by cGAS-STING and IFN signaling is necessary for ISG15 mono-mer and protein conjugate accumulation in response to HCMV.Accumulation of an interferon-induced gene product like ISG15 early in the viral life cycle is characteristic of cell-intrinsic innate responses. With this in mind, host signaling pathways necessary for HCMV-induced ISG15 accumulation and conjugation were examined. For HCMV,
FIG 2Preventing ISG15 accumulation enhances HCMV replication. (A) NHDFs were transfected with a nonsilencing (control) or ISG15-specific siRNA (ISG15 no. 1 or ISG15 no. 2). After 3 days, the cultures were mock infected or infected with HCMV (MOI⫽3 PFU/cell). Total protein was collected 24 hpi, fractionated by SDS-PAGE, and analyzed by immunoblotting with anti-ISG15 or-actin (loading control). (B) As in panel A, except the cells were infected at low multiplicity (MOI⫽0.05 PFU/cell), and photographs of living cells (phase-contrast and eGFP fluorescence) were captured 6 days postinfection to visualize virus replication and spread. (C) As in panel B, except infectious virus was quantified from supernatants using a TCID50assay. The error bars indicate SEM.**,Pⱕ0.01 by Student’s
ttest. (D) As in panel A, except using siRNAs specific for UBA7 (UBA7 no. 1 and UBA7 no. 2) or Herc5, individually or together. After 3 days, the cultures were infected with HCMV (MOI⫽3 PFU/cell). Total protein was collected 24 hpi, fractionated by SDS-PAGE, and analyzed by immunoblotting with anti-ISG15, -UBA7, -Herc5, or --actin (loading control). Herc5 was not detected in mock-infected cells but was readily detected in HCMV-infected cells, as it is encoded by an ISG. (E) NHDFs treated with siRNAs as in panel D were infected with HCMV (MOI⫽0.05 PFU/cell). After 6 days, infectious virus was quantified as in panel C. (F) NHDFs treated with siRNAs as in panel D were infected with HCMV, and infectious virus was quantified as in panel E.
on November 7, 2019 by guest
http://jvi.asm.org/
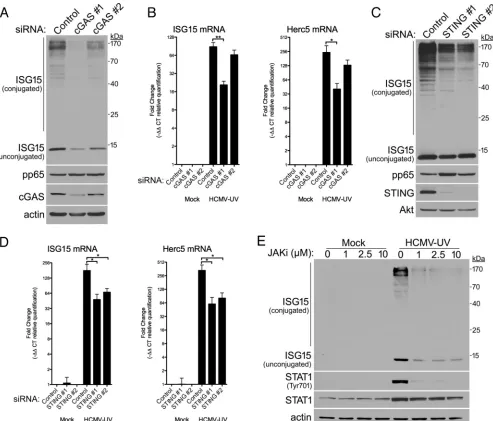
[image:5.585.43.552.69.451.2]engaging the cytoplasmic dsDNA sensor cGAS is responsible for activating the critical adaptor protein STING and triggering antiviral responses (34, 35). To determine if ISG15 accumulation was cGAS dependent, NHDFs transfected with control NS siRNA or cGAS-specific siRNAs were infected with HCMV, and ISG15 abundance was measured by immunoblotting. Compared to control NS siRNA-treated cultures, both cGAS siRNAs reduced overall levels of ISG15 monomer and protein conjugates detected at 48 hpi (Fig. 3A). cGAS protein levels were reduced more by cGAS-specific siRNA no. 1 than by siRNA no. 2, and the reduction in ISG15 monomer and conjugate accumulation by each siRNA was proportional to the extent of cGAS depletion. Control NS siRNA- and cGAS-specific-siRNA-treated cultures exhibited equivalent levels of HCMV pp65
accu-FIG 3Dependence of HCMV-induced ISG15 accumulation and conjugation upon cGAS, STING, and interferon signaling. (A) NHDFs were transfected with a nonsilencing (control) or cGAS-specific siRNA (cGAS no. 1 or cGAS no. 2) and either mock infected or infected with HCMV (MOI⫽3 PFU/cell). Total protein was collected 48 hpi, fractionated by SDS-PAGE, and analyzed by immunoblotting with antibodies specific for ISG15, HCMV pp65, cGAS, or-actin (loading control). Migration of molecular mass standards is shown on the right. (B) NHDFs transfected with siRNA as in panel A were infected with UV-inactivated HCMV (HCMV-UV) (MOI⫽3 PFU/cell). Total RNA was isolated at 24 hpi, and RT-qPCR was performed using primers specific for ISG15 or Herc5 mRNA. The error bars indicate SEM.*,Pⱕ0.05;**,Pⱕ0.01; Student’sttest. (C) As in panel A except using siRNAs specific for STING (STING no. 1 and STING no. 2) and antibodies specific for STING or Akt (loading control). (D) NHDFs transfected with siRNA as in panel C were infected with HCMV-UV (MOI⫽3 PFU/cell). Total RNA was isolated and analyzed as in panel B. The error bars indicate SEM.*,Pⱕ0.05 by Student’sttest. (E) NHDFs treated with pyridone 6, a small-molecule JAKi, at the indicated concentrations were mock infected or infected with HCMV-UV (MOI⫽3 PFU/cell). Total protein was collected 24 hpi, fractionated by SDS-PAGE, and analyzed by immunoblotting with antibodies specific for ISG15, STAT1, pTyr701-STAT1, or-actin (loading control). Migration of molecular mass standards is shown on the right.
on November 7, 2019 by guest
http://jvi.asm.org/
[image:6.585.46.539.71.492.2]mulation at 48 hpi, indicating that virus similarly infected the cultures. As cGAS-dependent DNA sensing likely occurs prior to viral gene expression, the impact of cGAS on ISG15 and the ISG15 E3 ligase Herc5 mRNA levels was examined by real-time PCR in cultures infected with UV-inactivated HCMV. Both ISG15 and Herc5 mRNAs were detected at low levels in uninfected cultures (Fig. 3B). Compared to HCMV-infected control NS siRNA-treated cultures, levels of both ISG15 and Herc5 mRNAs were con-sistently reduced in HCMV-infected cGAS-specific-siRNA-treated cultures, and cGAS siRNA no. 2 was less effective than siRNA no. 1 (Fig. 3B). Once activated by dsDNA sensing, cGAS produces the second messenger cGAMP, which in turn binds to and activates STING. Depletion of STING likewise reduced (i) ISG15 protein monomer and conjugate accumulation in HCMV-infected cells at 48 hpi (Fig. 3C) and (ii) ISG15 and Herc5 mRNA abundance in cells infected with UV-inactivated HCMV (Fig. 3D), demonstrating that the pathway contributes to ISG15 induction during HCMV infection.
Activated STING initiates a signaling cascade that culminates in IFN production, which acts in an autocrine and paracrine manner to induce expression of ISGs (36). However, alternative noncanonical ISG15 accumulation has been reported without IFN signaling in cells lacking subunits of the type I IFN receptor (17, 18). To determine if HCMV-induced ISG15 accumulation and conjugation were dependent upon IFN signal-ing, the impact of pharmacologically blocking IFN signal transduction was examined using a Janus kinase (JAK) inhibitor (JAKi) (pyridone 6). Compared to mock-infected cultures, infection of NHDFs with UV-inactivated HCMV potently stimulated the accu-mulation by 24 hpi of phosphorylated STAT1, a receptor-proximal marker of signaling through activated type I IFN receptors (Fig. 3E). ISG monomer and protein conjugate abundances were similarly increased in cells infected with UV-inactivated HCMV and in mock-infected cultures. The JAK inhibitor reduced phospho-STAT1, ISG15 monomer, and ISG15 protein conjugate overall abundance, demonstrating that ISG15 accumula-tion was dependent upon JAK activity (Fig. 3E). However, while concentraaccumula-tions of JAK inhibitor as high as 10 M further reduced phospho-STAT1 abundance, additional reductions in overall ISG15 levels were not detected (Fig. 3E). Thus, ISG15 monomer and protein conjugate accumulation in HCMV-infected cells primarily resulted from JAK-dependent interferon signaling. However, as some ISG15 monomer and conjugate accumulation persisted in cells treated with high concentrations of JAK inhibitor, the possibility remains that noncanonical induction of ISG15 also contributes to a much lesser extent.
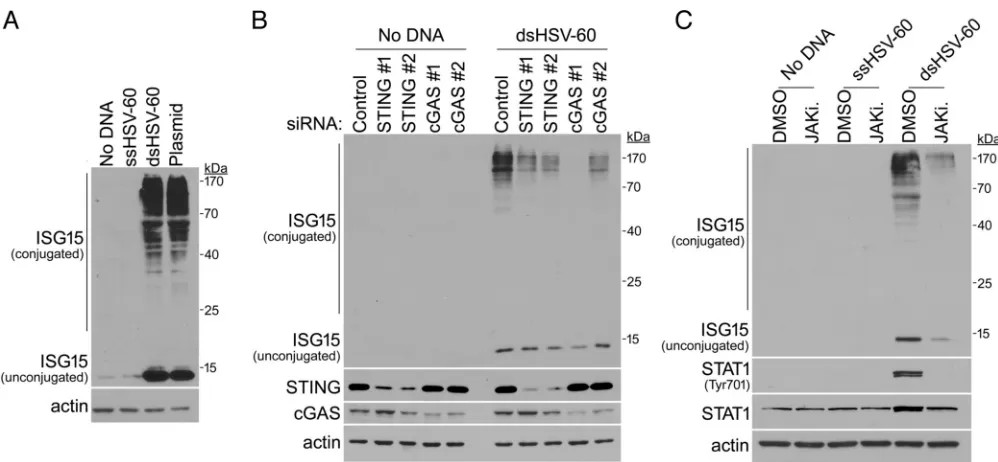
Regulation of ISG15 accumulation and ISGylation by dsDNA in normal, unin-fected cells.The dependence of ISG15 monomer and protein conjugate accumulation upon cGAS-STING in HCMV-infected cells suggested that viral DNA might trigger this response. The possibility that modification of the uninfected cell proteome by ISG15 might occur in normal cells exposed to dsDNA was therefore considered. To test this, uninfected NHDFs were mock transfected or transfected with single-stranded DNA (ssDNA) 60 nucleotides (nt) in length derived from herpes simplex virus 1 (HSV-1); a 60-bp dsDNA derived from HSV-1, one strand of which was identical to the 60-nt ssDNA sequence; or plasmid DNA. After 24 h, a substantial increase in ISG15 monomer and high-molecular-weight protein conjugate abundance was selectively observed only in samples exposed to 60-nt dsDNA from HSV-1 or plasmid DNA (Fig. 4A). Furthermore, the dsDNA-induced increase in ISG15 monomer and protein conjugate accumulation was impaired (i) when either STING or cGAS was depleted with two siRNAs (Fig. 4B) and (ii) in the presence of a small-molecule JAKi (Fig. 4C). Once again, phospho-STAT was no longer detected in cultures treated with JAKi, although ISG15 monomer and conjugate accumulation was reduced, but still detected (Fig. 4C). Thus, dsDNA-induced ISG15 monomer and protein conjugate accumulation in uninfected, normal cells is dependent upon signaling through cGAS, STING, and Janus kinase (JAK) activity.
Attenuation of dsDNA-induced ISG15 and ISGylated protein accumulation by HCMV IE1. Even though their accumulation was dependent upon dsDNA sensing in uninfected and HCMV-infected NHDFs, the virus-induced increase in ISG15 monomer and ISGylated protein levels was curtailed by a virus-encoded function expressed at
on November 7, 2019 by guest
http://jvi.asm.org/
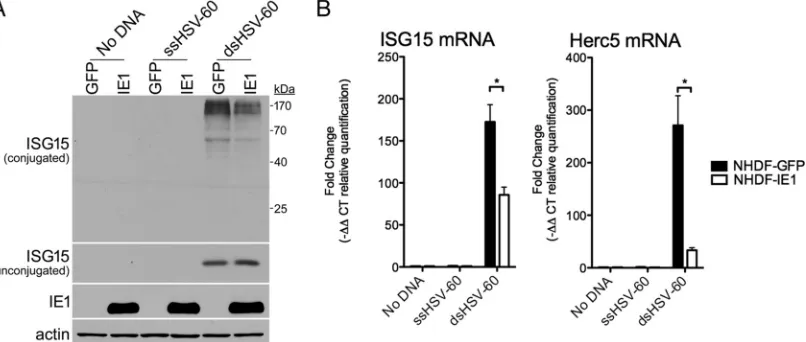
immediate-early (IE) or early (E) times postinfection (Fig. 1B and C). While this article was in preparation, a report demonstrating that a mutant virus with IE1 deleted (CR208) was less able to inhibit the accumulation of ISG15 monomer and conjugates than wild-type virus was published (33). Indeed, HCMV IE1 has been reported to inhibit ISG transcription by interacting with STAT1 and STAT2 to prevent their association with IFN-responsive gene promoters (37). As ISGylation requires four ISG products (UBA7, UBCH8, Herc5, and ISG15), the capacity of HCMV IE1 to suppress ISG15 accumulation and conjugation in response to dsDNA was investigated. NHDFs stably transduced with an eGFP-expressing or HCMV IE1-expressing lentivirus were transfected without DNA, with ssDNA, or with dsDNA, and overall levels of ISG15 monomer or protein conjugates were evaluated by immunoblotting. While ISG15 monomer and protein conjugates accumulated only in dsDNA-treated cultures, fewer ISGylated proteins were detected in HCMV IE1-expressing cultures compared to those expressing eGFP (Fig. 5A). However, ISG15 monomer levels remained similar in eGFP- and IE1-expressing cultures (Fig. 5A). Reduced levels of mRNA encoding ISG15 and the ISG15 E3 ligase Herc5 were observed in IE1-expressing cultures compared to eGFP-expressing cultures in response to dsDNA (Fig. 5B). Although the fold change in ISG15 mRNA levels was reduced by no more than a factor of 2 by IE1, the fold reduction in Herc5 mRNA abundance was far greater (approximately 10-fold), potentially accounting for the larger reduction in ISGylated protein levels by IE1 (Fig. 5A). Thus, ectopic HCMV IE1 expression in uninfected NHDFs was sufficient to limit ISG15 protein conjugate accumulation in response to dsDNA. This demonstrates that IE1 controls dsDNA-induced ISGylation and likely accounts for the attenuation of ISG15 monomer and protein conjugate accumulation by HCMV gene expression in virus-infected cells.
DISCUSSION
In spite of the multiple mechanisms by which HCMV blunts innate immune defenses (30), the abundance of ISG15 mRNA and protein increases in response to HCMV infection (31, 32). Until now, the virus-host interactions that precipitate ISG15 induction
FIG 4Triggering of cGAS-STING- and interferon signaling-dependent ISGylation in uninfected cells by dsDNA. (A) NHDFs were transfected with either no DNA, a 60-nt single-stranded (ssHSV-60) or 60-bp duplex (dsHSV-60) DNA derived from HSV, or plasmid DNA (pFLAG-CMV-5a; 4.7 kb). After 24 h, total protein was collected, fractionated by SDS-PAGE, and analyzed by immunoblotting with anti-ISG15 or anti--actin (loading control). Migration of molecular mass standards is shown on the right. (B) NHDFs were transfected with nonsilencing (control), cGAS-specific (cGAS no. 1 or cGAS no. 2), or STING-specific (STING no. 1 or STING no. 2) siRNAs. After 3 days, the cultures were transfected with or without a 60-bp DNA duplex (dsHSV-60). Total protein was collected 24 h after transfection with or without dsDNA, separated by SDS-PAGE, and analyzed by immunoblotting using the indicated antibodies as in panel A. (C) NHDFs treated with DMSO (vehicle control) or the small-molecule JAKi pyridone 6 (2.5M) were transfected without DNA, with a 60-nt single-stranded DNA (ssHSV-60), or with a 60-bp duplex DNA (dsHSV-60). After 24 h, total protein was collected and analyzed as for panel B.
on November 7, 2019 by guest
http://jvi.asm.org/
[image:8.585.42.541.68.299.2]and the impact of the ISG15 polypeptide itself on productive HCMV replication re-mained unexplored. Here, we establish that although ISG15 accumulation and conju-gation to protein targets are triggered by HCMV infection, they are subsequently tempered, but not completely abrogated by viral gene expression. Preventing HCMV-induced ISG15 accumulation stimulated productive viral growth by up to 20-fold. ISG15 monomer and protein conjugate accumulation in response to HCMV was dependent upon cGAS-STING and interferon receptor signaling. Significantly, exposure of unin-fected primary human cells to dsDNA was sufficient to induce cGAS/STING- and interferon signaling-dependent ISG15 monomer and protein conjugate accumula-tion. Moreover, accumulation of ISG15-conjugated proteins in uninfected cells treated with dsDNA was attenuated by HCMV IE1 expression. This demonstrates that the dsDNA-sensing components cGAS-STING trigger ISG15 monomer and protein conju-gate accumulation, which is predominately dependent upon interferon receptor sig-naling and restricts HCMV productive replication. Moreover, it establishes that dsDNA sensing is sufficient to trigger ISG15 accumulation and ISGylation of the proteome in uninfected cells that can be suppressed by the HCMV IE1 transactivator.
A variety of host DNA binding regulatory proteins have been implicated in cyto-plasmic dsDNA sensing; however, a nonredundant role for cGAS in controlling inter-feron production has emerged (1). Although inhibiting cGAS-STING stimulated HCMV replication (34), the relevant effector molecules responsive to cGAS-STING remained unknown. Here, we show that ISG15, a single host factor induced in response to dsDNA-dependent cGAS-STING activation in uninfected and infected cells, potently restricts HCMV replication. This establishes ISG15 itself as an effector molecule of cGAS-STING activation. While ISG monomer and protein conjugate accumulation re-sults predominately from cGAS-STING-stimulated IFN- production, it was not com-pletely abrogated when a JAK inhibitor inhibited IFN receptor signaling. Thus, we are unable to unambiguously exclude the possibility that a minor amount of ISG15 accumulates through a JAK inhibitor-insensitive noncanonical pathway. Such a path-way has been reported in IFN receptor-deficient cell lines (17, 18).
While depletion of ISG15 ligases is effective at partially limiting HCMV-induced ISG15 conjugation, we found a negligible impact on viral replication. Our results contrast with the reported 5- to 11-fold increase in HCMV replication upon Herc5 depletion (33). This could reflect differences in HCMV strains (AD169 versus Towne) and/or methods used to measure virus productive growth between the two studies. However, while our
FIG 5Suppression of dsDNA-induced ISGylation by HCMV IE1. NHDFs stably expressing eGFP or HCMV IE1 were transfected without DNA, with a 60-nt single-stranded DNA (ssHSV-60), or with a 60-bp duplex DNA (dsHSV-60). After 24 h, total protein was collected, fractionated by SDS-PAGE, and analyzed by immunoblotting using antibodies specific for ISG15, HCMV IE1, or-actin (loading control). Migration of molecular mass standards is shown on the right. (B) As in panel A, except total RNA was isolated and RT-qPCR was performed using primers specific for ISG15 or Herc5 mRNA. The error bars indicate SEM.*,Pⱕ0.05 by Student’s
ttest.
on November 7, 2019 by guest
http://jvi.asm.org/
[image:9.585.47.450.74.245.2]results are consistent with the minimal 2-fold increase in HCMV replication reported upon UBA7 depletion (33), our interpretations are at odds, reflecting the far greater potency of ISG15 itself than of ISG15 conjugation enzymes in restricting HCMV repli-cation. The existence of antiviral roles for both conjugated and unconjugated ISG15 provides another possible explanation for these differences. Depletion of UBA7 or Herc5 ligase resulted in increased abundance of unconjugated ISG15, potentially intensifying its antiviral role and masking the proviral impact of inhibiting ISG15 conjugation. Alternatively, siRNA depletion of these ligases, which act enzymatically, may not reduce their abundance enough to sufficiently stimulate viral replication, even though reduced ISGylation is detected by immunoblotting. While our data suggest a greater role for ISG15 itself in restricting HCMV replication, it might simply be a better candidate for depletion precisely because it is not an enzyme and largely acts stoichi-ometrically. Attempts to distinguish discrete roles for conjugated versus unconjugated ISG15 have so far been equivocal in HCMV-infected cells (33; C. Bianco and I. Mohr, unpublished observations). Finally, because ISGylation occurs cotranslationally (10), HCMV might further influence host proteins targeted for ISG15 conjugation by regu-lating which host mRNAs are associated with polyribosomes (22, 29). Excluding mRNAs from polyribosomes that code for antiviral proteins whose activities might be aug-mented by cotranslational ISGylation would limit the impact of ISG15 conjugation on HCMV replication.
Although diminished at late times postinfection, the persistence of ISG15 monomer and high-molecular-weight protein conjugate accumulation throughout the HCMV replication cycle is puzzling. Even though ISG15 limits HCMV replication and the virus encodes functions capable of attenuating ISG15 accumulation, ISG15 is clearly tolerated in HCMV-infected cell populations. Perhaps a subset of ISGs might be repurposed to facilitate viral replication while a tonic level of antiviral ISGs is tolerated and/or antagonized. Roles for IFI16 in promoting transcription of immediate-early viral genes (38) and for viperin in stimulating replication-required fatty acid biosynthesis (39), which reportedly occurs prior to the reduction in ISG15 levels, are consistent with this possibility. Another possibility is that ISG15 monomer and protein conjugate accumu-lation may inhibit ISG expression and/or the functions of ISG products at later times during infection. The ISG USP18, a negative regulator of interferon signaling (40), is stabilized by ISG15, and their abundances are correlated (41). USP18 is proviral toward many viruses, including HCMV (14). ISG15 accumulation early during infection might stabilize USP18, curtailing cell-intrinsic immune responses later in infection. This would allow HCMV to benefit from repurposed proviral ISG expression early in infection while reducing ISGylation later. Finally, our data on the population level may simply reflect stochastic decisions on the single-cell level. In this scenario, HCMV replication would be restricted in cells where ISG15 accumulates but would proceed to completion in those cells where ISG15 accumulation is antagonized by viral functions.
Our finding that dsDNA is sufficient to induce ISG15 monomer and protein conju-gate accumulation in uninfected cells has numerous important ramifications. How this DNA-sensing-induced ISG15 modification might impact experimental outcomes and the functionality of proteins expressed from transfected DNA in uninfected cells needs to be considered. This could have far-reaching consequences ranging from interpreting experimental results to understanding DNA vaccine efficacy. Likewise, stimulating dsDNA-sensing pathways could result in proteomes distinguished by constitutively elevated ISG15 monomer and protein conjugates and influence autoimmune disease pathogenesis (42, 43). Type I interferonopathies, like Aicardi-Gourierres syndrome (AGS) and systemic lupus erythematosus (SLE), are characterized by excessive type I interferon production and result from mutations in a number of genes, including one that limits cytoplasmic dsDNA accumulation, encoding the exonuclease TREX1 (44–46). Remark-ably, autoimmunity associated with TREX1 deficiency is dependent upon cGAS, further indicating a role for dsDNA-sensing disease pathology (47, 48). A modified proteome enriched with ISGylated proteins resulting from dsDNA-induced ISG15 accumulation might contribute to disease severity or progression.
on November 7, 2019 by guest
http://jvi.asm.org/
MATERIALS AND METHODS
Cell culture and viruses.Normal human dermal fibroblasts (Lonza) were cultured in Dulbecco’s modified Eagle medium (DMEM) (Corning; 10-013-CV) supplemented with 5% (vol/vol) fetal bovine serum (FBS) and 100 U/ml penicillin-100g/ml streptomycin (Corning; MT-30-002-CI) at 37°C in 5% CO2. The AD169GFP virus was a gift from Dong Yu and was propagated in NHDFs, as previously described (49). The virus was concentrated by centrifugation at 20,000 rpm in an SW28 rotor for 75 min at 18°C, followed by resuspension in 1.5% bovine serum albumin in DMEM. Titers of virus stocks were determined using a plaque assay. Where indicated, virus was UV inactivated with 6 pulses of 125 mJ/cm2of UV irradiation, as previously described (50). For experiments, virus titers were determined using a 50% tissue culture infective dose (TCID50) assay, as previously described (22, 51).
Chemicals and chemical treatments.Phosphonoacetic acid (Sigma; P6909) was dissolved in water and used at a concentration of 300g/ml. Pyridone 6/JAK inhibitor I (Millipore; 420099-500UG) was dissolved in dimethyl sulfoxide (DMSO). The indicated concentrations of chemicals were added to cultures at the time of infection.
qPCR.RNA was isolated from cells using TRIzol reagent (Invitrogen; 15596018) according to the manufacturer’s protocol. cDNA was synthesized from 250 ng of purified RNA using qScript cDNA Master Mix (Quanta; 95048) according to the manufacturer’s protocol. Quantitative PCR (qPCR) was performed in a Bio-Rad IQ5 thermal cycler using IQ Sybr green Supermix (Bio-Rad; 170-8882) with a 58°C annealing temperature and the following primers at 0.5M: ISG15-Forward (5=-AGATCACCC AGAAGATCG-3=), ISG15-Reverse (5=-TGTTATTCCTCACCAGGATG-3=), Herc5-Forward (5=-AAAATTGAG ACGGTGCAAGAG-3=), Herc5-Reverse (5=-TGTTGAAGAAGCTGCACAGG-3=), GAPDH-Forward (5=-TCTTT TGCGTCGCCAGCCGA-3=), and GAPDH-Reverse (5=-ACCAGGCGCCCAATACGACC-3=).
DNA transfections.DNA was transfected using Lipofectamine 2000 (Invitrogen; 11668019) accord-ing to the manufacturer’s protocol at a ratio of 3l Lipofectamine 2000 to 1g DNA. HSV-1 60-mer oligonucleotides (5=-AGTCGTAAAAAAGTTTTATCTCTTTCTCTCTTCGATGGTCTCACAAAAATATTAAACCTCTT TCTGATGG-3=and 5=-CCATCAGAAAGAGGTTTAATATTTTTGTGAGACCATCGAAGAGAGAAAGAGATAAAACT TTTTTACGACT-3=) were synthesized by Integrated DNA Technologies and annealed in annealing buffer (10 mM Tris-HCl, pH 7.5, 100 mM NaCl, and 1 mM EDTA). For plasmid DNA transfections, the plasmid pFLAG-CMV-5a in annealing buffer was used. For “no-DNA” transfections, annealing buffer with no nucleic acids was used.
siRNA transfections.siRNAs were transfected at 20 nM using RNAimax (Invitrogen; 13778075) as previously described (52). The sequences of siRNAs (from Sigma unless otherwise indicated) used in this study were as follows: ISG15 no. 1, CAGACCGUGGCCCACCUGA; ISG15 no. 2, GGUGUGGCGCUGCAGGACA; cGAS no. 1, GCUACUAUGAGCACGUGAA; cGAS no. 2, GCUGUAACACUUCUUAUUA; STING no. 1, GCCUC AUUGCCUACCAGGA; STING no. 2, GGCUUUAGCCGGGAGGAUA; UBA7 no. 1, CUAUGAUGGGCAAAUUGCA; UBA7 no. 2, CUAAUAAAGUGCUUGAGGA; Herc5 (Qiagen), GAGGAGAATGGTAATGTTCAA.
Immunoblotting and antibodies.Cells were lysed in Laemmli buffer (60M Tris-HCl, pH 6.8, 2% SDS, 10% glycerol, 710 mM 2-mercaptoethanol), and immunoblotting was performed as previously described (53). The antibodies used in this study were as follows: anti-ISG15 (Proteintech Group; 15981-1-AP), anti-STAT1 (Cell Signaling; 9172), anti-STAT1 Y701 (Cell Signaling; 7649), anti-actin (Cell Signaling; 3700), anti-cGAS (Proteintech Group; 26416-1-AP), anti-STING (Cell Signaling; 13647), anti-Akt (Cell Signaling; 9272), anti-UBA7 (Cell Signaling; 69023), anti-Herc5 (Enzo; BML-PW0920), anti-pp28 (Virusys; CA004-100), anti-pp65 (Virusys; CA003), anti-UL44 (Virusys; CA006), and anti-IE1/IE2 (Millipore; MAB810).
Lentiviruses. Lentiviral transfer vectors expressing GFP (pLJM1-EGFP; Addgene; 19319) and IE1 (pLJM1-IE1) were gifts from James Alwine (University of Pennsylvania). Lentivirus particles were produced by cotransfection of 293LTV cells (Cell Biolabs; LTV-100) with psPAX2 (Addgene; 12260), pMDG.2 (Addgene; 12259), and the appropriate transfer vector. Lentivirus-containing supernatants were har-vested 48 and 72 h posttransfection, pooled, filtered through a 0.45-m polyvinylidene difluoride (PVDF) filter, and used to transduce NHDFs; 72 h postransduction, the transduced cells were selected with puromycin (2g/ml).
ACKNOWLEDGMENTS
We thank members of the Mohr laboratory and Angus Wilson for stimulating discussions and Hannah Burgess for critically reading the manuscript.
This work was supported by grants from the NIH to I.M. (R01GM056927). C.B. was supported in part by NIH grants 5T32AI007180 and 5T32AI7647. The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication.
REFERENCES
1. Chen Q, Sun L, Chen ZJ. 2016. Regulation and function of the cGAS-STING pathway of cytosolic DNA sensing. Nat Immunol 17:1142–1149.
https://doi.org/10.1038/ni.3558.
2. Cai X, Chiu Y-H, Chen ZJ. 2014. The cGAS-cGAMP-STING pathway of cytosolic DNA sensing and signaling. Mol Cell 54:289 –296.https://doi .org/10.1016/j.molcel.2014.03.040.
3. Schoggins JW. 2014. Interferon-stimulated genes: roles in viral patho-genesis. Curr Opin Virol 6:40 – 46.https://doi.org/10.1016/j.coviro.2014 .03.006.
4. Fensterl V, Chattopadhyay S, Sen GC. 2015. No love lost between viruses and interferons. Annu Rev Virol 2:549 –572. https://doi.org/10.1146/ annurev-virology-100114-055249.
on November 7, 2019 by guest
http://jvi.asm.org/
5. Morales DJ, Lenschow DJ. 2013. The antiviral activities of ISG15. J Mol Biol 425:4995–5008.https://doi.org/10.1016/j.jmb.2013.09.041. 6. Zhao C, Collins MN, Hsiang TY, Krug RM. 2013. Interferon-induced ISG15
pathway: an ongoing virus-host battle. Trends Microbiol 21:181–186.
https://doi.org/10.1016/j.tim.2013.01.005.
7. Hermann M, Bogunovic D. 2017. ISG15: in sickness and in health. Trends Immunol 38:79 –93.https://doi.org/10.1016/j.it.2016.11.001.
8. Kerscher O, Felberbaum R, Hochstrasser M. 2006. Modification of pro-teins by ubiquitin and ubiquitin-like propro-teins. Annu Rev Cell Dev Biol 22:159 –180.https://doi.org/10.1146/annurev.cellbio.22.010605.093503. 9. Zhang D, Zhang D-E. 2011. Interferon-stimulated gene 15 and the
protein ISGylation system. J Interferon Cytokine Res 31:119 –130.https:// doi.org/10.1089/jir.2010.0110.
10. Durfee LA, Lyon N, Seo K, Huibregtse JM. 2010. The ISG15 conjugation system broadly targets newly synthesized proteins: implications for the antiviral function of ISG15. Mol Cell 38:722–732.https://doi.org/10.1016/ j.molcel.2010.05.002.
11. Okumura F, Zou W, Zhang D-E. 2007. ISG15 modification of the eIF4E cognate 4EHP enhances cap structure-binding activity of 4EHP. Genes Dev 21:255–260.https://doi.org/10.1101/gad.1521607.
12. Kim M-J, Hwang S-Y, Imaizumi T, Yoo J-Y. 2008. Negative feedback regulation of RIG-I-mediated antiviral signaling by interferon-induced ISG15 conjugation. J Virol 82:1474 –1483. https://doi.org/10.1128/JVI .01650-07.
13. Shi H-X, Yang K, Liu X, Liu X-Y, Wei B, Shan Y-F, Zhu L-H, Wang C. 2010. Positive regulation of interferon regulatory factor 3 activation by Herc5 via ISG15 modification. Mol Cell Biol 30:2424 –2436.https://doi.org/10 .1128/MCB.01466-09.
14. Speer SD, Li Z, Buta S, Payelle-Brogard B, Qian L, Vigant F, Rubino E, Gardner TJ, Wedeking T, Hermann M, Duehr J, Sanal O, Tezcan I, Man-souri N, Tabarsi P, ManMan-souri D, Francois-Newton V, Daussy CF, Rodriguez MR, Lenschow DJ, Freiberg AN, Tortorella D, Piehler J, Lee B, García-Sastre A, Pellegrini S, Bogunovic D. 2016. ISG15 deficiency and increased viral resistance in humans but not mice. Nat Commun 7:11496.https:// doi.org/10.1038/ncomms11496.
15. Malakhova OA, Zhang D-E. 2008. ISG15 inhibits Nedd4 ubiquitin E3 activity and enhances the innate antiviral response. J Biol Chem 283: 8783– 8787.https://doi.org/10.1074/jbc.C800030200.
16. Bogunovic D, Boisson-Dupuis S, Casanova J-L. 2013. ISG15: leading a double life as a secreted molecule. Exp Mol Med 45:e18.https://doi.org/ 10.1038/emm.2013.36.
17. Hasan M, Koch J, Rakheja D, Pattnaik AK, Brugarolas J, Dozmorov I, Levine B, Wakeland EK, Lee-Kirsch MA, Yan N. 2013. Trex1 regulates lysosomal biogenesis and interferon-independent activation of antiviral genes. Nat Immunol 14:61–71.https://doi.org/10.1038/nri3603. 18. Radoshevich L, Impens F, Ribet D, Quereda JJ, Nam Tham T, Nahori M-A,
Bierne H, Dussurget O, Pizarro-Cerdá J, Knobeloch K-P, Cossart P. 2015. ISG15 counteracts Listeria monocytogenes infection. eLife 4:e06848.
https://doi.org/10.7554/eLife.06848.
19. Lenschow DJ, Lai C, Frias-Staheli N, Giannakopoulos NV, Lutz A, Wolff T, Osiak A, Levine B, Schmidt RE, García-Sastre A, Leib DA, Pekosz A, Knobeloch K-P, Horak I, Virgin HW. 2007. IFN-stimulated gene 15 func-tions as a critical antiviral molecule against influenza, herpes, and Sind-bis viruses. Proc Natl Acad Sci U S A 104:1371–1376.https://doi.org/10 .1073/pnas.0607038104.
20. Jacobs SR, Stopford CM, West JA, Bennett CL, Giffin L, Damania B. 2015. Kaposi’s sarcoma-associated herpesvirus viral interferon regulatory fac-tor 1 interacts with a member of the interferon-stimulated gene 15 pathway. J Virol 89:11572–11583.https://doi.org/10.1128/JVI.01482-15. 21. Jan E, Mohr I, Walsh D. 2016. A cap-to-tail guide to mRNA translation
strategies in virus-infected cells. Annu Rev Virol 3:283–307.https://doi .org/10.1146/annurev-virology-100114-055014.
22. McKinney C, Zavadil J, Bianco C, Shiflett L, Brown S, Mohr I. 2014. Global reprogramming of the cellular translational landscape facilitates cyto-megalovirus replication. Cell Rep 6:9 –17.https://doi.org/10.1016/j.celrep .2013.11.045.
23. Britt W. 2008. Manifestations of human cytomegalovirus infection: pro-posed mechanisms of acute and chronic disease, p 417– 470.InShenk TE, Stinski MF (ed),Human cytomegalovirus. Springer, Berlin, Germany. 24. Boeckh M, Geballe AP. 2011. Cytomegalovirus: pathogen, paradigm, and
puzzle. J Clin Invest 121:1673–1680.https://doi.org/10.1172/JCI45449. 25. Ljungman P, Hakki M, Boeckh M. 2010. Cytomegalovirus in
hematopoi-etic stem cell transplant recipients. Infect Dis Clin North Am 24:319 –337.
https://doi.org/10.1016/j.idc.2010.01.008.
26. Razonable RR, Humar A, AST Infectious Diseases Community of Practice. 2013. Cytomegalovirus in solid organ transplantation. Am J Transplant 13:93–106.https://doi.org/10.1111/ajt.12103.
27. Cannon MJ, Schmid DS, Hyde TB. 2010. Review of cytomegalovirus seroprevalence and demographic characteristics associated with infec-tion. Rev Med Virol 20:202–213.https://doi.org/10.1002/rmv.655. 28. Manicklal S, Emery VC, Lazzarotto T, Boppana SB, Gupta RK. 2013. The
“silent” global burden of congenital cytomegalovirus. Clin Microbiol Rev 26:86 –102.https://doi.org/10.1128/CMR.00062-12.
29. Tirosh O, Cohen Y, Shitrit A, Shani O, Le-Trilling VTK, Trilling M, Fried-lander G, Tanenbaum M, Stern-Ginossar N. 2015. The transcription and translation landscapes during human cytomegalovirus infection reveal novel host-pathogen interactions. PLoS Pathog 11:e1005288.https://doi .org/10.1371/journal.ppat.1005288.
30. Marshall EE, Geballe AP. 2009. Multifaceted evasion of the interferon response by cytomegalovirus. J Interferon Cytokine Res 29:609 – 619.
https://doi.org/10.1089/jir.2009.0064.
31. Nicholl MJ, Robinson LH, Preston CM. 2000. Activation of cellular interferon-responsive genes after infection of human cells with herpes simplex virus type 1. J Gen Virol 81:2215–2218.https://doi.org/10.1099/ 0022-1317-81-9-2215.
32. McFarlane S, Aitken J, Sutherland JS, Nicholl MJ, Preston VG, Preston CM. 2011. Early induction of autophagy in human fibroblasts after infection with human cytomegalovirus or herpes simplex virus 1. J Virol 85: 4212– 4221.https://doi.org/10.1128/JVI.02435-10.
33. Kim YJ, Kim ET, Kim Y-E, Lee MK, Kwon KM, Kim KI, Stamminger T, Ahn J-H. 2016. Consecutive inhibition of ISG15 expression and ISGylation by cytomegalovirus regulators. PLoS Pathog 12:e1005850.https://doi.org/ 10.1371/journal.ppat.1005850.
34. Lio C-WJ, McDonald B, Takahashi M, Dhanwani R, Sharma N, Huang J, Pham E, Benedict CA, Sharma S. 2016. cGAS-STING signaling regulates initial innate control of cytomegalovirus infection. J Virol 90:7789 –7797.
https://doi.org/10.1128/JVI.01040-16.
35. Paijo J, Döring M, Spanier J, Grabski E, Nooruzzaman M, Schmidt T, Witte G, Messerle M, Hornung V, Kaever V, Kalinke U. 2016. cGAS senses human cytomegalovirus and induces type I interferon responses in human monocyte-derived cells. PLoS Pathog 12:e1005546.https://doi .org/10.1371/journal.ppat.1005546.
36. Akira S, Uematsu S, Takeuchi O. 2006. Pathogen recognition and innate immunity. Cell 124:783– 801.https://doi.org/10.1016/j.cell.2006.02.015. 37. Paulus C, Krauss S, Nevels M. 2006. A human cytomegalovirus antagonist
of type I IFN-dependent signal transducer and activator of transcription signaling. Proc Natl Acad Sci U S A 103:3840 –3845.https://doi.org/10 .1073/pnas.0600007103.
38. Cristea IM, Moorman NJ, Terhune SS, Cuevas CD, O’Keefe ES, Rout MP, Chait BT, Shenk T. 2010. Human cytomegalovirus pUL83 stimulates activity of the viral immediate-early promoter through its interaction with the cellular IFI16 protein. J Virol 84:7803–7814.https://doi.org/10 .1128/JVI.00139-10.
39. Seo J-Y, Cresswell P. 2013. Viperin regulates cellular lipid metabolism during human cytomegalovirus infection. PLoS Pathog 9:e1003497.
https://doi.org/10.1371/journal.ppat.1003497.
40. Malakhova OA, Kim KII, Luo J-K, Zou W, Kumar KGS, Fuchs SY, Shuai K, Zhang D-E. 2006. UBP43 is a novel regulator of interferon signaling independent of its ISG15 isopeptidase activity. EMBO J 25:2358 –2367.
https://doi.org/10.1038/sj.emboj.7601149.
41. Zhang X, Bogunovic D, Payelle-Brogard B, Francois-Newton V, Speer SD, Yuan C, Volpi S, Li Z, Sanal O, Mansouri D, Tezcan I, Rice GI, Chen C, Mansouri N, Mahdaviani SA, Itan Y, Boisson B, Okada S, Zeng L, Wang X, Jiang H, Liu W, Han T, Liu D, Ma T, Wang B, Liu M, Liu J-Y, Wang QK, Yalnizoglu D, Radoshevich L, Uzé G, Gros P, Rozenberg F, Zhang S-Y, Jouanguy E, Bustamante J, García-Sastre A, Abel L, Lebon P, Notarangelo LD, Crow YJ, Boisson-Dupuis S, Casanova J-L, Pellegrini S. 2015. Human intracellular ISG15 prevents interferon-␣/over-amplification and auto-inflammation. Nature 517:89 –93.https://doi.org/10.1038/nature13801. 42. Stetson DB, Ko JS, Heidmann T, Medzhitov R. 2008. Trex1 prevents
cell-intrinsic initiation of autoimmunity. Cell 134:587–598. https://doi .org/10.1016/j.cell.2008.06.032.
43. Gao D, Li T, Li X-D, Chen X, Li Q-Z, Wight-Carter M, Chen ZJ. 2015. Activation of cyclic GMP-AMP synthase by self-DNA causes autoimmune diseases. Proc Natl Acad Sci U S A 112:E5699 –E5705.https://doi.org/10 .1073/pnas.1516465112.
44. Crow YJ, Hayward BE, Parmar R, Robins P, Leitch A, Ali M, Black DN, van Bokhoven H, Brunner HG, Hamel BC, Corry PC, Cowan FM, Frints SG,
on November 7, 2019 by guest
http://jvi.asm.org/
Klepper J, Livingston JH, Lynch SA, Massey RF, Meritet JF, Michaud JL, Ponsot G, Voit T, Lebon P, Bonthron DT, Jackson AP, Barnes DE, Lindahl T. 2006. Mutations in the gene encoding the 3⬙-5⬙DNA exonuclease TREX1 cause Aicardi-Goutières syndrome at the AGS1 locus. Nat Genet 38:917–920.https://doi.org/10.1038/ng1845.
45. Lehtinen DA, Harvey S, Mulcahy MJ, Hollis T, Perrino FW. 2008. The TREX1 double-stranded DNA degradation activity is defective in domi-nant mutations associated with autoimmune disease. J Biol Chem 283: 31649 –31656.https://doi.org/10.1074/jbc.M806155200.
46. Fye JM, Orebaugh CD, Coffin SR, Hollis T, Perrino FW. 2011. Dominant mutation of the TREX1 exonuclease gene in lupus and Aicardi-Goutieres syndrome. J Biol Chem 286:32373–32382.https://doi.org/10.1074/jbc .M111.276287.
47. Ablasser A, Hemmerling I, Schmid-Burgk JL, Behrendt R, Roers A, Hor-nung V. 2014. TREX1 deficiency triggers cell-autonomous immunity in a cGAS-dependent manner. J Immunol 192:5993–5997.https://doi.org/10 .4049/jimmunol.1400737.
48. Grieves JL, Fye JM, Harvey S, Grayson JM, Hollis T, Perrino FW. 2015. Exonuclease TREX1 degrades double-stranded DNA to prevent
sponta-neous lupus-like inflammatory disease. Proc Natl Acad Sci U S A 112: 5117–5122.https://doi.org/10.1073/pnas.1423804112.
49. McKinney C, Perez C, Mohr I. 2012. Poly(A) binding protein abundance regulates eukaryotic translation initiation factor 4F assembly in human cytomegalovirus-infected cells. Proc Natl Acad Sci U S A 109:5627–5632.
https://doi.org/10.1073/pnas.1202829109.
50. Walsh D, Perez C, Notary J, Mohr I. 2005. Regulation of the translation initiation factor eIF4F by multiple mechanisms in human cytomegalovirus-infected cells. J Virol 79:8057– 8064.https://doi.org/10 .1128/JVI.79.13.8057-8064.2005.
51. Reed LJ, Muench H. 1938. A simple method of estimating fifty per cent endpoints. Am J Epidemiol 27:493– 497. https://doi.org/10.1093/ oxfordjournals.aje.a118408.
52. Burgess HM, Mohr I. 2015. Cellular 5=-3=mRNA exonuclease Xrn1 con-trols double-stranded RNA accumulation and anti-viral responses. Cell Host Microbe 17:332–334.https://doi.org/10.1016/j.chom.2015.02.003. 53. Walsh D. 2004. Phosphorylation of eIF4E by Mnk-1 enhances HSV-1
translation and replication in quiescent cells. Genes Dev 18:660 – 672.
https://doi.org/10.1101/gad.1185304.