0022-538X/01/$04.00⫹0 DOI: 10.1128/JVI.75.23.11827–11833.2001 Copyright © 2001, American Society for Microbiology. All Rights Reserved.
A Single Amino Acid in the Reverse Transcriptase Domain of
Hepatitis B Virus Affects Virus Replication Efficiency
XU LIN,1ZHENG-HONG YUAN,1LI WU,1† JIAN-PING DING,2ANDYU-MEI WEN1*
Department of Molecular Virology, Medical Center of Fudan University, Shanghai 200032,1and Shanghai Institute of
Biochemistry and Cell Biology, Chinese Academy of Sciences, Shanghai 200031,2People’s Republic of China
Received 12 June 2001/Accepted 28 August 2001
To explore functional domains in the hepatitis B virus (HBV) polymerase, two naturally occurring HBV isolates (56 and 2-18) with 98.7% nucleic acid sequence homology but different replication efficiencies were studied. After transfection into HepG2 cells, HBV DNA isolated from intracellular virus core particles was much higher in 56-transfected cells than in cells transfected with 2-18. The structural basis for the difference in replication efficiency between these two isolates was studied by functional domain gene substitution. The complete polymerase (P) gene and its gene segments coding for the terminal protein (TP), spacer (SP), reverse transcriptase (RT), and RNase H in 2-18 were separately replaced with their counterparts from 56 to construct full-length chimeric genomes. Cell transfection analysis revealed that substitution of the complete P gene of 2-18 with the P gene from 56 slightly enhanced viral replication. The only chimeric genome that regained the high replication efficiency of the original 56 isolate was the one with substitution of the RT gene of 2-18 with that from 56. Within the RT region, amino acid differences between isolates 2-18 and 56 were located at positions 617 (methionine versus leucine), 652 (serine versus proline), and 682 (valine versus leucine). Point mutation identified amino acid 652 as being responsible for the difference in replication efficiency. Homologous modeling studies of the HBV RT domain suggest that the mutation of residue 652 from proline to serine might affect the conformation of HBV RT which interacts with the template-primer, leading to impaired polymerase activity.
Hepatitis B virus (HBV) infection results in a broad spec-trum of clinical manifestations, including asymptomatic car-riage, acute self-limited hepatitis, chronic hepatitis, and fulmi-nant hepatitis. Variability in host immune response is mainly responsible for the diverse clinical manifestations, but viral mutations may also affect the clinical course of HBV infection (11). Enhanced replication efficiency of HBV variants could lead to increased viral virulence associated with high virus load in the host, which may influence the response to treatment. Despite containing a DNA genome, HBV replicates via a re-verse transcription process, using the polymerase encoded by its own genome (4). This polymerase, designated P, is com-posed of four domains (18). Starting from the N terminus, they are (i) the terminal protein (TP), which becomes covalently linked to the negative-strand DNA to initiate reverse transcrip-tion; (ii) the spacer, which has not been associated with a specific function and is tolerant to mutations; (iii) the reverse transcriptase (RT), which contains the YMDD consensus mo-tif that is also found in human immunodeficiency virus type 1 (HIV-1) reverse transcriptase; and (iv) RNase H, which digests the RNA in RNA-DNA hybrids. Studies have shown that the replication of the HBV genome is a complex process involving a priming reaction, translocation of complementary sequences,
reverse transcription, and second-strand DNA synthesis (16, 19, 20).
Two main experimental approaches have been used to in-vestigate the functions of HBV polymerase. One is the molec-ular approach, which consists of developing systems for direct analysis of full-length polymerase function in the absence of viral replication and other viral proteins (2, 3, 13), and transcomplementation studies between functional segments of HBV polymerase (14, 15). The other is the clinical approach, which consists of structural and functional analysis of HBV P genes in various naturally occurring HBV strains from infected patients. A nonreplicative HBV mutant with a single amino acid change in the TP domain of HBV polymerase has been reported (5). Different types of HBV splice variants have been described in HBV-infected patients under immunosuppressive treatment (10), and HBV core internal deletion mutations that inhibited the replication of wild-type virus have also been de-scribed (22).
In cells, replication of HBV not only requires a functional active polymerase, but also depends on host factors that can interact with various elements in the viral genome and viral proteins. Therefore, the most authentic system in which to study HBV polymerase function would involve full-length ge-nome replication in liver-derived cells. The approach of am-plifying full-length HBV genomes from serum or tissues of HBV-infected patients followed by hepatic cell line transfec-tion (10) provided an excellent platform for studying the func-tional diversity of HBV polymerase from naturally occurring strains.
In a study of the biological properties of various HBV iso-lates, we have come across two isolates with high nucleotide * Corresponding author. Mailing address: Department of Molecular
Virology, Medical Center of Fudan University (formerly Shanghai Medical University), 138 Yi Xue Yuan Road, Shanghai 200032, Peo-ple’s Republic of China. Phone: 86-21-64041900, ext. 2116/2523. Fax: 86-21-64174578. E-mail: [email protected].
† Present address: HIV Drug Resistance Program, National Cancer Institute, Frederick, MD 21702.
11827
on November 9, 2019 by guest
http://jvi.asm.org/
sequence homology but marked difference in their replication efficiency (21). In the current study, we used gene segment substitution to generate chimeric HBV genomes and identified an amino acid in the RT domain which affected the efficiency of intracellular replication of HBV.
MATERIALS AND METHODS
Full-length HBV genome cloning.Six HBV DNA-positive serum or liver tissue samples from chronic hepatitis B patients or hepatocellular carcinoma patients were collected (14, 16, 2-18, 20, 56, and 97). Full-length HBV genomes were separately amplified by PCR, followed by cloning into vector pUC18 via theSstI restriction site as described by Gu¨nther et al. (10). These six HBV DNA genomes were sequenced separately (GenBank accession numbers: 2-18, AF100308; 56, AF100309; 14, 16, 20, and 97 have been submitted and accession numbers are pending).
Cell culture and transfection.HepG2 cells were cultured in Dulbecco’s mod-ified Eagle’s medium with penicillin (100 U/ml) and streptomycin (100g/ml), supplemented with 10% fetal calf serum. Recombinant plasmid DNA used for transfection was extracted and purified with the Qiagen Maxiprep kits, and HBV DNA was released from the recombinant plasmids by digestion withSapI at 1 U of enzyme/g of DNA for 16 h, followed by extraction and purification. HBV DNA (25g) from each clone was used to transfect HepG2 cells in 60-mm plates using the calcium phosphate precipitation method as reported previously (21). Duplicate plates were used for all samples, and 10g of reporter plasmid DNA expressing secreted alkaline phosphatase (SEAP) was cotransfected into each culture as an internal control to normalize the transfection efficiency among plates. Cells were collected separately 72 h after transfection, which has been shown previously to be the appropriate time for monitoring replication efficiency and expression of antigens by HBV isolates in this laboratory. Vector pUC18 DNA was used as a mock transfection control. Transfection experiments were done twice, repeated on separate days.
Purification of HBV DNA from intracellular core particles.As reported by Gu¨nther et al. (10), with some modifications, cells were washed twice with chilled phosphate-buffered saline buffer and lysed with 600l of lysing buffer (10 mM Tris-HCl [pH 7.9], 1 mm EDTA, 1% NP-40, 8% sucrose). After centrifugation at 12,000⫻gfor 2 min, cleared supernatants were collected, and 6l of 1 M magnesium acetate (MgOAc), 12l of 5-mg/ml DNase I, and 3l of 20-mg/ml RNase A were added, followed by incubation at 37°C for 30 min to digest remaining DNA and RNA. After a quick spin at 12,000⫻gfor 1 min,
super-natants were collected, 16l of 0.5 M EDTA and 130l of 35% polyethylene glycol 8000 in 1.75 M NaCl were added, and the mixture was kept on ice for 1 h to precipitate core particles. After spinning at 9,000⫻gfor 5 min, pellets were resuspended in 100l of buffer with DNase I (10 mM Tris-HCl [pH 7.9], 6 mM MgOAc, 0.1g of DNase I perg) and incubated at 37°C for 10 min to ensure digestion of any remaining DNA used for transfection. The resuspended core particles were digested with proteinase K at 1 mg/ml in 0.5% sodium dodecyl sulfate at 37°C for 5 h. Nucleic acids were extracted with phenol-chloroform (1:1) once and chloroform once and precipitated with ethanol.
Southern blot and HBV DNA hybridization. The amount of HBV DNA extracted from intracellular cores from each plate of transfected cells was nor-malized using the expression of SEAP by enzyme-linked immunosorbent assay as an internal control. After electrophoresis in a 1.2% agarose gel, HBV core DNA was blotted onto a Hybond N⫹nylon membrane. Twenty-five nanograms of cloned full-length HBV DNA (adr subtype) was labeled by random primer with [32P]dCTP and used as the probe for hybridization. The signals were detected by autoradiography and scanned with densitometry for semiquantification of the intensity of signals.
Source of HBV DNA used for construction of chimeric genomes.Two full-length HBV genomes (56 and 2-18) which showed 98.7% nucleotide sequence homology but high (no. 56) and low (no. 2-18) replication efficiencies were used for the construction of chimeric genomes.
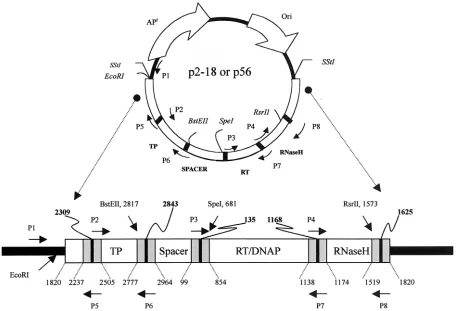
Construction of chimeric genomes based on p2-18 with the polymerase gene or its functional domain fragments replaced with counterparts from p56. (i) Search for the boundaries of functional regions.The P gene sequences of 2-18 and 56 were analyzed for DNA homology by Vector NTI Suite 5.5 AlignX software to identify boundaries of the functional regions to be replaced. In order to detect a significant change in the replication efficiency of the chimeric constructs, replace-ment of genes from the high-replication-efficiency isolate into the low-replica-tion-efficiency isolate was preferred. Therefore, p2-18 (a pUC18 backbone con-taining the full-length HBV DNA from strain 2-18 with low replication efficiency) was used as the acceptor, while p56 with high replication efficiency was used as the donor. For identification of the roles played by each of the four functional regions of the P gene, putative segments coding for terminal protein, spacer, RT/DNA polymerase, and RNase H in 2-18 were individually replaced with their counterparts from 56. The schematic diagram of the strategy used for replacement is shown in Fig. 1.
After the search, because of the limited number of restriction sites available, we adopted fusion PCR combined with enzyme digestion to achieve gene re-placement in most cases.
FIG. 1. Functional region of the 2-18 polymerase gene (open bars), HBV region flanking the polymerase gene of 2-18 (checkered bars), functional region of the 56 polymerase gene (shaded bars), and HBV region flanking the polymerase gene of 56 (stippled bars). 2-18P56, 2-18SP56, 2-18TP56, 2-18RNaseH56, and 2-18RT56are 2-18 chimeric genomes in which the complete P gene or each of the functional regions of the P gene, namely, spacer, TP, RNase H, and RT, was replaced with either the complete P gene or each of its counterparts from 56. 56RT2-18is a 56 HBV chimeric genome in which the RT region of the P gene was replaced with its 2-18 counterpart.
on November 9, 2019 by guest
http://jvi.asm.org/
(ii) Construction of chimeric genomes.To construct recombinant vector p2-18TP56, in which the TP region of the P gene of 2-18 was replaced with the TP region from 56, two pairs of primers were used: primer P1 (pUC reverse uni-versal primer), 5⬘-AGCGGATAACAATTTCACACAGGA-3⬘, and primer P2, 5⬘-AGACCACCAAATGCCCCTATC-3⬘(nucleotides [nt] 2299 to 2319); and primer P5, 5⬘-GATAGGGGCATTTGGTGGTCT-3⬘(nt 2319 to 2299, comple-mentary to primer P2), and primer P6, 5⬘-CTGAGTTGGCTTTGAAGCAG G-3⬘(nt 2971 to 2948). Primers P1 and P5 were used to amplify the 5⬘-flanking region of the P gene of p2-18 DNA, and primers P2 and P6 were used to amplify the TP region (nt 2309 to 2843) of p56 DNA. The two amplified fragments were annealed and fused by PCR with primers P1 and P6. The fused fragment was digested withEcoRI andBstEII and used to replace theEcoRI-BstEII fragment of p2-18 (Fig. 2.).
For the construction of p2-18P56, in which the complete P gene of 2-18 was replaced with that of 56, theBstEII-RsrII (nt 1573 to 2817) fragment of the P gene of p56 was recovered and ligated into p2-18P from which theBstEII-RsrII fragment had been removed (Fig. 2).
For the construction of p2-18SP56, in which the spacer region of 2-18 was replaced with the spacer region of 56, theBstEII-SpeI (nt 2817 to 681) fragment of the P gene of p2-18 was removed and replaced with its counterpart from p56 (Fig. 2).
For the construction of p2-18RT56, in which the reverse transcriptase region of the P gene of 2-18 was replaced with the same region of 56, two sets of primers were used: primer P3, 5⬘-CAAGGTATGTTGCCCGTTTGTCCTC-3⬘(nt 457 to 481); primer P4, 5⬘-CCTTTACCCCGTTGCTC-3⬘(nt 1139 to 1158); primer P7, 5⬘-GAGCAACGGGGTAAAGGTTC-3⬘ (nt 1158 to 1139, complementary to primer P4); and primer P8, 5⬘-GTTCACGGTGGTCTCCAT-3⬘ (nt 1610 to
1627). Primers P4 and P8 were used to amplify the RNase H region of the P gene of p2-18 DNA, while primers P3 and P7 were used to amplify the RT region (nt 854 to 1183) of p56 DNA. The two amplified fragments were annealed and fused by PCR with primers P3 and P8. The fused fragment was digested withSpeI and RsrII and used to replace theSpeI-RsrII fragment of p2-18 (Fig. 2).
The procedure used to construct p2-18RNaseH56, in which the RNase H region of the P gene of 2-18 was replaced with that of 56, was similar to the procedure for constructing p2-18RT56. However, primers P4 and P8 were used to amplify the RNase H region of the P gene of p2-18 DNA, and primers P3 and P7 were used to amplify the RT region (nt 854 to 1183) of p56 DNA.
Construction of chimeric genomes based on p56 with the RT domain replaced with its counterpart from p2-18.To confirm the critical role played by the RT region of the P gene, the RT region of 56 was replaced with its counterpart from 2-18. The procedure for construction of recombinant p56RT2-18, in which the RT region of 56 was replaced with that of 2-18, was similar to that for p2-18RNaseH56.
Replacement of amino acid in RT region of 2-18 with the 56 counterpart.
Based on the results obtained by substitution of the RT gene in p2-18 with that from p56 (see Results below), three site-directed mutants were generated. To construct recombinant p2-18617Met3Leuat amino acid 617, p2-18652Ser3Proat amino acid 652, and p2-18682Val3Leu
at amino acid 682, PCR-based site-directed mutagenesis was used to create the mutations at the corresponding nucleotides, nt 942, 1047, and 1137, respectively. The sequences of the primers used and their positions are shown in Table 1.
[image:3.587.65.528.77.388.2]Homologous modeling studies of HBV RT.In order to predict the difference in replication efficiency between isolates 56 and 2-18 at the three-dimensional structural level, homologous modeling studies of HBV polymerase were based FIG. 2. p2-18 and p56 are pUC18 recombinant plasmids containing either full-length HBV strain 2-18 or 56 cloned at theSstI site. The polymerase gene, ranging from nt 2309 to 1625, consists of four functional regions, TP (terminal protein), spacer, RT/DNA polymerase (DNAP), and RNase H. The 5⬘border of the TP region, 3⬘border of TP/5⬘border of spacer, 3⬘border of spacer/5⬘border of RT/DNA polymerase, 3⬘border of RT/DNA polymerase/5⬘border of RNase H, and 3⬘border of the RNase H region are located at nt 2309, 2843, 135, 1168, and 1625, respectively, and are indicated by vertical bars. Gray boxes indicate consensus regions flanking the borders of functional regions, and the nucleotide positions are shown. P1 is the pUC18 reverse universal primer, and P2 to P8 are primers located in the consensus regions. P1, P2, P5, and P6 were used for construction of the TP chimera of the P gene, whereas P3, P4, P7, and P8 were used for the construction of the RT/DNA polymerase and RNase H chimeras. The sequences of these primers are given in the text.
on November 9, 2019 by guest
http://jvi.asm.org/
on the work reported by Das et al. (7), but the sequence alignment between HBV RT and HIV RT was modified. A homologous model of HBV RT using HIV-1 RT (13), Moloney murine leukemia virus RT (9), and hepatitis C virus polymer-ase (1, 6) as modeling templates was constructed, which covered amino acid residues 319 to 700, comprising the entire RT domain of HBV polymerase. The HBV RT was modeled in a conformation similar to that of other viral poly-merases or RTs, with the nucleic acid substrate-binding cleft defined by struc-tural elements from the fingers, palm, and thumb subdomains. The DNA tem-plate-primer was docked into the model based on the crystal structure of the HIV-1 RT/DNA binary complex (8) and HIV-1 RT/DNA/deoxynucleoside triphosphate ternary complex.
RESULTS
Replication efficiency of six different HBV isolates.The rep-lication efficiency of the six HBV isolates was compared by the semiquantification of scanned intensity of signals detected by autoradiography. Setting the value for the isolate with the lowest replication efficiency (2-18) at 1, the relative intensities of the other isolates, 14, 16, 20, 56, and 97, were 32, 32, 21, 32, and 41, respectively.
Because 56 and 2-18 showed 98.7% nucleotide sequence homology (21), and restriction sites between these two isolates were matched, these two isolates were selected to construct chimeric genomes for structural analysis of differences in rep-lication efficiency.
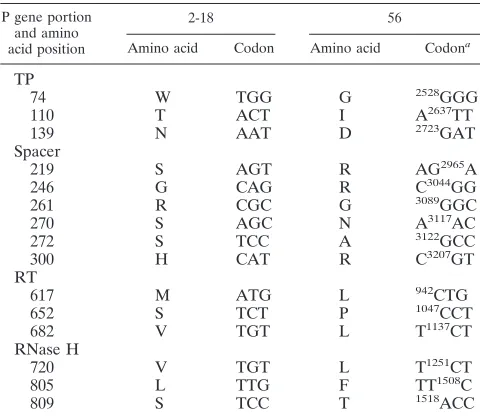
DNA homology and consensus boundary analysis of P gene between 56 and 2-18. DNA homology analysis (Vector NTI Suite 5.5 AlignX software) indicated that there were 15 pre-dicted amino acid differences between the polymerase genes of 2-18 and 56 (Table 2).
Consensus regions for both isolates flanking the boundaries of all four functional regions of the polymerase gene are shown in Fig. 2. Three restriction sites within these consensus regions suitable for cloning wereBstEII (nt 2817),SpeI (nt 681), and RsrII (nt 1573). Based on this analysis, procedures to generate chimeric HBV genomes were designed, and five chimeric ge-nomes that replaced either the complete P gene or each of its functional domain gene fragments were successfully con-structed and confirmed by restriction enzyme analysis and se-quencing.
Replication efficiency of 2-18 HBV chimeric genomes with functional domain genes replaced with 56 counterparts.The hybridization signals of HBV DNA isolated from intracellular core particles in transfected cells were markedly different among the five constructs used for transfection (Fig. 3). As controls, 56 showed strong hybridization signals indicating a high replication efficiency, while with 2-18 the hybridization
signal could barely be seen, indicating a low replication effi-ciency. When the spacer region of the P gene or the complete P gene of 2-18 was replaced with that of 56, the chimeric genome showed slightly enhanced replication compared to the original 2-18 (ratio of intensity, 5 versus 1). However, the HBV DNA hybridization signal was markedly increased in cells transfected with the chimeric genome 2-18RT56, in which the
[image:4.587.301.541.92.301.2]gene fragment coding for the reverse transcriptase region of 2-18 was replaced with the counterpart from 56 (ratio of in-tensity, 26 versus 1). Other chimeric genome constructs
[image:4.587.42.283.93.194.2]FIG. 3. 2-18 and 56 are original isolates with low and high rep-licative efficiencies, respectively. 2-18P56, 2-18SP56, 2-18TP56, 2-18RNaseH56, and 2-18RT56 are 2-18 HBV chimeric genomes in which the full-length P gene and functional regions of the P gene (spacer, TP, RNase H, and RT) were replaced with their counterparts from 56. Intensity of hybridization signal was measured by densitom-etry, and the relative intensity of these samples is indicated at the bottom. Lane M, molecular size standards.
TABLE 1. Sequences and nucleotide positions of primers used for PCR-based site-directed mutagenesis
Primer Sequencea
(5⬘33⬘) Nucleotide positions
PMD CGCAAAATACCTATGGGAGTG 1602–1622
PMU GTTCACGGTGGTCTCCATGCG 630–650
PA617 AAAACACAGⴱTTGATTTTTTGTACAATATG 921–951 PS617 AAATCAAACTGTGTTTTAGGAAACTTCCTG 939–969 PA652 TAAAGCAGGATATCCACATTGCGTGAAAG 1027–1057 PS652 GTGGATATCCTGCTTTAATGCCTTTATATG 1040–1070 PA682 AGGTTCAGATACTGTTTACTTAGAAAGGC 1117–1147 PS682 AACAGTATCTGAACCTTTACCCCGTTGCTC 1129–1159
aMutated nucleotides are underlined.
TABLE 2. Differences in nucleotides and predicted amino acids of HBV polymerase between isolates 2-18 and 56
P gene portion and amino acid position
2-18 56
Amino acid Codon Amino acid Codona
TP
74 W TGG G 2528GGG
110 T ACT I A2637TT
139 N AAT D 2723GAT
Spacer
219 S AGT R AG2965A
246 G CAG R C3044GG
261 R CGC G 3089GGC
270 S AGC N A3117AC
272 S TCC A 3122GCC
300 H CAT R C3207GT
RT
617 M ATG L 942CTG
652 S TCT P 1047CCT
682 V TGT L T1137CT
RNase H
720 V TGT L T1251CT
805 L TTG F TT1508C
809 S TCC T 1518ACC
a
The position of the altered nucleotide is indicated as a superscript to the left of the changed base.
on November 9, 2019 by guest
http://jvi.asm.org/
[image:4.587.306.537.485.652.2]showed no enhanced replication in this study. These results indicate that the RT gene of 56 was critical for the high rep-lication efficiency of this isolate.
Replication efficiency of 56 HBV chimeric genome with RT replaced with its 2-18 counterpart.Conversely, the replication efficiency of 56RT2-18, in which the RT region of the P gene of
56 was replaced with the 2-18 counterpart, was reduced com-pared to 56 (Fig. 4). The ratio of intensity of hybridization signals between 56RT2-18and 56 was 1:3.5, as assayed by
den-sitometry.
Replication efficiency of site-directed RT domain mutants. Among three site-directed mutants (2-18RT617, 2-18RT652,
and 2-18RT682) in which each of the amino acids at the 617,
652, and 682 position of the RT region of 2-18 was replaced with its 56 counterpart and studied by cell transfection, only 2-18RT652showed enhanced replication efficiency, with an
in-tensity of hybridization signal similar to that of 2-18RT56(Fig.
5).
Structural analysis of mutations in HBV RT domain. Com-puter-based structural analysis predicted that residue 617 was located at the loop connecting the palm subdomain and the thumb domain and would have no contact with any important structural element of the protein and the DNA substrate. Sim-ilarly, residue 682 was predicted to be located in the middle of the third ␣-helix of the subdomain and oriented toward the outer surface of the protein, which would not have significant impact on the interaction between RT and DNA. In contrast, residue 652 was predicted to be located at the connecting loop between two␣-helices. Alteration of residue 652 from proline to serine might affect the precise conformation of the two helices and their interactions with the DNA template-primer, which might impair the polymerase activity (Fig. 6).
DISCUSSION
Functional domains of the HBV P gene product have been identified by mutational analysis and transient expression in cells by Radziwill et al. (18). Alignment of the amino acid sequences of the three enzymatic domains (TP, RT, and
RNase H) with similar enzymes from other retroviruses and hepadnaviruses revealed conserved and consensus regions which were considered important motifs. To date, all attempts at expression and production of large amounts of functionally active forms of HBV polymerase in animal cells or prokaryotic cells have been unsuccessful. Therefore, detailed functional analysis of HBV P protein has been hampered.
Since HBV replication has a unique strategy and molecular mechanism, it is speculated that there may be specific func-tional domains in HBV P protein which are not shared by other viruses. To explore novel functional domains or residues of HBV P protein, we used the chimeric genomic cell trans-fection analysis method. Among six full-length naturally occur-ring HBV isolates, we selected isolates 2-18 and 56 because they had highly homologous nucleotide sequences, making it possible to perform the domain-swapping experiments. Studies were designed to use only the HBV genome for cell transfec-tion, without any exogenous plasmid sequences which might interfere with the replication efficiency of isolates. In addition, substitutions of the P gene or the gene fragments coding for the putative functional domains were all carried out between two naturally occurring HBV genomes. Therefore, the results obtained in this cell transfection study would correlate with the replication efficiency of the virus in vivo and could reveal pos-sible novel functional residues in HBV P protein.
[image:5.587.61.260.74.226.2]In this study we have shown that when amino acid 652 was changed from serine to proline, HBV replication efficiency was enhanced, and replication efficiency was decreased by the con-verse change. In the four other isolates (14, 16, 20, and 97) that we studied, the amino acid at 652 was always proline. Hase-gawa et al. (12) have reported enhanced replication of an HBV mutant associated with an epidemic of fulminant hepatitis. The amino acid at 652 in that strain of HBV was proline. A search of GenBank for HBV RT sequences showed that the consen-sus residue at 652 was proline. The counterpart of 652 is also FIG. 4. 56RT2-18 is an HBV chimeric genome in which the RT
region of the P gene was replaced with its 2-18 counterpart. Intensity of hybridization signal was measured by densitometry, and the relative intensity of samples is indicated at the bottom. Lane M, molecular size standards.
FIG. 5. 2-18617Met3Leu, 2-18652Ser3Pro, and 2-18682Val3Leuare 2-18 HBV chimeric genomes in which the nucleotide sequences coding for amino acids at positions 617, 652, and 682 within the RT region were each mutated to change the amino acid to the corresponding amino acid of isolate 56. Intensity of hybridization signal was measured by densitometry. The intensity of signal of 2-18RT652Ser3Prowas similar to that of 2-18RT56, in which the RT region of 2-18 was replaced with its 56 counterpart. Lane M, molecular size standards.
on November 9, 2019 by guest
http://jvi.asm.org/
[image:5.587.309.535.487.653.2]proline in HIV-1 RT. Therefore, one may consider 56 as rep-resenting a wild-type virus and 2-18 as a mutant with a substi-tution of proline by serine at amino acid 652. This substisubsti-tution resulted in decreased replication efficiency.
Interestingly, this substitution only regulated the relative replication efficiency of the virus strain, in contrast to a muta-tion in TP, which abolished virus replicamuta-tion (5). It is intriguing that a virus strain with decreased replication efficiency could persist during natural infection. Since host immune responses are highly relevant in HBV infections, this virus strain could persist in a host with decreased immune responses. Unfortu-nately, it was not possible to study the immune responses in the patient.
In this study, when the complete P gene of 56 replaced the complete P gene of 2-18, although enhanced replication was observed, it was not as marked as that when only the RT gene fragment was replaced. It is tempting to speculate that there might be replication-inhibitory functional domains in frag-ments of the HBV P gene which counteracted the replication-enhancing effect of amino acid 652 in the RT region. Besides, in this study, substitution of the spacer region from 56 for that of 2-18 also slightly enhanced replication efficiency. Recently, Landford et al. (15) reported that a portion of the spacer domain was required for both TP and RT function and resi-dues 300 to 334 were identified as the minimal portion of the spacer domain. A substitution was found at residue 300 in the spacer region between 2-18 and 56 (Table 2, from histidine to arginine), which could be associated with the slightly enhanced replication efficiency.
The mechanism of the change in replication efficiency caused by changing the residue at amino acid position 652 is not clear. However, computer modeling suggested that residue 652 was located at the connecting loop between two␣-helices of the thumb subdomain, equivalent to␣-helices H and I in HIV-1 RT. In HIV,␣-helices H and I interact extensively with
the DNA template-primer substrate (8). It was suggested that these two helices form part of a translocation track that con-trols and modulates the relative movements of polymerase. During the polymerization reaction, the template-primer has to translocate after incorporation of each nucleotide. The poly-merase should have the flexibility to allow the translocation of the template-primer but still maintain the precise positioning of the template-primer relative to the polymerase active site. It is speculated that in the HBV RT, proline has a rigid confor-mation with less flexibility, and this rigidity might be necessary to constrain the relative conformation and/or positions of the two helices. Mutation from proline to serine at residue 652 could disrupt this constraint, allowing more flexibility and thus affecting the polymerase activity.
Although HBV is recognized as a noncytopathic virus, en-hanced viral replication could contribute to the pathogenesis of liver disease in several aspects. First, enhanced virus repli-cation can lead to an increased number of hepatocytes being infected, resulting in more severe hepatocellular injury medi-ated by host immune responses. Second, antiviral-drug-resis-tant muantiviral-drug-resis-tants could be selected in higher frequency among patients with a high virus load. Third, a higher potential could arise for emergence of viral mutants with altered biological characteristics and for higher incidences of random integration of viral DNA into the host genome. In general, enhanced replication has been associated with increased virulence with other viruses. Therefore, studies on the regulatory mechanisms of the replication efficiency of HBV are of importance.
[image:6.587.113.473.72.271.2]The polymerase of HBV has been the essential target for development of anti-HBV drugs, and most studies have fo-cused on the common and conserved functional domains at the active site of RT shared with other known viruses. Our finding of the replication-enhancing or -decreasing effect of amino acid 652 in the RT region provides the clue that there could be other functional domains outside the A-E functional domains FIG. 6. Ribbon diagram showing the three-dimensional model of the RT domain of HBV polymerase (residues 319 to 700). HBV RT is composed of three subdomains, fingers (319 to 400 and 451 to 515, in blue), palm (401 to 450 and 516 to 610, in red), and thumb (611 to 700, in green). A docked template-primer double-stranded DNA is shown as a ribbon, with the template in magenta and the primer in cyan. The three catalytically essential aspartic acid residues, Asp429, Asp551, and Asp552, are shown with side chains. The locations of the mutations are indicated with gold balls.
on November 9, 2019 by guest
http://jvi.asm.org/
predicted by amino acid alignment (17). Novel functional do-mains revealed in naturally occurring HBV strains could better resemble the situation in patients and thus could become new targets for development of antiviral drugs. Whether changes from proline to other amino acids at 652 would also affect virus replication is currently under study. Research aimed at eluci-dating the functional domains at and around this residue is also being conducted.
ACKNOWLEDGMENTS
This work was supported by Chinese State Basic Research Founda-tion Grant G1999054105. X.L. is a Ph.D. student supported by the Chinese Ministry of Education.
We are grateful to Christopher Burrell for reviewing the paper. The excellent technical help of Xin Yao and Zhang-Mei Ma is much ap-preciated.
REFERENCES
1.Ago, H., T. Adachi, A. Yoshida, M. Yamamoto, N. Habuka, K. Yatsunami, and M. Miyano.1999. Crystal structure of the RNA-dependent RNA poly-merase of hepatitis C virus. Struct. Fold Des.7:1417–1426.
2.Ayola, B., P. Kanda, and R. E. Lanford.1993. High level expression and phosphorylation of hepatitis B virus polymerase in insect cells with recom-binant baculoviruses. Virology194:370–373.
3.Bartenschlager, R., C. Kuhn, and H. Schaller. 1991. Expression of the P-protein of the human hepatitis B virus in a vaccinia virus system and detection of the nucleocapsid-associated P-gene product by radiolabelling at newly introduced phosphorylation sites. Nucleic Acids Res.20:195–202. 4.Bavand, M., M. Feitelson, and O. Laub.1989. The hepatitis B
virus-associ-ated reverse transcriptase is encoded by viralpolgene. J. Virol.63:1019– 1021.
5.Blum, H. E., E. Galun, and T. J. Liang.1991. Naturally occurring missense mutation in the polymerase gene terminating hepatitis B virus replication. J. Virol.65:1836–1842.
6.Bressanelli, S., L. Tomei, A. Roussel, I. Incitti, R. L. Vitale, M. Mathieu, R. De Francesco, and F. A. Rey.1999. Crystal structure of the RNA-dependent RNA polymerase of hepatitis C virus. Proc. Natl. Acad. Sci. USA96:13034– 13039.
7.Das, K., X. Xiong, H. Yang, C. E. Westland, C. S. Gibbs, S. G. Sarafianos, and E. Arnold.2001. Molecular modeling and biochemical characterization reveal the mechanism of hepatitis B virus polymerase resistance to lamivu-dine (3TC) and emtricitabine (FTC). J. Virol.75:4771–4779.
8.Ding, J., K. Das, Y. Hsiou, S. G. Sarafianos, A. D. Clark, A. Jacobo-Molina,
C. Tantillo, S. H. Hughes, and E. Arnold.1998. Structure and functional implications of the polymerase active site region in a complex of HIV-1 RT with a double-stranded DNA template-primer and an antibody Fab fragment at 2.8 Å resolution. J. Mol. Biol.284:1095–1111.
9.Georgiadis, M. M., S. M. Jessen, C. M. Ogata, A. Telesnitsky, S. P. Goff, and W. A. Hendrickson.1995. Mechanistic implications from the structure of a catalytic fragment of Moloney murine leukemia virus reverse transcriptase. Structure3:879–892.
10.Gu¨nther, S., B. C. Li, S. Miska, D. H. Kru¨ger, H. Meisel, and H. Will.1995. A novel method for efficient amplification of whole hepatitis B virus ge-nomes permits rapid functional analysis and reveals deletion mutants in immunosuppressed patients. J. Virol.69:5437–5444.
11.Gunther, S., L. Fisher, J. Pult, M. Sterneck, and H. Will.1999. Naturally occurring variants of hepatitis B virus. Adv. Virus Res.22:25–137. 12.Hasegawa, K., J. Huang, S. A. Rogers, H. E. Blum, and T. J. Liang.1994.
Enhanced replication of a hepatitis B virus mutant associated with an epi-demic of fulminant hepatitis. J. Virol.68:1651–1659.
13.Kim, S. S., H. J. Shin, and H. M. Rho.2000. Expression of stable hepatitis B viral polymerase associated with GRP94 in E.coli. Arch. Virol.145:1305– 1320.
14.Landford, R. E., L. Notvall, and B. Beames.1995. Nucleotide priming and reverse transcriptase activity of hepatitis B virus polymerase expressed in insect cells. J. Virol.69:4431–4439.
15.Landford, R. E., Y. H. Kim, H. Lee, L. Notvall, and B. Beames.1999. Mapping of the hepatitis B virus reverse transcriptase TP and RT domains by transcomplementation for nucleotide priming and by proteprotein in-teraction. J. Virol.73:1885–1893.
16.Miller, R. H., P. L. Marion, and W. S. Robinson.1984. Hepatitis B viral DNA-RNA hybrid molecules in particles from infected liver are converted to viral DNA molecules during an endogenous DNA polymerase reaction. Virology139:64–72.
17.Poch, O., I. Sauvager, M. Delarue, and N. Tordo.1989. Identification of four conserved motifs among the RNA-dependent polymerase encoding ele-ments. EMBO. J.12:3867–3874.
18.Radziwill, G., W. Tucker, and H. Schaller.1990. Mutational analysis of the hepatitis virus P gene product: domain structure and RNase H activity. J. Virol.64:613–620.
19.Wang, G. H., and C. Seeger.1992. The reverse transcriptase of hepatitis B virus acts as a protein primer for viral DNA synthesis. EMBO J.71:663–670. 20.Will, H., W. Reiser, T. Weimer, E. Pfaff, M. Bu¨scher, R. Sprengel, R. Cat-taneo, and H. Schaller.1987. Replication strategy of human hepatitis B virus. J. Virol.61:904–911.
21.Wu, L., Z. H. Yuan, L. F. He, W. L. Jiang, S. X. Qiu, and Y. M. Wen.2000. Functional comparison of two full-length genome of hepatitis B virus iso-lated from two chronic hepatitis B virus patients. Chin. J. Virol.16:16–22. 22.Yuan, T. T., M. H. Lin, D. S. Chen, and C. Shih.1998. A defective
interfer-ence-like phenomenon of human hepatitis B virus in chronic carriers. J. Vi-rol.72:578–584.