Virology, Jan. 0022-538X/83/010354-13$02.00/0
Copyright C) 1983,AmericanSocietyforMicrobiology
Genetic
Analysis
of
Temperature-Sensitive
Mutants
Which
Define the Gene for the Major Herpes Simplex Virus Type 1
DNA-Binding Protein
SANDRA K. WELLER, KATHRYN J. LEE, DENNIS J. SABOURIN,ANDPRISCILLA A. SCHAFFER* Laboratory ofTumor Virus Genetics, SidneyFarber CancerInstitute,andDepartment of Microbiologyand
Molecular Genetics, Harvard MedicalSchool, Boston, Massachusetts 02115
Received 22 June1982/Accepted 15September1982
We haveassigned eighttemperature-sensitive mutantsofherpes simplex virus
type 1 tocomplementationgroup1-1. Members of thisgroupfailto complement
mutantsinherpes simplex virustype2complementationgroup2-2.The mutation ofone member ofgroup1-1, tsHAl of strain mP, has been shown to map inor nearthe sequence which encodes the major herpes simplex virus type 1
DNA-bindingprotein (Conley et al., J. Virol. 37:191-206, 1981). The mutations of five other members ofgroup 1-1 map in or near the sequence in which the tsHAl mutationmaps, asequencewhichliesnearthecenterofUL between thegenesfor
the viral DNApolymerase and viral glycoprotein gAgB. These mutants can be divided into two groups; the mutations ofone group map between coordinates 0.385 and0.398, and the mutations of the othergroup map betweencoordinates 0.398 and0.413. At thenonpermissivetemperature mutantsingroup 1-1 areviral
DNAnegative, and mutant-infected cells failto reactwithmonoclonalantibodyto the130,000-dalton DNA-binding protein. Taken together,these data indicate that mutants in complementation groups 1-1 and 2-2 define the gene for the major
herpes simplex virus DNA-binding protein, an early gene product required for
viral DNA synthesis.
At least 50 virus-specific polypeptides are
synthesized in cells infected withherpessimplex
virus type 1 (HSV-1) (19). Although
consider-able progress has been made in mapping viral
transcripts and polypeptides on the HSV-1
genome, specific functions have been assigned
to fewer than 10 viral polypeptides. The most
direct approachto elucidating the roles of viral
proteins in the replicative process has been to
isolate mutantswhich exhibit alterations in the
synthesis
oractivity of these proteins.The availability of HSV-1 mutants has
facili-tated both the identification of the functional roles of the following HSV-1 proteins and the map locations of the genes which specify them: HSV-1 thymidine kinase (2, 48); HSV-1 DNA
polymerase (1, 6); the immediate-early
regula-tory protein (molecular weight, 175; ICP4 or VP175) (12, 34); HSV-1 glycoprotein gAgB (24,
39, 40; N. DeLuca, personal communication);
and the major DNA-binding protein (11, 33). Mutantswhichexhibit altered distribution of the latterprotein arethe subject of this study.
The major DNA-binding protein, a
polypep-tide of approximately 130,000 daltons (130K)
belongingtotheearlykinetic class of viral gene
products, was described first for HSV-1 by
Baylissetal.(3)andsubsequently for HSV-2 by
Purifoy and Powell (36). The major HSV-1
DNA-binding protein, ICP8 (19) or VP143 (15),
corresponds to HSV-2-infected cell protein
ICSP 11/12 (33).Todate,weknowthat the 130K
protein binds preferentially to single-stranded
DNA, isprobably required for HSVDNA
syn-thesis (34), and may act to stabilize DNA
poly-meraseactivity (32). Interestingly, antisera
pre-pared against HSV-1- or HSV-2-infected cell
proteinscross-reactwith DNA-binding proteins
having approximate molecular weights of
130,000 in cells infected withbovinemammilitis
virus, pseudorabies virus, and equine abortion
virus, indicating that the gene for the 130K
protein has been conserved evolutionarily (25,
49).
Themaplocationof the 130Kproteinwasfirst
suggested by the studies of Conley et al., who
isolated aDNA-negative, temperature-sensitive
mutant, tsHAl, which maps between coordi-nates 0.385 and 0.402 (11). The smallest DNA
fragment able to rescue tsHAl selected an
mRNA species from HSV-1-infected cells
which, when translated in vitro, encoded a
poly-peptide having a molecular weight of 128,000.
Thesimplest explanationfor these results is that
354
on November 10, 2019 by guest
http://jvi.asm.org/
the mutation in tsHAl lies in or nearthe gene coding for the 130K DNA-binding protein. It is
interestingthattheHSV-1 DNA fragment which
rescued tsHAl (coordinates 0.385to0.402) (11) lies within the region of the HSV-1 genome which has been shownto induce morphological
transformation of mouse and hamster cells in
vitro (5, 38), suggesting apotential role for this protein in morphologicaltransformation.
The availability of tsHAl, whose mutation
maps near the coding sequences for the 130K protein, prompted the genetic analysis described below. These studies demonstrated that tsHAl isamember ofalargergroupofmutantswhich constitute complementation group 1-1. In addi-tion, mutants in group 1-1 fail to complement
mutants in HSV-2 complementation group 2-2, whose members are characterized by altered cellulardistribution of the HSV-2 130K DNA-bindingprotein (1la).
In this paper we describe the physical map-ping of themutationsoffive members of comple-mentationgroup 1-1 and demonstratethat these mutants, like their HSV-2 counterparts, exhibit altered distribution of the major DNA-binding protein. Our findings confirm the relationship between mutants in group 1-1 and the HSV-1
DNA-binding protein and provide a more pre-cisedefinition of thelimits of thegeneencoding thisprotein.
MATERIALS AND METHODS
Cells and cell culture.Humanepidermoidcarcinoma
no.2(HEp-2), primary rabbit kidney(RK),and
Afri-cangreenmonkey kidney(Vero)cellswere propagat-ed in the Dulbecco modification of Eagle minimal
essential mediumcontaining10%newborn calfserum, 0.03% glutamine, and 0.25% NaHCO3. Cells were
maintained in the same medium containing 5%
new-born calf serum. Virus stocks were grown and
as-sayed,andcomplementationtestswere performed in
Vero cells. RK cells were used for marker rescue
studies, and viral DNA was isolated from infected
HEp-2cells.
Virusand virusassays. Theoriginsof the 11 HSV-1 mutantsandfour HSV-2mutantsusedin thisstudyare
shown in Table 1. The wild-type virus strains and mutagensused fortheinductionofthesemutantsare
alsoshown in Table1.Theresults ofprevious comple-mentation tests placed HSV-1 mutants tsAl, tsA15, tsA16, ts656, and tsN103 into one complementation group, group1-1 (41, 42). Mutants tsC7and tsD9are
members of complementation groups 1-3 and 1-4, respectively,andexhibit thermolabile DNA
polymer-aseactivity(1, 35, 37, 42).TheHSV-1 mutanttsJ12is
amember ofcomplementationgroup1-9 and exhibits
altered synthesisofglycoprotein gAgBatthe nonper-missive temperature (24, 42). The HSV-2 mutants
tsH9,tsAl, and tsA8werepreviously assignedto two
complementationgroups(groups2-2 and2-7);
howev-er,a recent study hasdemonstrated that in all likeli-hood these mutantsare membersofacommongroup designated group 2-2 (lla). The HSV-2 mutanttsD6, which is able to synthesize viral DNAat the nonper-missivetemperature, isincomplementationgroup2-15 (14, 42).
Virus stocks were prepared and assayedas previ-ously described(42). The permissiveand nonpermis-sive temperatures usedwere34and39°C, respective-ly, unlessotherwisespecified. All virus stocks used in this studyexhibited efficiencies ofplatinglessthan or equalto10-4. Ingeneral,mutantsexhibited low levels of leak and reversion (<10-5);however,one mutant,
tsA15, consistently exhibited a slightly higher
rever-sion frequency(10-4to
10-')
thanthe othermutants.Complementation.Complementationtests were con-ducted in Vero cellsas describedpreviously(41).
Viral DNA phenotype. Viral DNA phenotypes of mutantsweredeterminedasdescribedbyAron etal.
(1).
Immunofluorescence tests. Indirect immunofluores-cence tests were conducted in Vero cells by the method ofPorter et al.(30). Monoclonal antibodyto the HSV-1 130K DNA-binding protein was kindly provided by MartinZweig(National Cancer Institute, Frederick, Md.) (44). Fluorescein isothiocyanate-labeledrabbit anti-mouseimmunoglobulinGwasused todetectmonoclonalantibodyonfixed monolayers.
RecombinantDNAplasmids containing HSV-1DNA. Recombinant DNAplasmids containing fragments of the genome of HSV-1 strain KOS were generously provided by Myron Levine andRozanne Sandri-Gol-din(UniversityofMichigan,AnnArbor), DavidKnipe (Harvard Medical School, Boston, Mass.), Wai-Choi Leung (McMaster University, Hamilton, Ontario, Canada), Neal DeLuca(Pennsylvania State Universi-ty, University Park), and Richard Dixon and Donald Coen (Sidney Farber Cancer Institute, Boston, Mass.).Therecombinantplasmidsused allcontained HSV-DNA sequences located between map coordi-nates 0.312 and 0.422 on the physical map of the genome (see Fig. 2). A brief description of these plasmid clones follows.
pSG18wasconstructedbyinsertingEcoRIfragment
F(map coordinates 0.315to0.422) into the EcoRI site of pBR325 (17). Sall subfragments of pSG18 were
inserted into the Sall site ofpBR325,generatingclones pSG18-SA, pSG18-SD, and pSG18-SE (D. Knipe, personal communication). PlBIl containsBglII
frag-ment I(map coordinates 0.312 to 0.415)inserted into
theBglII site of pKC7 (23). pKEF-Pl, pKEF-P2, and
pKEF-P3 contain strain KOS DNAextending from the PstI sitesat mapcoordinates 0.398, 0.388, and 0.370, respectively,totheEcoRIsite at mapcoordinate 0.422 (N. DeLuca, D.J. Bzik, V. C. Bond, S. Person, and W. Snipes, Virology, in press). pKOS10 and pKOS15 containBamHIfragment G (map coordinates 0.345 to 0.398)inserted intotheBamHIsite ofpBR322 (10). In addition to BamHI fragment G, pKOS15 contains strain KOS BamHI fragment W (Weller and Lee, unpublished data).
DNAisolation.HSV-1strainKOS waspropagated in HEp-2 cells,andviral DNAwasobtained from partial-lypurifiedvirionsas describedbyParriset al.(29).
Plasmid DNA was prepared as follows. Bacteria containing plasmids were grownin Frazier medium, and plasmid DNA was extracted by the method of VOL.45,1983
on November 10, 2019 by guest
http://jvi.asm.org/
Clewell and Helinski (9). PlasmidDNAwas purified
by equilibrium centrifugation inasolution containing
50%(wt/wt) CsCland 1mgof ethidiumbromideperml for 48 hat45,000rpminaBeckman 6OTirotor. After centrifugationtheplasmidbandwasextracted with
n-butanol, dialyzed against 0.1x SSC (0.015 M NaCl plus 0.0015 M sodium citrate, pH 7.0),ethanol precip-itated,andstoredin 0.1x SSCor1x TE(0.01M Tris plus0.001 M EDTA[trisodiumsalt], pH 7.4).
Mapping of restrictionendonucleasecleavage sites in viral and plasmid DNAs. Viral andplasmidDNAswere
digested to completion with restriction enzymes EcoRI, Sall, HindIll, KpnI, PstI, and HpaI (New England Biolabs, Beverly, Mass.) and with BamHI and BgIII (Biotec, Madison, Wis.) by using the
pre-scribed buffers.Electrophoresiswascarriedoutin 0.5
to 1.5% agarose horizontal slab gels (5 mm thick) containing0.5,ugof ethidium bromideperml. A DNA
marker mixture containing bacteriophage lambda DNA digested withHindlIl andpBR322DNA digest-ed with BstNIwasusedtomakeaccuratesize determi-nations of the DNAfragments generatedafter restric-tion enzyme digestion. The sizes of the standards
ranged from 100 base pairsto27kilobases(kb).DNA
wastransferredtonitrocellulose filters by the method ofSouthern(46) andwashybridizedwithHSV-1BglII fragment I labeled by nick translation with 32p(see below). Autoradiography was carried out at -80°C with Kodak X-OmatX-rayfilm, using Cronex enhanc-ingscreens.
Isolation of DNA fragments. Restriction enzyme-generatedDNAfragmentswereisolatedfromagarose gels in one of two ways. The first method was a
modification ofthe glass powder elution procedure described byVogelstein and Gillespie (47; B. Pater-son, personal communication). Briefly, agarose gel slicescontaining restrictionfragmentsweredissolved at37°Cin6.1 M sodium iodide saturated with sodium sulfite. The DNAwasabsorbedtofineparticlesof
325-mesh silica glass for 2 h at 0°C. The glass was
recovered by centrifugation and washed once with sodiumiodide solution and twice with 50%ethanol-10 mMTris (pH 7.5)-0.1 M NaCl-1 mM EDTA at0to
4°C. DNAwaseluted with 1x TEat37°Cfor 30min. Alternatively, electrophoresis was carried out in low-melting-pointagaroseasdescribedbyParker and Seed (28). The DNA fragment was obtained free of agarose by chromatographyon a benzoyl-naphthoyl-DEAE-cellulose column, using amodification of the procedureof Sedatetal.(43).
Preparation ofnick-translated viral DNA hybridiza-tionprobe. DNA fromplasmid plBI1 waspurifiedas
describedabove,exceptthat the DNAwas extracted
twice with redistilled phenol containing 1.6 mg of hydroxyquinoline per ml and saturated with 10 mM Tris-hydrochloride (pH 7.3)andoncewith
chloroform-isoamyl alcohol (24:1) before dialysis. The HSV-1 BglII fragmentIinsertionfromthe plBI1clone (map coordinates 0.314to0.415ontheHSV-1 genome)was
purifiedafterdigestionwithBglII bypreparative low-melting-point agarose electrophoresis as described
above. The purified BgII fragmentIwasnick translat-edbytheprocedureof Maniatisetal.(27).[32P]dCTP and[32P]dGTPwereobtained from AmershamCorp.,
Arlington Heights,Ill.
DNAinfectivityassays. DNAsampleswereassayed forinfectivity bythe calciumphosphate co-precipita-tionmethod of Graham and Van der Eb(18),with the followingmodification. RK cells were transfected in
suspensionandplated as infectious centers by using uninfected RK cells as indicators, as described by
Parrisetal.(29).
Marker rescue. Marker rescue experiments were
performed by cotransfection of RK cells with intact infectious HSV-1 DNAfromatemperature-sensitive
(ts) mutant and cloned wild-type DNAfragments as
plasmids. Before transfection, plasmidDNAwas di-gested witharestrictionenzyme which either
linear-TABLE 1. Mutantsof HSV-1 and HSV-2used in this study
VirusVirus Wild-typestrain
~~Mutanta
Muat Mutagenbuae'GopReference
Group investigatorortype strain ivsiao
HSV-1 KOS tsAl BUdR 1-1 41
tsA15 NTG 1-1 41
tsA16 NTG 1-1 41
tsA24 UVlight ' P. A.Schaffer
tsA42 HA S. P. Little
tsC7 BUdR 1-3 41
tsD9 BUdR 1-4 41
tsJ12 NTG 1-9 41
KOS1.1 ts656 BUdR 1-1 20
HFEM tsN103 NA 1-1 A.Buchan
mP tsHAl HA 11
HSV-2 186 tsH9 UVlight 2-2 8
tsAl BUdR 2-2d 14
tsA8 BUdR 2-2d 14
tsD6 BUdR 2-15 14
aNomenclature from Schafferet al.(42).
bBUdR, 5-Bromodeoxyuridine; NTG, nitrosoguanidine; NA, nitrous acid; HA, hydroxylamine.
-,Notpreviously reported.
dHSV-2mutants
tsAl
and tsA8 were previously assignedto complementation group 2-7; however, recentevidence (lla) indicates thatthese mutants belong tocomplementation group 2-2 (see text).
J. VIROL.
on November 10, 2019 by guest
http://jvi.asm.org/
[image:3.489.50.445.465.628.2]HSV-1 MUTANTS 357
ized theplasmidorexcised theinserted HSV-1DNA; the restrictionenzyme wasinactivated either by
incu-bationat60°Cfor 10minorbyphenol extraction and
ethanolprecipitation. Eachtransfection mixture
con-tained 1 ,ug of digested plasmid DNA, 200 to 1,000 PFU of infectiousHSV-1DNA, and12,ug ofRK DNA in 0.6 ml ofHEPES (N-2-hydroxyethylpiperazine-N'-2-ethanesulfonic acid)-buffered saline. Transfections
were performed as described previously (29). When generalized cytopathic effects were observed, virus
was harvested, and infectious progeny viruswas
as-sayedin Verocells at34 and39°C. Theefficiency of platingwascalculatedasfollows:plaque-forming units
permilliliterat39°C/plaque-forming unitspermilliliter
at34°C.
RESULTS
Properties oftsmutants. (i) Viral DNA pheno-types and viral DNA polymerase activities. Mu-tants tsAl, tsA15, tsA16, tsC4, and tsD9 have beendescribedpreviouslyastotheir viralDNA phenotypes and viral DNA polymerase activities (1). The following two new mutants of strain KOS are described below: tsA24 and tsA42.
Mutant tsA42was isolated afterhydroxylamine
mutagenesis of a mixture of XbaI fragments
(S. P. Little, personal communication), and tsA24 was isolated after UV mutagenesis
(Schaffer, unpublished data). No viral DNA
synthesis was detected in lysates of cultures infected with anyof themutants ingroupA, C, or Dat39°C (Table 2) (1).
When mutants tsC4 and tsD9weretested for induction of DNA polymerase activity at 39°C, they exhibited a marked reduction in
polymer-aseactivity (1). Moreover, Purifoyetal.(35, 37) have shown that the purified polymerases
syn-thesized by tsC7 (another member of comple-mentationgroup 1-3) and tsD9 are temperature sensitive. Incontrast, mutantstsAl, tsA15, and tsA16 induce wild-type levels ofpolymerase at
39°C (1). Moreover, the mutations of tsC4 and
tsD9mapto aregionof DNA whichdefines the
polymerase locus (6), whereas the mutations of tsAl, tsA15, and tsA16 donot (seebelow).
(ii) Immunofluorescence tests: the 130K
poly-peptide. Because physical mapping studies
placed members ofcomplementationgroup 1-1
within the region of the HSVgenome spanning coordinates 0.312to0.422,the region thoughtto
containthe structuralgeneforthe130Kprotein
(11; S. K. Weller, W. R. Sacks, D. M. Coen, and P. A. Schaffer, submitted for publication), we
were interested in knowing whether mutants
with mutations mapping in this region were affected in the expression ofthis protein. For
thispurpose cells infected with wild-type virus
and mutants in complementation group 1-1, as well as tsJ12 (group 1-9) and tsD9 (group 1-4), were tested for reactivity with a monoclonal antibodytotheHSV-1 130K protein by
immuno-fluorescence as described above. Photomicro-graphs of typical fluorescence reactions for strainKOS and tsAlat34 and39°Careshownin Fig. 1. Whereas cells infected with strain KOS exhibitedbright nuclearfluorescenceatboth 34 and 39°C (Fig. 1A and 1B), cells infected with tsAl exhibited nuclear fluorescence at 34°C (Fig. 1C) but no specific fluorescence in the cytoplasm or nucleus at 39°C (Fig. 1D). Other members ofcomplementationgroup 1-1 (tsA15 and tsA16) resembled tsA1 in that no specific nuclear or cytoplasmic fluorescence was
ob-served at 39°C (Table 2). Importantly, cells infected with mutantstsA24, tsA42, and tsHAl resembled cells infected with establishedgroup 1-1 mutants. Incontrast, tsJ12-and tsD9-infect-ed cells exhibited nuclear fluorescence at both 34 and 39°C (Table 2). Thepresence of nuclear fluorescence in tsD9-infected cellsat39°C dem-onstrated that the absence of nuclear
fluores-cence in cells infected with group 1-1 mutants wasnotsimply duetotheabsence of viralDNA
TABLE 2. ViralDNAsynthesis and fluorescentstainingwith anti-130K monoclonalantibody ofwild-type virus andts mutantsof HSV-1 at34 and 39°C
Viral DNAsynthesis' Nuclearfluorescence with
anti-Virus 130K monoclonal antibody
34°C 39°C
34°C
39°C
KOS 100 100 + +
tsAlb 44 0 +
-tsAlSb 19 0 +
-tsA16b 33 0 +
-tsA24 93 0 +
-tsA42c 93 0 +
-tsJ12 95 92 + +
tsC7b 27 0 + +
tsD9b 57 0 + +
a Results areexpressedaspercentages of viralDNA inwild-type virus-infectedcultures.
b Data on viral DNAsynthesis andDNApolymeraseactivityweretaken fromAronetal. (1). c Assay of viralDNAsynthesiswasperformedby S.P.Little(personalcommunication).
on November 10, 2019 by guest
http://jvi.asm.org/
[image:4.489.52.448.524.645.2]FIG. 1. Immunofluorescent staining of HSV-1 strain KOS- and tsAl-infected Vero cells with monoclonal antibodytothe130KDNA-binding protein: photomicrographsof cells infected with HSV-1 strainKOSat34°C (A) and 39°C (B) and with mutant tsAl at 34°C (C) and 39°C (D) andtreated successively with monoclonal antibody tothe HSV-1 130Kprotein and fluorescein isothiocyanate-labeled rabbit anti-mouse immunoglobulin G.
synthesis at this temperature, as tsD9 is DNA
negative. It should be emphasized that the
fail-ure of mutants in group 1-1 to exhibit specific
nuclearfluorescence whentheyweretested with monoclonal antibody to the 130K protein indi-cateseither that theproteinwas notsynthesized
at 39°C orthat theprotein was synthesized in a
configuration
not detectable by themonoclonal reagent. Ineither case, we conclude thatmem-bers of complementation group1-1 aremutated
in afunctionaffectingthecellular distribution of the 130K DNA-binding
protein.
Complementation. Because members of
com-plementation group 1-1, as well as mutants tsA24, tsA42, and tsHAl, exhibited altered expression of the 130K protein and because the tsHAl mutation has been shown to map in or near sequences specifying this protein (11), we were interested in determining the relationship among tsHAI, tsA24, tsA42, and members of
complementation group 1-1. Therefore, we
con-ducted complementation tests with these six mutants. Mutants tsJ12, tsC4, and tsD9 were
included as controls. Table 3 shows the results
ofquantitative complementation tests with these mutants.
J. VIROL.
on November 10, 2019 by guest
http://jvi.asm.org/
[image:5.489.83.409.69.475.2]TABLE 3. Complementation amongnine ts mutantsofHSV-1
Complementationindices' Mutant
tsAl tsA15 tsA16 tsA24 tsA42 tsHAI tsJ12 tsC7 tsD9
tsAl 1.2b 0.6 0.9 0.9 0.3 11 2,250c 170
tsA15 1.9 6.0 7.3 1.3 20 190C 35
tsAl6 0.3 0.7 0.7 68 19.5' 2,424
tsA24 0.7 0.5 583 488 554
tsA42 0.7 2,684 NDd 831
tsHAl 186 68.1 80
tsJ12 61' 23
tsC7 76
tsD9
aComplementationtests wereconductedas described previously(42).
bBoldfacevalues considered to be negative for complementation.
C Thecomplementation test was performed with tsC4, another member of complementation group 1-3.
dND,Notdone.
Inprevious studies,acomplementation index of 2was used to signify positive complementa-tion (41, 42). However, the levels of complemen-tation whichrepresentinter- and intragenic com-plementation were not known at the time that this arbitrary value was set. We now feel that complementation indices between 2 and 10 should be regarded as ambiguous in that they mayrepresenteither intra-orintergenic
comple-mentation, whereas values of 10or greater are
more likely to reflect intergenic complementa-tion (S. K. Weller, W. R. Sacks, D. M. Coen, and P. A. Schaffer, submitted for publication).
tsHA1 and the newmutants tsA24 and tsA42 failed to complement tsAl, tsA15, and tsA16, indicating that all six of thesemutantsare mem-bers ofcomplementation group 1-1 (Table 3). Mutants tsA24 andtsA42 gaveambiguous
com-plementation indices of 6.0 and 7.3, respective-ly, when they were paired with mutant tsA15; however, these mutants yielded complementa-tion indices of less than 2.0 when they were
tested with other members of thegroup,suchas tsAl and tsA16. Inaddition, mutantstsA24 and
tsA42failed tocomplement twoother members ofcomplementationgroup1-1,ts656 andtsN103 (datanotshown). All members of complementa-tiongroup1-1,includingts656 and tsN103, com-plemented the control mutants tsC4, tsD9, and tsJ12 efficiently (i.e., indiceswere10orgreater). Thus, we conclude that the five strain KOS mutants tsAl, tsA15, tsA16, tsA24, and tsA42, as well as the tsHA1 mutant of strain mP, the ts656 mutant of strain HFEM, and the tsN103
mutantof strain KOS1-1,belongto complemen-tationgroup 1-1.
Intertypic complementation. Preliminary inter-typic complementationtests (R. Dixon, unpub-lished data) indicated that mutants in HSV-1
group1-1and HSV-2group2-2failedto
comple-ment each other. To confirm this finding, we performed intertypic complementation tests
with six HSV-1 mutants from group 1-1 and threeHSV-2mutantsfromgroup2-2at38°C, the nonpermissive temperature for HSV-2 ts mu-tants. HSV-1mutantstsJ12 and tsC4 and HSV-2
mutant tsD6 were included as controls. The resultsof these testsareshown in Table 4. This
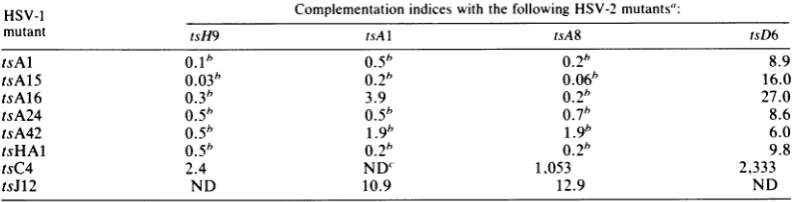
TABLE 4. Intertypiccomplementation amongeight mutantsof HSV-1 and fourmutantsofHSV-2
HSV-1 Complementation indices with the following HSV-2mutants":
mutant tsH9 tsA1 tsA8 tsD6
tsAl 0.1" 0.5" 0.26 8.9
tsA15 0.03" 0.2" 0.06" 16.0
tsA16 0.3" 3.9 0.2" 27.0
tsA24 0.5" 0.5" 0.7" 8.6
tsA42 O.5" 1.9" 1.9* 6.0
tsHAl 0.5" 0.260.2" 9.8
tsC4 2.4 ND' 1,053 2,333
tsJ12 ND 10.9 12.9 ND
aComplementationtests were conductedasdescribedpreviously (42).
bValues considered to benegative forcomplementation. ' ND,Notdone.
VOL. 45,1983
on November 10, 2019 by guest
http://jvi.asm.org/
[image:6.489.51.450.546.647.2]table shows that the five HSV-1 mutants in group 1-1 failed to complement the three HSV-2 mutants in group 2-2. However, ambiguous re-sults were obtained with the pair tsA16 and
tsA1. Incontrast, the HSV-1 mutants in group
1-1complementedHSV-2mutanttsD6 efficiently.
It should be notedthat lower complementation
indicesareroutinely obtainedin intertypic
com-plementation tests due in part toexcessive leak
of HSV-1 ts mutants at 38°C. Therefore, the
complementation indices between 6 and 10
ob-served intestswithtsD6 were thought toreflect
positive complementation. Withone exception,
HSV-2 mutants tsH9, tsAl, and tsA8
comple-mented HSV-1 mutants tsC4and tsJ12
efficient-ly.Thecomplementation indexgenerated bythe
pair tsH9 and tsC4 was 2.4, which is low and
thus ambiguous. We conclude from these
stud-ies that the mutations of members of groups
1-1 and 2-2 very likely represent a function
commontoboth HSV-1 and HSV-2.
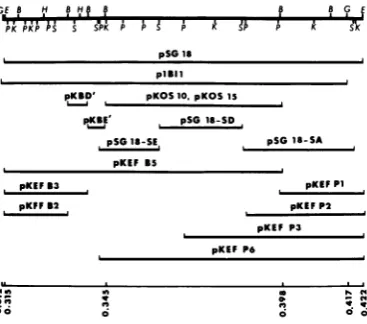
Restriction enzyme mappingofEcoRIfragment
F. As mentioned above, preliminary marker
rescue tests with mutants in complementation
group 1-1 indicated that the mutations ofthese mutants lie in EcoRI fragment Fof strain KOS
(Weller et al., submitted for publication). To
mapthe mutations of these mutants morefinely,
it was first necessary to construct a more
de-tailed restriction map of this region of the
genome. The region of strain KOS DNA
con-tained in EcoRI fragment F (map coordinates
0.315 to0.422) was mapped with respect to the
cleavage
sites of thefollowing
restriction en-zymes: BglII, KpnI, Sall, PstI, HpaI, andBamHI. Strain KOS DNA and plasmid
sub-clones containing portions of EcoRI fragment F
were digested, subjected to agarose gel
electro-phoresis, transferred to nitrocellulose filters,
andhybridizedtonick-translatedBglII fragment
I DNA (map coordinates 0.312 to 0.415). The
plasmid subclones used for restriction enzyme
mappingare showninFig.2.Single, double,and
triple digestions permitted relative ordering of
cleavage sitesof each restriction enzyme within
this region. Thephysical map summarizing the
results of these studies is shown in Fig. 3. Our
restriction map of this region of strain KOS
DNAis identical to the mapconstructedby N.
DeLuca et al.for the sameregionof strain KOS DNA(DeLuca et al., in press). However, there areminordifferencesbetween the maps of strain
KOSEcoRIfragment F and the equivalent
frag-mentsof HSV-1 strainsJustin,F(26), and 17(7).
Withoneexception(theBglIIandEcoRIsites at
the far leftendofEcoRIfragment F are reversed on our map of strain KOS and the previously
publishedmap [7] of strain 17), the
EcoRI,
BglII,HpaI,
andBamHIsites which lie between coor-dinates 0.312 and 0.422 appeartobe conservedamongthese four strains ofHSV-1. It should be noted that severalofthestrain KOSBamHIand HpaI fragments have designations which differ from thoseof strains F andJustin; however, the positions of the sites are conserved. On the other hand, the KpnI and Sall sites withinstrain KOS EcoRI fragment F are similar (but not
identical) to the KpnI and Sall sites in the
equivalent regions of strains 17 and Justin (7,
26); more significant differences exist between strains KOS and F (26). The PstI sites forother strains are not available for comparison.
Deletedsequences in cloned HSV DNA.Several investigators have observed that HSV-1 DNA fragments consisting ofsequences which lie
be-tweencoordinates 0.407 and 0.413 contain
dele-tions whenthey are cloned into Escherichia coli (31; N. DeLuca, personal communication; N. Frenkel, personalcommunication; C. Gray, per-sonal communication; D. Knipe, personal com-munication).
We made similarobservations in the course of mapping the KpnI and BamHI cleavage sites
withinsubclones ofEcoRIfragment F. Thus, we
found substantial differences between the
elec-trophoretic mobilities ofuncloned fragments of
strain KOS DNA and their cloned counterparts. One such comparison is shown in Fig. 4. The DNAs of strain KOS, pSG18, pKEF-P1, and pKEF-P2 weredigestedwithKpnIandanalyzed
as described in the legend to Fig. 3. Figure 4A
shows the ethidium bromide-stained gel, and
Fig.4B showsanautoradiogramof the same gel
afterhybridization. KpnIfragmentd(map
coor-PKPKPAi S SPKP P S P
pSG 18
pKBD' pKOS10, pKOS 15
pKBE' pSG18-SD,
pSG ls-SE pSG18-SA
pKEF BS
pKEF 33 pKEF P1
pKFFS2 pKEF P2
pKEFP3
pKEFP6
I I
a 0
10 bt4.0
I
0
FIG. 2. Recombinant DNA plasmids containing HSV-1DNA.The lineatthe topisacompositemapof the restriction endonuclease sites taken from Fig. 3. TheHSV-1 DNA insertions in eachrecombinant plas-mid described in the text are shownbelowthis map. Thenumbersat the bottom areselected map
coordi-nates. B, BamHI;E, EcoRL; G, BgII;H, HpaI; K, KpnI;P,PstI;S,SalIl.
J.VIROL.
on November 10, 2019 by guest
http://jvi.asm.org/
[image:7.489.258.442.437.599.2]M b UL b'd:anc'U a
0.0 0.1 0.2 0.3 0 0.5 0.6 0.7 08:5 0.9 1.0
9- -- i I~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~~
M
AP U E' F' 0 v Aa
AI T As
d f * c b a 9
kh i i c f 9 d b * a
f ce a b d 9
A
B
-c
E
F
G
H
0
00n
*~~~~~
JIL
o 00 0
[image:8.489.101.381.71.324.2]d
FIG. 3. Restriction map of HSV-1 strain KOS EcoRI fragment F. Line A represents the sequence
arrangementofHSV-1DNA.Thenumbersonline Barethephysicalmapcoordinates of thegenome.In lines C throughI,EcoRI fragment F (mapcoordinates0.315to0.422) isexpandedtoshow theEcoRI,BglII, BamHI, HpaI,SalI,PstI,andKpnIcleavagesites withincoordinates0.315to0.422. Inthecasesofenzymesfor which theentire strain KOS mapis known (EcoRI, BglII, BamHI, andHpaI), fragments are designated by capital
letters corresponding to the fragments generated after cleavage of total strain KOS DNA (45; J. Skare, unpublished data; Wellerand Lee, unpublished data). Theopen triangles indicate that only a portion ofthe
designated fragment is contained within EcoRI fragment F. In the cases ofenzymes Sall, PstI, and KpnI, fragments aredesignated with lower-case letters corresponding tothe sizes of the fragmentsgenerated after cleavageofEcoRIfragmentF. Line Jshowsselectedmapcoordinates within EcoRIfragmentF.
dinates0.407to0.420)of each DNA is indicated withan arrow. Plasmids pSG18, pKEF-P1, and pKEF-P2 each produced a KpnI fragment d
which was smaller than strain KOS KpnI frag-ment d. The size of the deletion in these plas-midsvaried fromapproximately 80 base pairs in the1.82-kbspecies inpSG18to660 basepairs in pKEF-P2. Substantial heterogeneity was also observed in BamHI fragment V (map
coordi-nates 0.398 to 0.413), which shares sequences
with KpnI fragment d (Fig. 3). Hence, we con-clude that the deleted sequences are located betweentheKpnI siteatthe b-djunction and the BamHI site at the V-Qjunction (map
coordi-nates 0.407 to 0.413). The presence of these
deletionshadnoapparenteffectontheability of thesubclones torescue tsmutations in physical mapping studies (see below). It should be noted that the region of HSV-1 DNArepresented by deletion-prone BamHI fragment Vis contained within the DNA sequences represented by a class ofdefective DNAs which are thought to
includeanorigin of viral DNA synthesis (16, 21, 38). The difficultiesexperienced byusand other
workers in cloning these sequencesin an unde-leted formmayberelatedtothepresenceof this
putative origin.
Marker rescue. The fine-structure physical
map locations of the mutations of five strain
KOS mutantsinHSV-1 complementationgroup
1-1 within EcoRI fragment F were determined by markerrescue.Aseries ofplasmidsubclones of EcoRI fragment F (Fig. 2) were used in marker rescue experiments with infectious DNAs from tsAl, tsA15, tsA16, tsA24, and tsA42. Inaddition, pKEF-P1 was digestedwith BamHI, and BamHI fragment V was isolated aftergel electrophoresis.
Theresults of markerrescueexperimentswith tsAl, tsA15, tsA16, tsA24, and tsA42areshown inTable 5. The mapcoordinates of each ofthe subcloned fragmentsarealso shownin Table 5. Intheseexperiments rescue efficienciesgreater than 1.0 were considered positive. Mutants tsAl, tsA15, and tsA42 wererescued efficiently with digested DNAs from pSG18, pKOS10,
pKOS15, pKEF-B5, and pSG18-SA. Fragments which rescued these mutants shared sequences
AM Eco RI
-Bgl 11
-SamHi
HpaI
SalI
PsiI
KpnI
,0'--'-
Don November 10, 2019 by guest
http://jvi.asm.org/
362 WELLER ET AL.
A.
0^. 0. C)
; 0 y Ye
1 I a
B.
oo X CL
U-u w 0. C.
1 2 3 4
kb
.~
1.91. 82 "40
1.78
1.244
FIG. 4. Analysis ofKpnI-digested strain KOS and plasmid DNAs. Strain KOS and plasmid pSG18, pKEF-P1, and pKEF-P2 DNAs (lanes 1 through 4, respectively) were digested with KpnI and subjected toelectrophoresison a0.7%agarose gel. DNAswere
transferred to nitrocellulose and hybridized to 32p-labeledBgIII fragment I DNAasdescribedinthetext. (A) Ethidium bromide-stainedgel. (B) Autoradiograph of the same gel. A DNA markermixture containing bacteriophage A DNA digested with HindIll and
pBR322 DNA digested with BstNI was subjected to electrophoresisonthesamegel; the sizes of fragments areshown ontheright. Thefragments corresponding
to KpnI fragment d for each DNA preparation are
markedwitharrows.ThestrainKOSfragment (lane 1) migratedatapositioncorrespondingto1.9 kb.
Diges-tion ofpSG18 with KpnlgeneratedtwoKpnI fragment
dbands whichmigrated slightly faster than the Kpnl fragment d from strain KOS (1.82 and 1.78 kb) (lane 2).
The Kpnl-digested plasmids pKEF-P1 and pKEF-P2 (lanes 3 and 4) each contained a single band
corre-sponding to KpnI fragment d which also migrated fasterthan the fragment from strain KOS (1.78 and 1.24kb,respectively).
that mapped between the Sall site at the b-a junction and the BamHI siteatthe G-Vjunction (map coordinates 0.385 to 0.398) (Fig. 3). All other tests yielded plating efficiencies less than 1.0. Inexperiments with tsA15, unusually large quantities of ts+ viruswereobserved with
plas-J. VIROL.
mids pKEF-P1, pSG18-SD, and pSG18-SE,
BamHI fragment V, and a "no fragment"
con-trol, resulting in plating efficiencies that were
approximately 10-fold lower than the plating
efficiencies observed with pKOS10 and
pKOS15, which rescued most efficiently.
Be-causethereversion frequencyof tsA15 is higher
(10-4
to10-3)
thanthatof othermutants(10-5
to10-4),
we conclude that the high backgroundobserved intestswith tsA15 DNA wasprobably
due to a high frequency of reversion of the ts
lesion.
Mutants tsA16 and tsA24 were rescued
effi-ciently by pSG18, pSG18-SA, and pKEF-P1
(Table 5). In addition, BamHI fragment V
res-cued bothtsA16andtsA24efficiently. The
frag-ments which rescued tsA16 and tsA24 shared sequences whichlaywithin BamHIfragment V
between coordinates0.398 and 0.413.
Selected progeny of marker rescue
experi-ments with tsAl, tsA24, and tsA42 were tested
for plating efficiency at 34 and 39°C to confirm
their ts+ phenotypes (i.e., the ts lesion was
rescued). All plaque isolates produced large
plaques at 39°C. Thus, these plaques were ts+
and resembledwild-type plaques at both 34 and
39°C, suggesting that mutants
tsA1,
tsA24, andtsA42 each contain a single ts mutation which can berescuedefficiently togenerate infectious
wild-type progeny.
The progeny of marker rescue tests with tsA16 and tsA15 (Table 5) produced small
plaques at 39°C. When these plaques were
picked,grown, and assayed at 34 and 39°C, 15 of
15 tsA16 and 21of21tsA15 plaqueisolateswere
ts+ and produced very small plaques at 39°C
(data not shown). Weconclude, therefore, that
tsA15andtsA16 aredouble mutants containing a
tslesion andanadditionalsmall plaquemutation
present elsewhere in the genome. It is notable
that both tsA15 and tsA16 were isolated after
mutagenesis by
N-methyl-N'-nitro-N-nitroso-guanidine, a mutagen noted for its ability to
induce multiple mutations (4).
DISCUSSION
Complementationanalysis. We have described
the resultsof intra- andintertypic
complementa-tion tests with a series of DNA-negative ts mutants ofHSV-1 and HSV-2. HSV-1 mutants
tsAl, tsA15, tsA16, tsA24, tsA42,tsHAl, ts656,
and tsN103 are members of complementation
group 1-1. In a similar study, Dixon et al. (lla) haveshown that HSV-2 mutants tsH9,tsAl,and tsA8, which were used in this study, as wellas
four other HSV-2 mutants, are members ofa
singleHSV-2 complementation group, group
2-2. Because HSV-1 mutants ingroup 1-1 failed to
complement mutants ingroup2-2, we conclude
that the mutants in the two groups very likely
**: _*
on November 10, 2019 by guest
http://jvi.asm.org/
[image:9.489.45.244.59.375.2]TABLE 5. Marker rescue oftsAl, tsA15,tsA16, tsA24, andtsA42a
Plasmid or Map Marker rescue efficiencies (x103) with the following mutantDNAs:
fragmentb coordinates tsAl tsA15 tsA16 tsA24 tsA42
pSG18 0.315-0.422 25.1 35.0 10.0 12.1 20.8
pSG18-SA 0.385-0.419 4.0 2.2 10.2 7.9 3.7
pSG18-SD 0.360-0.385 NDC' 0.2d <0.005d ND <0.01d
pSG18-SE 0.342-0.360 ND 0.4d 0.005d ND <0.003d
pKOS10 0.345-0.398 32.0 13.4 <0.005d <0.002d 1.8
pKOS15 0.345-0.398 11.0 17.6 <0.005d <0.01d ND
pKEF-B5 0.315-0.398 4.0 2.1 <0.005d <0.01d 3.8
pKEF-P1 0.397-0.422 <0.03d 0.5d 7.8 1.0
<0.02d
BamHI-V 0.398-0.413 0.56d 0.8d 4.1 11.0 ND
None <0.01d <0.005d <0.005"d <0.05d
<0.02d
a Results areexpressed as platingefficiencies, which were determinedasfollows:PFU per milliliterat39°C/ PFU per milliliter at 34°C.
bPlasmidspSG18, pKEF-B5, andpKEF-P1 were digested with EcoRI, and the enzymewasinactivatedby incubation at 60°C for 10 min before transfection. Plasmids pSG18-SA, pSG18-SD, and pSG18-SE were digested with Sall, andpKOS10 and pKOS15 were digested withBamHI.The plasmids digested with Sall orBamHl were phenol extracted and ethanol precipitated before transfection. BamHl fragmentVwaselutedfrom an agarose gel by glass powder elution, as described in the text.
C ND, Notdone.
dValues considered to be negativefor marker rescue.
rF
el ti
Si
G
T of
ts
1-(I
K
epresent afunctioncommon toboth HSV-1 and maps between the Sall site at the b-ajunction
ISV-2. and the BamHI site at the G-V junction (map
Mappingstudies. In
preliminary
tests wedem- coordinates0.385to0.398)
(Fig.
5).
Conley
etal.nstrated that the mutations ofmutantsincom- have reported that mutant tsHAl was rescued
lementation
group 1-1 mapped to EcoRI frag-by
aSallsubfragment
obtained from aplasmid
ientF(mapcoordinates 0.315 to0.422)(Weller
(pRB102)
containing
BamHIfragment
G oftal., submitted forpublication). The fine-struc- HSV-1 strainF(11).
Although
therearediscrep-are mapping ofthe mutations of five members ancies between the locations of the KpnI and
If complementation group 1-1 reported here Sall sites in
pRB102
and theKpnI and Sall siteshows that the DNA fragments which rescue
reported
for strainF DNAby
LockerandFren-sAl,
tsA15, and tsA42 share a sequencewhich kel(26)
and for strainKOSDNAby
us(Fig. 3),
itappears thatthelocations of the ts mutations in
tsHAl, tsAl,
tsA15,and tsA42lie within ther, 0 ,, ,,, ^ b same 1.7-kb DNA sequence between the SalI
lkb ° 0° siteatthe b-a
junction
and theBamHI
siteattheG-Vjunction (Fig. 3 and5). Mutants tsA16 and
tsA24 were rescued by DNA fragments which
uE
B H BHB aGE
map withinBamHI
fragment
V(map
coordi-PKPKP PS S SPK P P S P KS
KS
nates 0.398 to 0.413) (Fig. 5). Taken together,thefragments whichrescue mutantsingroup 1-1
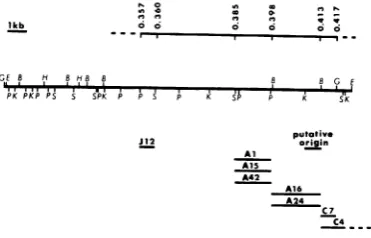
J12
putative
spanapproximately 4 kb on the map of theHSV-Al
A15 1 genome (map coordinates 0.385 to 0.413).
A42 Limits of the gene for the 130KDNA-binding
A24 C7 protein.The nearest marker in strain KOS to the C4. leftof the mutations in theDNA-binding protein
(i.e., mutants in group 1-1) is the tsJ12 marker FIG. 5. Physicalmap of eight HSV-1 strain KOSts (group 1-9), which maps between coordinates
utantsand aputative origin of viralDNAsynthesis. 0.357 and 0.360 (N. DeLuca, personal communi-he line at tcommuni-hetop shows the physical mapcoordinates cation) and probably lies in the structural gene ftheregion. The map locations of the tsJW2, tsAl, for viralglycoprotein gAgB (Fig. 5). This marker
{A1S, tsAl6, tsA24, tsC4,
and tsC7mutations
are liesapproximately 4 kb to the left of the locationhown.ThemutationoftsJ12(complementation group
-9) was mapped by marker rescue by Neal DeLuca
of
themutations
tsA1,
tsAl5,
andtsA42in
group
)ersonal communication), and the mutations of tsC4 1-1. Althoughwe do not know the precise physi-nd tsC7 weremappedbyanalysis of intertypic recom- cal limits of the genes forgAgB and the 130K inants(6). B,
BamHI;
E,EcoRI;
G,BglIl;
H, Hpal;protein,
there is sufficientgenetic
information>,
KpnI;
P,PstI;
S,Sal,.
between the tsJ12 (gAgB) mutation and theVOL. 45,1983
on November 10, 2019 by guest
http://jvi.asm.org/
[image:10.489.52.238.452.571.2]mutations of members of complementation
group 1-1 for at least one additional gene.
In-deed, in the HSV-2 system a gene coding fora
late function maps in this region (lla). The
nearest markerstothe right ofthemutations in
group 1-1 are in the DNA polymerase locus
represented by mutants tsC4 andtsC7 (Fig. 5).
The mutations in the polymerase gene map
between coordinates 0.398 and 0.438 (6; D.
Coen, personal communication).
Relationship of mutants in complementation group 1-1 to the 130K DNA-binding protein.
Several pieces of evidence indicate that the
mutations of members of groups 1-1 and 2-2
define thegenefor the 130K DNA-binding
pro-tein (11, lla; this paper). First, members of
group 1-1 fail to complement tsHAl, a mutant
which was isolated by Conley et al. (11) and
whose mutation maps in or near sequences
specifying a 130K DNA-binding protein (i.e.,
ICP8). The complementation and mapping
re-sults described in this study areconsistent with
this interpretation.
Second, thepurified 130Kproteinfrom
HSV-2tsH9-infectedcellsexhibitsareduced ability to
denaturepolydeoxyadenylic
acid-polydeoxythy-midylic acid helices at 40°C compared with
purified
130K protein from wild-typevirus-infected cells (32).
Third, immunofluorescencetests have shown
that, whereas cells infected with strain KOS
exhibit bright nuclear fluorescence at both 34
and 39°C when theyarereacted with anti-130K
monoclonal antibody, cells infected withtsHAl,
tsAl,
tsA15, tsA16, and tsA42 exhibit nuclearfluorescence at 34 but not 39°C. In addition,
cellsinfected withtsmutants in HSV-2
comple-mentationgroup 2-2also failedtoexhibit
detect-able nuclear fluorescence at 38°C in tests in
which thewild-type virus (strain 186)exhibited
brilliant
nuclearfluorescence(hla).
Importantly,immunofluorescencetestswith
polypeptide-spe-cific antiserum to the 130K protein confirmed
the absence of this protein in nuclei of cells
infected with selected members of
complemen-tationgroups 1-1 and 2-2 (R.Courtney,personal
communication). These observations indicate
that all members of groups 1-1 and 2-2 bear
mutations which affect the distribution of the
130KDNA-binding protein at thenonpermissive
temperature. Takentogether, these data
strong-ly suggest that mutants in HSV-1 and HSV-2
complementationgroups 1-1 and2-2define
anal-ogous gene(s) for the 130K DNA-binding
pro-tein.
Significance of the DNA-binding protein in
replication and transformation. To date, we
knowthat the 130K protein is required for HSV
DNA synthesis and that it may act to stabilize
DNA polymerase activity (K. Powell, personal
communication). Antiserum to theHSV-2
DNA-binding protein (ICSP11/12) has been shownto
react with biopsy material from patients with
severe dysplasia and carcinoma of the cervix,
squamous cell carcinoma in situ, and severe
dysplasia ofthe vulva (13, 22). Moreover, the
expression of the antigen in hamster cells
trans-formed in vitro by UV-inactivated HSV (15) has
beencorrelated with the tumorigenicityof these
cells when theyareinjected into newborn
ham-sters (unpublished data).
Thepossibility thatthe DNA-binding protein
mayplayarole inmorphologicaltransformation
andtumorigenicity is currently under
investiga-tion. Mutants with mutations affecting the
expression of the DNA-binding protein should
prove useful in elucidatingthefunctions ofthis
protein both in the viral replicative process and
in transformation. The region of the genome
between map coordinates 0.385 and0.460
con-tainsavariety of essentialreplicative functions,
including thegenefortheDNA-binding protein,
a putative origin of DNA replication, and the
HSVDNApolymerase gene. Thepossible
anal-ogy between HSV and the papovaviruses, in
which the essential transforming gene (a gene
encoding an early protein required for viral
DNA synthesis) maps adjacent to the origin of
DNA synthesis, deserves careful consideration.
ACKNOWLEDGMENTS
Wethank NealDeLucaandDavid Knipe for communicat-ing unpublished data andB.Pancake, D. Coen,L.Cohen,D. Knipe, and R. Respess for useful comments on the manu-script.Wealso thank M. Datz formanuscriptpreparation.
This investigationwassupported by Public Health Service grants CA20260 and CA21082 from the National Cancer Institute and grant MV-77 from the American Cancer Society. S.K.W. is the recipient of Public Health Service fellowship CA06836 fromtheNationalCancerInstitute, and D.J.S.was supported by Public Health Service training grant CA09031 from the National Cancer Institute.
LITERATURECITED
1. Aron, G.M.,D.J.M.Purifoy,and P. A.Schaffer. 1975. DNA synthesisand DNApolymeraseactivityofherpes
simplex virus type 1 temperature-sensitive mutants. J. Virol. 16:498-507.
2. Aron,G. M., P. A.Schaffer,R.J.Courtney,M. Benyesh-Melnick, and S. Kit. 1973. Thymidinekinaseactivityof herpes simplex virustemperature-sensitive mutants. In-tervirology1:96-109.
3. Bayliss, G. J.,H.S.Marsden, andJ. Hay. 1975.Herpes
simplexvirusproteins:DNA-bindingproteinsin infected cells and in the virusstructure. Virology68:124-134. 4. Botstein, D., and E. W.Jones.1969.Nonrandom
mutagen-esis oftheEscherichia coligenomebynitrosoguanidine. J. Bacteriol.98:847-848.
5. Camacho, A.,and P.G.Spear. 1978.Transformation of hamsterembryo fibroblastsbyaspecific fragment ofthe herpessimplex virus genome. Cell 15:993-1002. 6. Chartrand, P., C. S. Crumpacker, P. A. Schaffer, and
N. M.Wilkie.1980. Physicalandgeneticanalysisof the herpes simplex virus DNA polymerase locus. Virology 103:311-326.
7. Chartrand, P., N. D. Stow,M.C. Timbury, and N.M. Wilkie. 1979. Physical mapping ofpaar mutations of
on November 10, 2019 by guest
http://jvi.asm.org/
herpes simplex virus type 1 and type 2 by intertypic markerrescue.J.Virol.31:265-276.
8. Chu,C.-T., and P. A. Schaffer.1975. Qualitative comple-mentationtestfortemperature-sensitive mutantsof
her-pessimplex virus.J. Virol. 16:1131-1136.
9. Clewell, D. B., and D. R. Helinski. 1969. Supercoiled circularDNA-protein complex in Escherichiacoli: purifi-cation and induced conversiontoan opencircularDNA
form.Proc.Natl. Acad.Sci. U.S.A. 62:1159-1166. 10. Coen, D. M., R. A. F. Dixon, S.W. Ruby, and P. A.
Schaffer. 1980. Genetics of acycloguanosine resistance and the thymidine kinasegene in HSV-1, p. 581-590. In
B. N. Fields, R. Jaenisch, and C. F. Fox (ed.), Animal
virus genetics. ICN-UCLA Symposia on Molecular and
Cellular Biology, vol. 18. Academic Press, Inc., New York.
11. Conley, A. J., D. M. Knipe, P. C.Jones,andB.Roizman. 1981. Molecular genetics of herpes simplex virus. VII.
Characterization ofa temperature-sensitive mutant
pro-duced by in vitro mutagenesis and defective in DNA synthesis and accumulation ofy polypeptides. J. Virol.
37:191-206.
11a.Dixon,R.A. F., D. J.Sabourin, andP. A.Schaffer. 1983.
Genetic analysisoftemperature-sensitive mutantswhich define thegenesfor the majorherpessimplexvirustype2
DNA-binding protein anda new late function. J. Virol. 45:343-353.
12. Dixon,R. A. F., andP. A.Schaffer. 1980. Fine-structure
mapping and functional analysis oftemperature-sensitive
mutants in the gene encoding the herpes simplex virus
type 1 immediate early protein VP175. J. Virol.
36:189-203.
13. Dreesman, G. R., J. Burek, E. Adam, R. H. Kaufman, J. L. Melnick, K. L. Powell, and D. J. M.Purifoy. 1980.
Expression of herpesvirus-induced antigens in human
cervicalcancer. Nature(London) 283:591-593.
14. Esparza, J., D. J. M. Purifoy, P. A. Schaffer, and M.
Benyesh-Melnick. 1974. Isolation, complementation and
preliminary phenotypic characterization of
temperature-sensitivemutantsof herpes simplexvirustype2.Virology
57:554-565.
15. Flannery, V. L., R. J.Courtney,andP.A.Schaffer.1977. Expression ofan early, nonstructural antigen of herpes
simplex virus in cells transformed in vitro by herpes simplex virus.J. Virol. 21:284-291.
16. Frenkel, N.,H.Locker, andD.A.Vlazny.1980.Studies of
defective herpes simplex viruses. Ann. N. Y. Acad.Sci. 354:347-370.
17. Goldin,A.L., R. M.Sandri-Goldin, M.Levine, and J.C. Glorioso. 1981. Cloning of herpes simplex virus type 1
sequences representing the whole genome. J. Virol.
38:50-58.
18. Graham, F. L., and A. J. Van der Eb. 1973. A new
techniquefor theassayofinfectivity of humanadenovirus
5DNA. Virology52:456-467.
19. Honess,R. W., and B.Roizman. 1973. Proteinsspecified
by herpes simplex virus. XI. Identification and relative
molar rates ofsynthesis of structural and nonstructural
herpes virus polypeptides in the infected cell. J. Virol.
12:1347-1365.
20. Hughes, R. G.,and W. H. Munyon. 1975.
Temperature-sensitivemutantsofherpessimplex virustype1defective inlysisbut notintransformation. J. Virol. 16:275-283.
21. Kaerner, H. C.,I.B.Maichle, A.Ott, andC. H.Schroder.
1979. Originoftwo different classes ofdefective HSV-1
Angelotti DNA. Nucleic Acids Res. 6:1467-1478. 22. Kaufman, R. H., G. R. Dreesman, J. Burke, M. 0.
Korhonen, D.0. Matson, J.L. Melnick, K.L. Powell,
D. J. M. Purifoy, R. J. Courtney, and E. Adam. 1981. Herpesvirus-induced antigens in squamous-cell
carcino-mainsituofthe vulva. N. Engl.J. Med. 305:483-488.
23. Leung, W.-C., B. Fong, J. Zwaagstra, and M. Leung.
1981. MolecularcloningofHSV-1specificcx polypeptide
ICP4 and glycoprotein gA/gB genes, p. 46. In A. S. Kaplan, M. LaPlaca, F. Rapp. and B. Roizman (ed.),
Internationalworkshoponherpesviruses. Esculapio Pub-lishingCo., Bologna,Italy.
24. Little, S. P., J. T. Jofre, R. J. Courtney, and P. A. Schaffer. 1981. Avirion-associatedglycoproteinessential for infectivity of herpes simplex virus type 1. Virology 115:149-160.
25. Littler, E., J. Yeo, R. A. Killington, D.J.M.Purifoy,and K. L. Powell.1981.Antigenic and structural conservation of herpes virus DNA-binding proteins. J. Gen. Virol. 56:409-419.
26. Locker, H., and N. Frenkel. 1979. BainI,KptlI.andSall restrictionenzyme mapsofthe DNAs ofherpes simplex virus strains Justin and F: occurrenceofheterogeneities in defined regions oftheviral DNA. J.Virol. 32:429-441. 27. Maniatis, T., G. K. Sim, A. Efstratiatis, and F.Kafatos.
1976. Amplification and characterization of a ,B-globin gene synthesized in vitro. Cell 8:163-182.
28. Parker, R. C., and B. Seed. 1980.Two-dimensional agar-ose gel electrophoresis "SeaPlaque" agarose dimension. Methods Enzymol. 65:358-363.
29. Parris, D. S., R. A. F. Dixon, and P.A. Schaffer. 1980. Physical mapping ofherpes simplexvirus type 1 ts mu-tants by marker rescue: correlation ofthe physical and genetic maps. Virology 100:275-287.
30. Porter, D. D., I. Wimberly, and M. Benyesh-Melnick. 1969. Prevalence of antibodies to EB virus and other herpesviruses. J.Am. Med.Assoc. 208:1675-1679. 31. Post, L. E., A. J.Conley,E. S.Mocarski,and B. Roizman.
1980. Cloningof reiterated andnonreiterated herpes sim-plex virus 1 sequences as BamHI fragments. Proc. Natl. Acad. Sci. U.S.A.77:4201-4205.
32. Powell, K. L., E. Littler, R. A. Killington, andD. J. M. Purifoy. 1981. Herpes simplex virus major DNA binding protein, p. 14. Itn A. S. Kaplan. M.LaPlaca, F.Rapp,and B. Roizman (ed.). International workshop on herpesvi-ruses. Esculapio Publishing Co., Bologna.Italy. 33. Powell, K. L., E. Littler, and D.J. M. Puritoy. 1981.
Nonstructural proteins of herpessimplex virus. II. Major virus-specific DNA-binding protein.J. Virol.39:894-902. 34. Preston, C. M. 1979.Control of herpessimplex virustype 1 mRNAsynthesis in cellsinfectedwithwild-typevirus or the temperature-sensitive mutant tsK. J. Virol. 29:275-284.
35. Purifoy, D. J. M., R. B. Lewis, and K. L. Powell. 1977. Identification ofthe herpessimplex virus DNA polymer-ase gene. Nature (London)269:621-623.
36. Purifoy, D. J. M.,and K.L. Powell. 1976. DNA-binding proteinsinducedbyherpes simplexvirustype 2 inHEp-2 cells.J. Virol. 19:717-731.
37. Purifoy, D. J.M., and K. L. Powell. 1981. Temperature-sensitive mutantsintwo distinctcomplementation groups ofherpes simplex virustype 1 specifythermolabile DNA polymerase.J. Gen. Virol. 54:219-222.
38. Reyes, G. R., R. LaFemina, S.D. Hayward, and G. S. Hayward. 1980. Morphological transformation by DNA fragments of human herpesviruses: evidence for two distincttransforming regionsinherpessimplexvirus types 1 and 2and lack ofcorrelation withbiochemical transfer of thethymidine kinase gene. Cold SpringHarbor Symp. Quant. Biol. 44:629-641.
39. Ruyechan, W. T., L. S. Morse, D. M. Knipe, and B. Roizman. 1979. Molecular genetics of herpes simplex virus.II. Mappingofthe major viralglycoproteins and of thegeneticloci specifying the social behavior ofinfected cells. J. Virol.29:677-697.
40. Sarmiento, M.,M. Haffey, andP. G. Spear. 1979. Mem-brane proteins specified by herpes simplex viruses. III. RoleofglycoproteinVP7(B2)invirioninfectivity.J.Virol. 29:1149-1158.
41. Schaffer, P.A., G. M. Aron,N. Biswal, and M. Benyesh-Melnick. 1973. Temperature-sensitive mutants ofherpes simplex virus type 1: isolation, complementation and partial characterization. Virology 52:57-71.
42. Schaffer, P. A., V. C. Carter,and M.C. Timbury. 1978. Collaborative complementation study of
on November 10, 2019 by guest
http://jvi.asm.org/
sensitivemutantsofherpes simplex virustypes1and 2.J. Virol. 27:490-504.
43. Sedat, J. W., R. B. Kelly, and R. L. Sinsheimer. 1967. Fractionation of nucleic acidon
benzoylated-naphthoylat-edDEAEcellulose.J.Mol. Biol. 26:537-540.
44. Showalter,S. D., M.Zweig, andB.Hampar.1981. Mono-clonalantibodiestoherpes simplex virustype1proteins, including the immediate-early protein ICP4. Infect.
Im-mun.34:684-692.
45. Skare, J., and W. C. Summers. 1977. Structure and function ofherpesvirus genomes. II. EcoRI, XbaI, and HindIII endonuclease cleavage siteson herpes simplex
virustype1 DNA.Virology 76:581-595.
46. Southern, E. M. 1975. Detection of specific sequences amongDNA fragments separated by gel electrophoresis. J. Mol. Biol. 98:503-517.
47. Vogelstein, B., and D. Gillespie. 1979. Preparative and analyticalpurification ofDNAfromagarose.Proc. Natl. Acad. Sci. U.S.A. 76:615-619.
48. Wigler, M., S. Silverstein, L.-S. Lee, A. Pellicer, Y.-C. Cheng, and R. Axel. 1977. Transfer of purified herpes virus thymidinekinasegenetoculturedmousecells. Cell
11:223-232.
49. Yeo, J., R. A. Killington, D. H. Watson, and K. L. Powell. 1981. Studiesoncross-reactive antigens in the
herpesvi-ruses.Virology108:256-266.
J. VIROL.